

The Effect of Arginine on the Phase Stability of Aqueous Hen Egg-White Lysozyme Solutions


Abstract
1. Introduction
2. Results and Discussion
2.1. Added Arginine Increments the Phase Stability of HEWL Solutions—Experimental Observation
2.2. MD Simulations Show That Arginine Molecules Have a Strong Tendency toward Self-Association
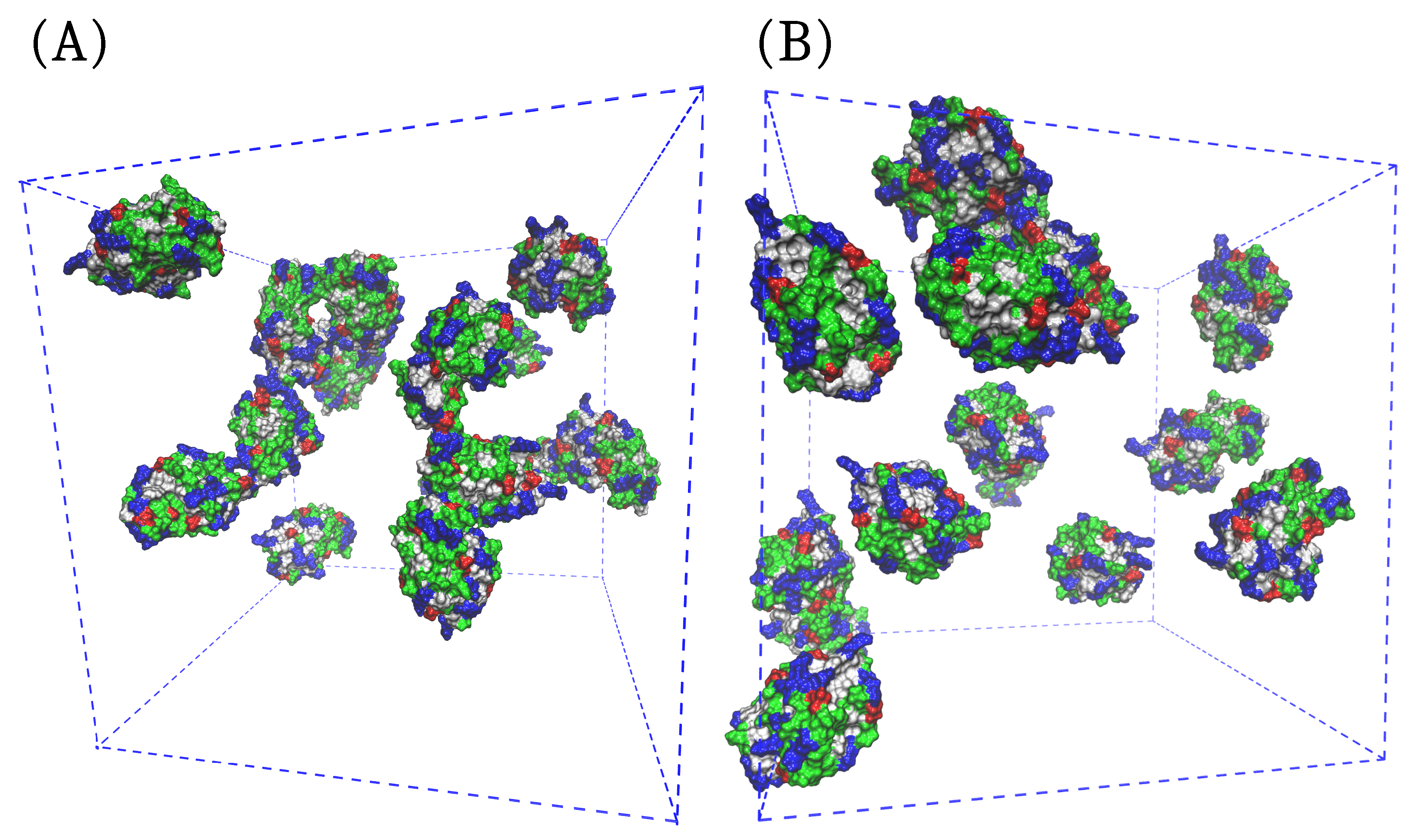
2.2.1. Addition of Arginine Reduces the Self-Association of HEWL
2.2.2. Changing the Force Field in Our Case Does Not Affect the Influence of Arginine on HEWL
3. Materials and Methods
3.1. Materials
3.2. Experimental Methods
3.2.1. NaBr-ACES Arginine Solutions
3.2.2. HEWL-ACES Solutions
3.2.3. Cloud-Point Measurements
3.3. MD Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACES | 2-[(2-Amino-2-oxoethyl)amino]ethane-1-sulfonic acid |
| COM | center of mass |
| HEWL | hen egg-white lysozyme |
| MD | molecular dynamics |
| PDB | Protein Data Bank |
| PME | particle-mesh Ewald |
| cloud-point temperature |
References
- van der Linden, E.; Venema, P. Self-assembly and aggregation of proteins. Curr. Opin. Colloid Interface Sci. 2007, 12, 158–165. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 2005, 289, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Ahmed, S.; Sharma, N.; Kumar, N.; Debas, A.; Matsumura, K. Review of the current state of protein aggregation inhibition from a materials chemistry perspective: Special focus on polymeric materials. Mater. Adv. 2021, 2, 1139–1176. [Google Scholar] [CrossRef]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation—Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Roberts, C.J. Protein aggregation—Mechanisms, detection, and control. Int. J. Pharm. 2018, 550, 251–268. [Google Scholar] [CrossRef]
- Baynes, B.M.; Wang, D.I.C.; Trout, B.L. Role of Arginine in the Stabilization of Proteins against Aggregation. Biochemistry 2005, 44, 4919–4925. [Google Scholar] [CrossRef]
- Lange, C.; Rudolph, R. Suppression of Protein Aggregation by L-Arginine. Curr. Pharm. Biotechnol. 2009, 10, 408–414. [Google Scholar] [CrossRef]
- Shukla, D.; Trout, B.L. Interaction of Arginine with Proteins and the Mechanism by Which It Inhibits Aggregation. J. Phys. Chem. B 2010, 114, 13426–13438. [Google Scholar] [CrossRef]
- Schneider, C.P.; Shukla, D.; Trout, B.L. Arginine and the Hofmeister Series: The Role of Ion–Ion Interactions in Protein Aggregation Suppression. J. Phys. Chem. B 2011, 115, 7447–7458. [Google Scholar] [CrossRef]
- Březina, K.; Duboué-Dijon, E.; Palivec, V.; Jiráček, J.; Křižek, T.; Viola, C.M.; Ganderton, T.R.; Brzozowski, A.M.; Jungwirth, P. Can Arginine Inhibit Insulin Aggregation? A Combined Protein Crystallography, Capillary Electrophoresis, and Molecular Simulation Study. J. Phys. Chem B 2018, 122, 10069–10076. [Google Scholar] [CrossRef]
- Mamsa, S.S.A.; Meloni, B.P. Arginine and Arginine-Rich Peptides as Modulators of Protein Aggregation and Cytotoxicity Associated with Alzheimer’s Disease. Front. Mol. Neurosci. 2021, 14, 759729. [Google Scholar] [CrossRef]
- Das, U.; Hariprasad, G.; Ethayathulla, A.S.; Manral, P.; Das, T.K.; Pasha, S.; Mann, A.; Ganguli, M.; Verma, A.K.; Bhat, R.; et al. Inhibition of Protein Aggregation: Supramolecular Assemblies of Arginine Hold the Key. PLoS ONE 2007, 2, e1176. [Google Scholar] [CrossRef]
- Tomita, S.; Yoshikawa, H.; Shiraki, K. Arginine controls heat-induced cluster–cluster aggregation of lysozyme at around the isoelectric point. Biopolymers 2011, 95, 695–701. [Google Scholar] [CrossRef]
- Brudar, S.; Gujt, J.; Spohr, E.; Hribar-Lee, B. Studying the mechanism of phase separation in aqueous solutions of globular proteins via molecular dynamics computer simulations. Phys. Chem. Chem. Phys. 2021, 23, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Brudar, S.; Hribar-Lee, B. Effect of Buffer on Protein Stability in Aqueous Solutions: A Simple Protein Aggregation Model. J. Phys. Chem. B 2021, 125, 2504–2512. [Google Scholar] [CrossRef]
- Nikolić, M.; Brudar, S.; Coutsias, E.; Dill, K.A.; Lukšič, M.; Simmerling, C.; Hribar-Lee, B. BioMThermDB 1.0: Thermophysical Database of Proteins in Solutions. Int. J. Mol. Sci. 2022, 23, 5371. [Google Scholar] [CrossRef]
- Janc, T.; Kastelic, M.; Bončina, M.; Vlachy, V. Salt-specific effects in lysozyme solutions. Condens. Matter Phys. 2016, 19, 23601. [Google Scholar] [CrossRef]
- Čančar, H.D.; Vivod, M.B.; Vlachy, V.; Lukšič, M. Phase stability of aqueous mixtures of bovine serum albumin with low molecular mass salts in presence of polyethylene glycol. J. Mol. Liq. 2022, 349, 1–19. [Google Scholar] [CrossRef]
- Mittal, J.; Best, R.B. Tackling Force-Field Bias in Protein Folding Simulations: Folding of Villin HP35 and Pin WW Domains in Explicit Water. Biophys. J. 2010, 99, L26–L28. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, K.A.; Lin, Y.S.; Das, R.; Pande, V.S. Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements. J. Chem. Theory Comput. 2012, 8, 1409–1414. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force Fields for Protein Simulations. In Protein Simulations; Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 2003; Volume 66, pp. 27–85. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic Validation of Protein Force Fields against Experimental Data. PLoS ONE 2012, 7, e32131. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Pritchard, R.B.; Hansen, D.F.; Gervasio, F.L. Importance of the Force Field Choice in Capturing Functionally Relevant Dynamics in the von Willebrand Factor. J. Phys. Chem. Lett. 2019, 10, 1928–1934. [Google Scholar] [CrossRef]
- Okur, A.; Strockbine, B.; Hornak, V.; Simmerling, C. Using PC clusters to evaluate the transferability of molecular mechanics force fields for proteins. J. Comput. Chem. 2003, 24, 21–31. [Google Scholar] [CrossRef]
- Aune, K.C.; Tanford, C. Thermodynamics of the denaturation of lysozyme by guanidine hydrochloride. II. Dependence on denaturant concentration at 25°. Biochemistry 1969, 8, 4586–4590. [Google Scholar] [CrossRef] [PubMed]
- Artymiuk, P.; Blake, C.C.F.; Rice, D.W.; Wilson, K.S. The Structures of the Monoclinic and Orthorhombic Forms of Hen Egg-White Lysozyme at 6 Å Resolution. Acta. Cryst. 1982, B38, 778–783. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2020, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Taratuta, V.G.; Holschbach, A.; Thurston, G.M.; Blankschtein, D.; Benedek, G.B. Liquid-liquid phase separation of aqueous lysozyme solutions: Effects of pH and salt identity. J. Phys. Chem. 1990, 94, 2140–2144. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ResID | ResNAME | SimTIME/% |
|---|---|---|
| 101 | ASP | 89.8 ± 2.3 |
| 18 | ASP | 77.8 ± 6.5 |
| 19 | ASN | 74.8 ± 5.9 |
| 128 | ARG | 71.5 ± 4.8 |
| 62 | TRP | 70.3 ± 5.9 |
| 73 | ARG | 68.5 ± 6.4 |
| 35 | GLU | 62.8 ± 7.3 |
| 21 | ARG | 57.7 ± 6.4 |
| 44 | ASN | 54.0 ± 4.6 |
| 61 | ARG | 53.5 ± 8.5 |
| 7 | GLU | 53.4 ± 6.9 |
| 14 | ARG | 53.1 ± 3.9 |
| 45 | ARG | 51.8 ± 6.4 |
| 125 | ARG | 51.7 ± 7.3 |
| 129 | LEU | 50.7 ± 5.7 |
| 68 | ARG | 50.2 ± 6.6 |
| 46 | ASN | 46.0 ± 8.6 |
| 48 | ASP | 43.9 ± 7.2 |
| 52 | ASP | 43.2 ± 9.7 |
| 112 | ARG | 42.9 ± 6.8 |
| ResNAME | TotalTIME/% | NumRESIDUES |
|---|---|---|
| GLU | 58.1 ± 7.1 | 2 |
| ARG | 52.2 ± 6.6 | 11 |
| ASP | 46.4 ± 6.1 | 7 |
| ASN | 26.1 ± 4.5 | 14 |
| PRO | 23.9 ± 4.1 | 2 |
| TRP | 20.8 ± 3.5 | 6 |
| GLN | 20.3 ± 6.5 | 3 |
| LYS | 19.4 ± 4.1 | 6 |
| GLY | 16.8 ± 3.3 | 12 |
| THR | 15.6 ± 3.5 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brudar, S.; Hribar-Lee, B. The Effect of Arginine on the Phase Stability of Aqueous Hen Egg-White Lysozyme Solutions. Int. J. Mol. Sci. 2023, 24, 1197. https://doi.org/10.3390/ijms24021197
Brudar S, Hribar-Lee B. The Effect of Arginine on the Phase Stability of Aqueous Hen Egg-White Lysozyme Solutions. International Journal of Molecular Sciences. 2023; 24(2):1197. https://doi.org/10.3390/ijms24021197
Chicago/Turabian StyleBrudar, Sandi, and Barbara Hribar-Lee. 2023. "The Effect of Arginine on the Phase Stability of Aqueous Hen Egg-White Lysozyme Solutions" International Journal of Molecular Sciences 24, no. 2: 1197. https://doi.org/10.3390/ijms24021197
APA StyleBrudar, S., & Hribar-Lee, B. (2023). The Effect of Arginine on the Phase Stability of Aqueous Hen Egg-White Lysozyme Solutions. International Journal of Molecular Sciences, 24(2), 1197. https://doi.org/10.3390/ijms24021197