
Bioinformatics and Connectivity Map Analysis Suggest Viral Infection as a Critical Causative Factor of Hashimoto’s Thyroiditis


Abstract
1. Introduction
2. Results
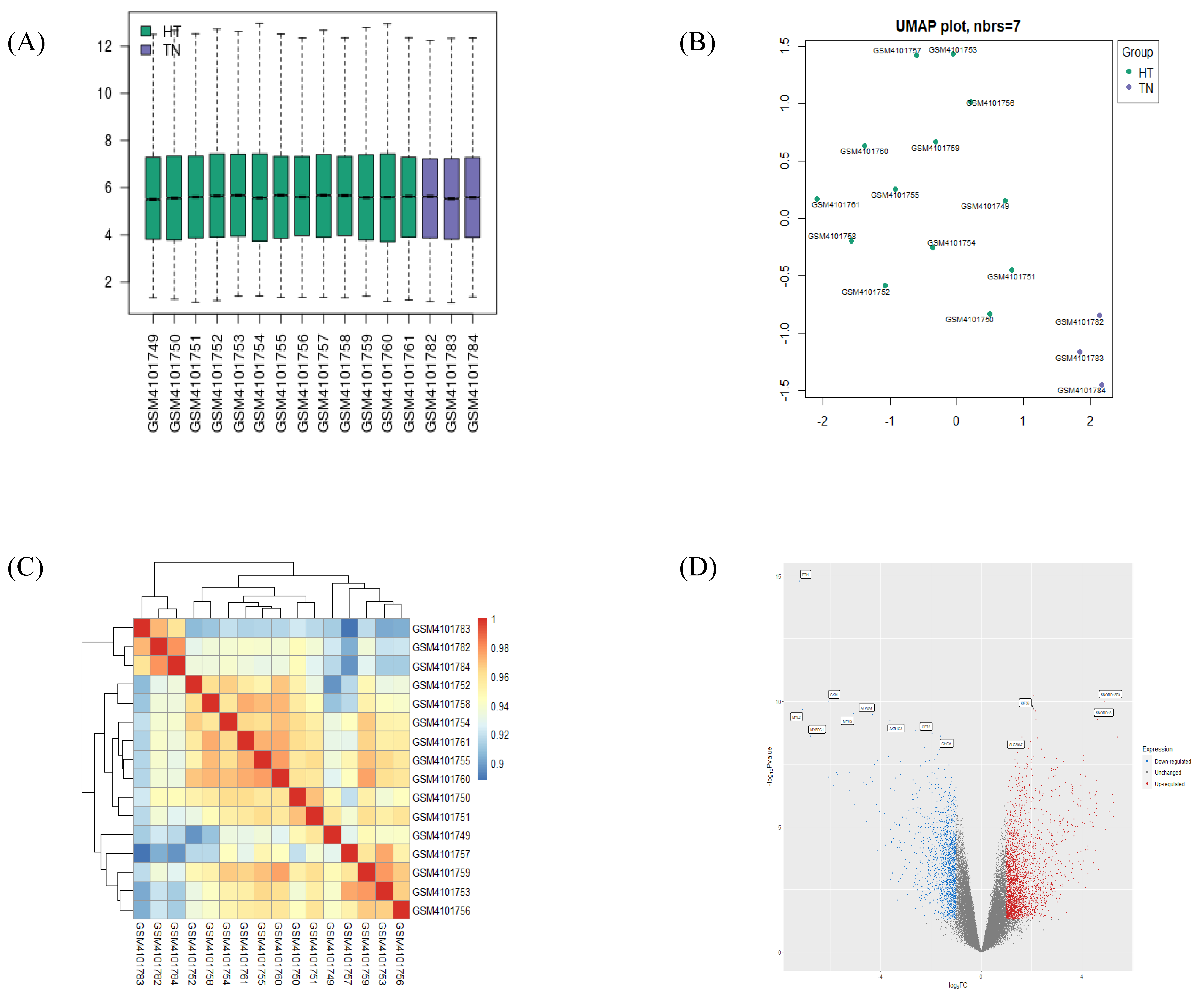
2.1. Validation of GEO Data
2.2. Identification of DEGs Extracted from HT
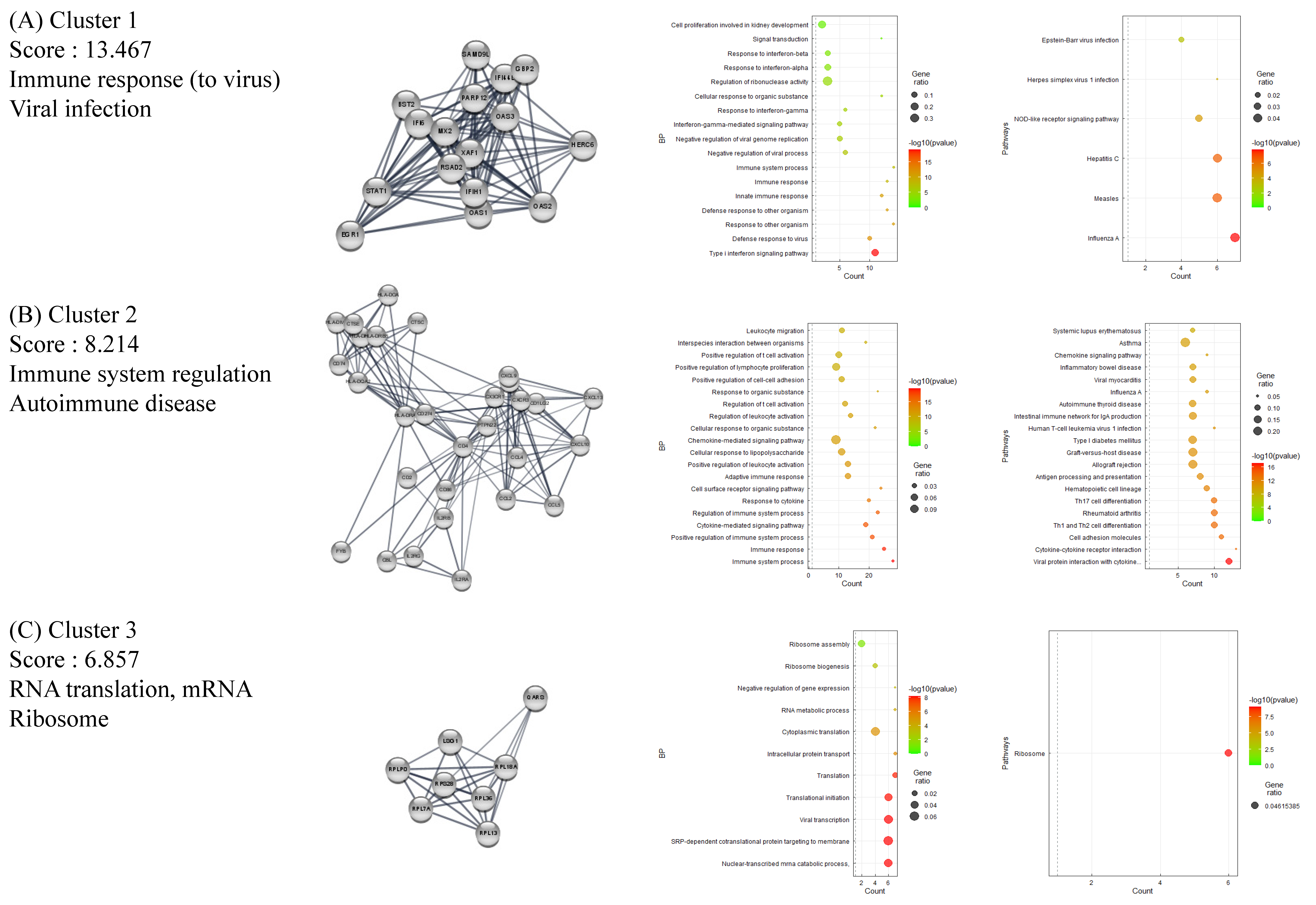
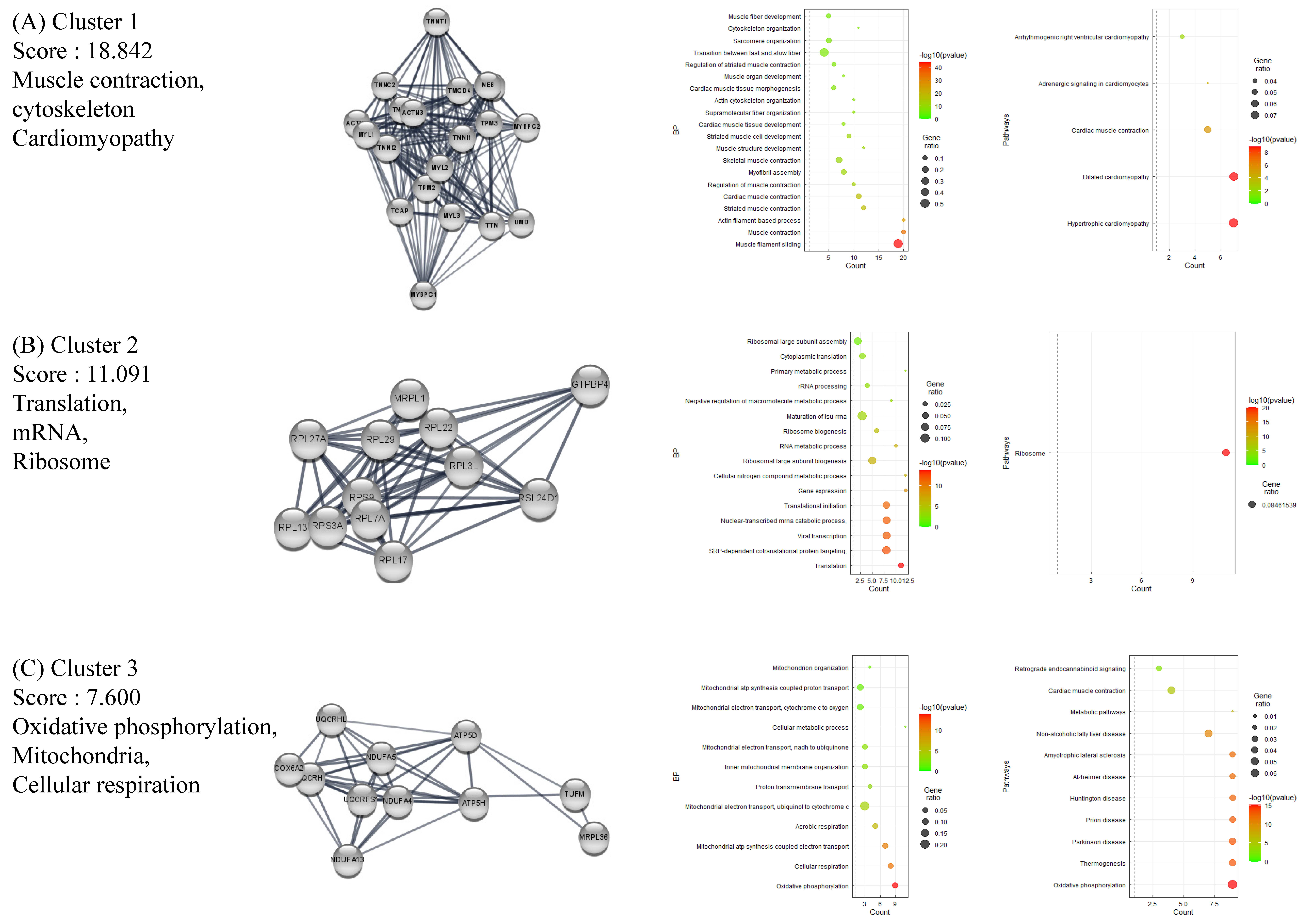
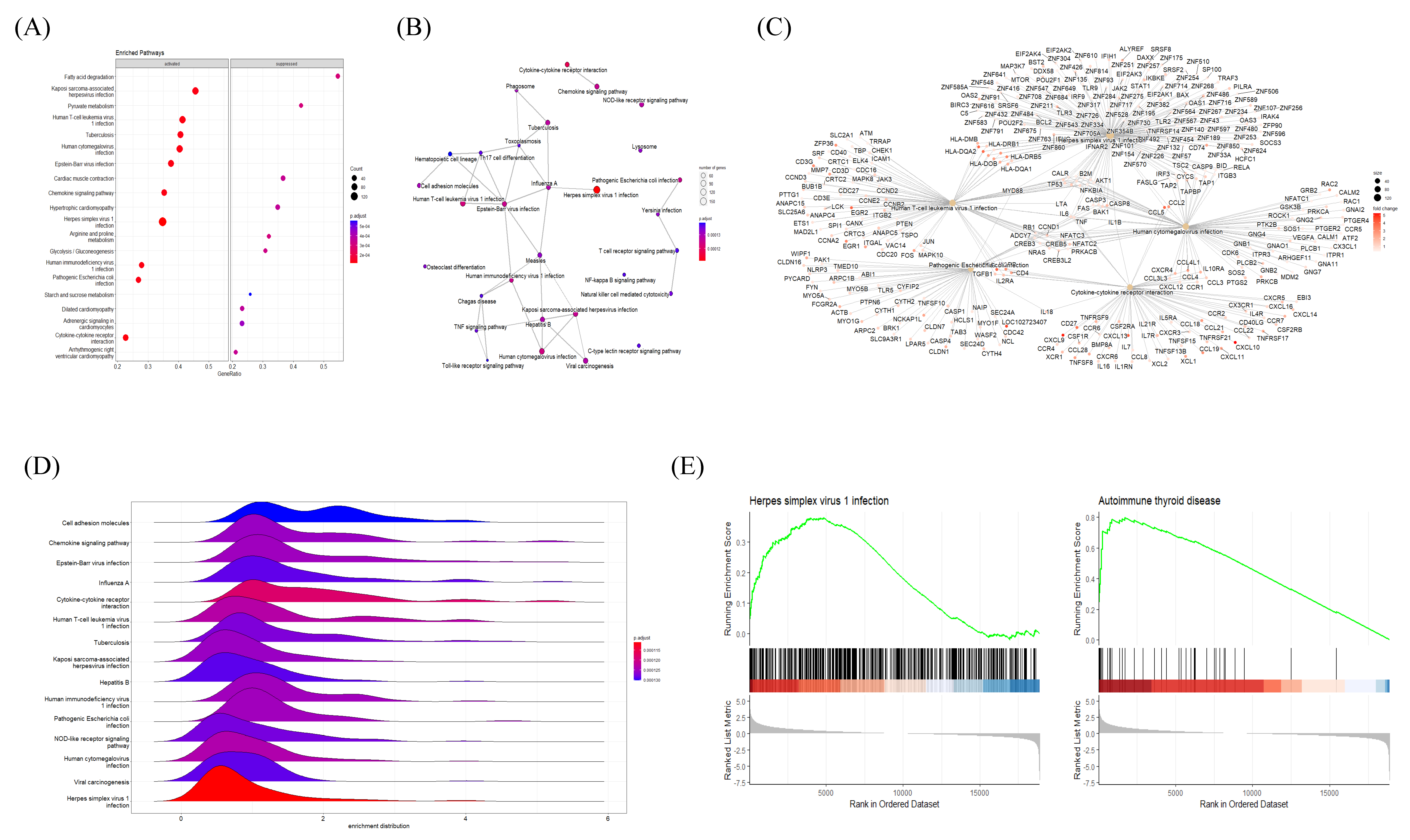
2.3. Functional Clustering Analysis of DEGs Reveals the Pathological Characteristics of HT
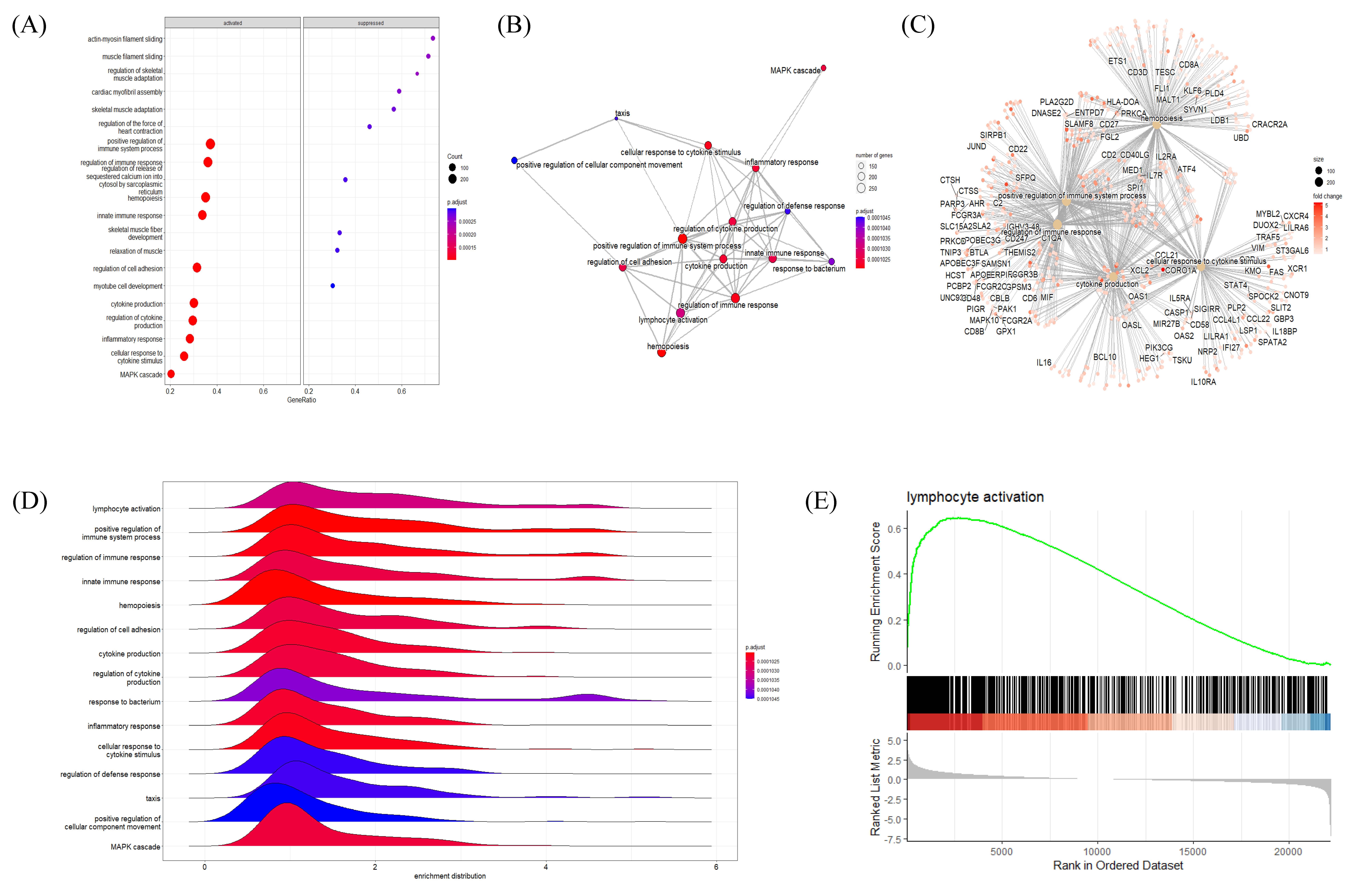
2.4. BP GSEA Results Revealed a Strong Connection between HT and Immune Regulation
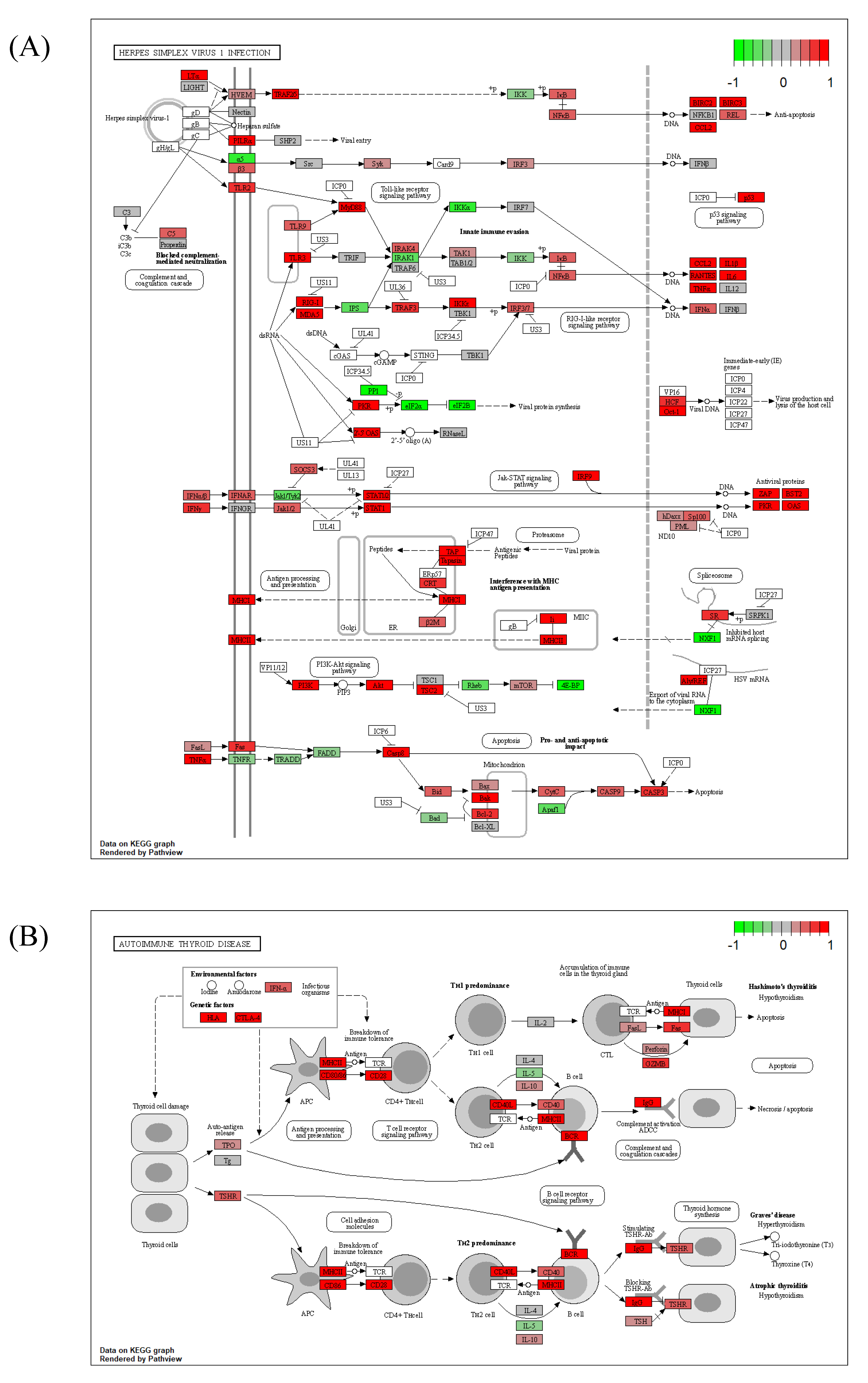
2.5. GSEA Results Suggested a Link between Autoimmune Thyroiditis and Viral Infections
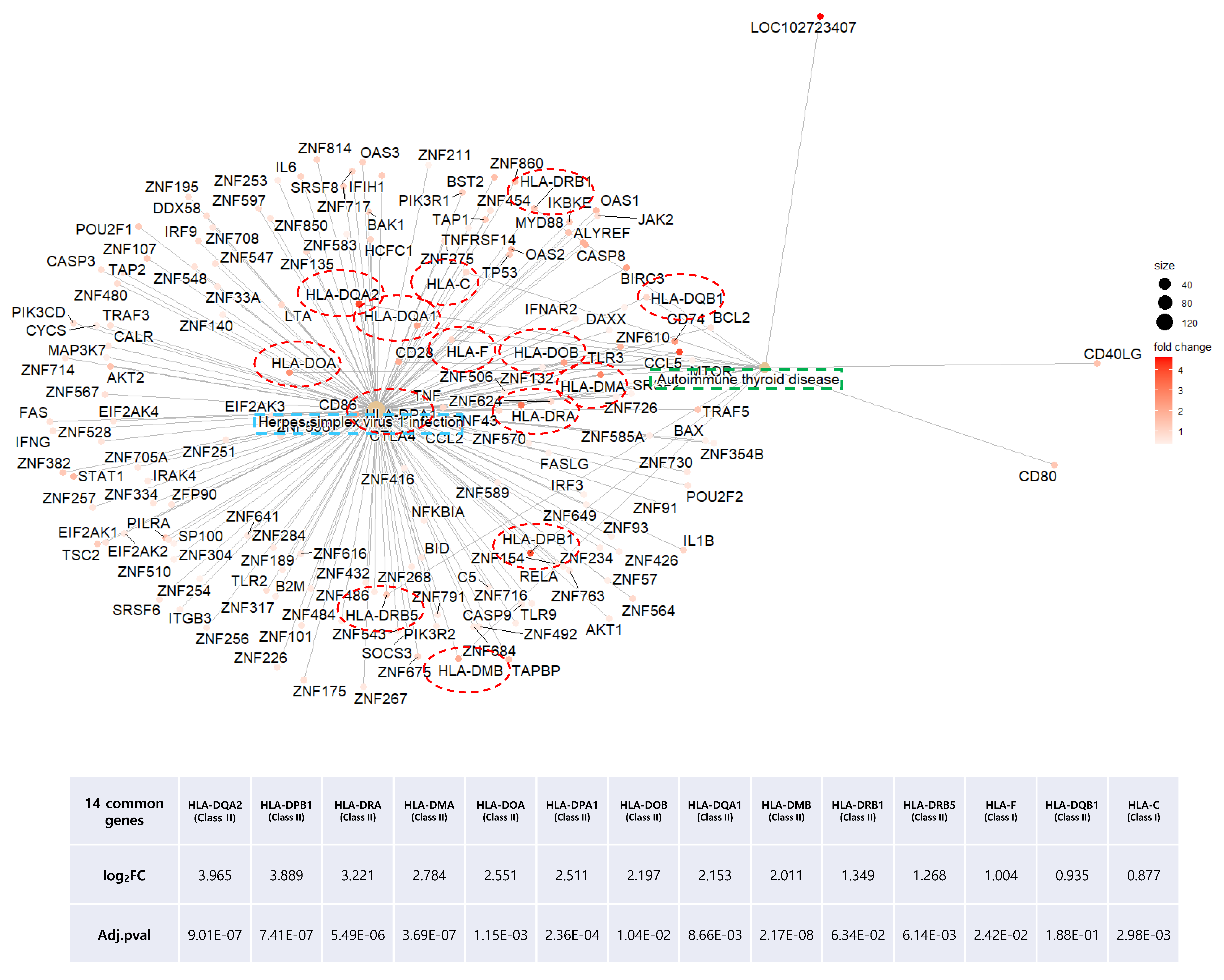
2.6. Common DEGs Involved in Two Intuitive Pathways Provide Insight into Disease Etiology
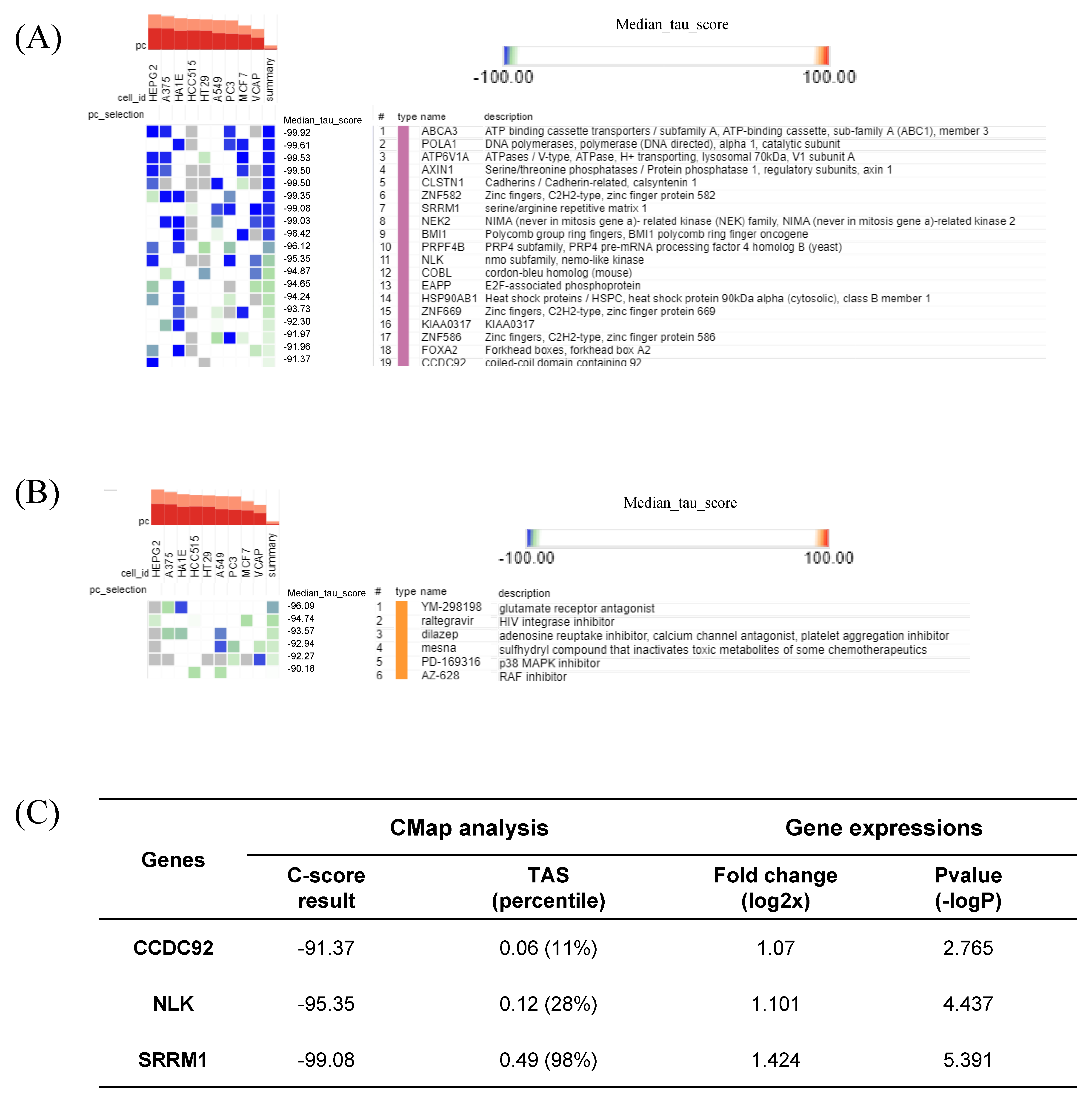
2.7. CMap Analysis Revealed Potential Markers and Drugs Based on the HT Gene Signature
3. Discussion
4. Materials and Methods
4.1. Data Resources and Processing
4.2. Screening of DEGs
4.3. Construction of PPI Network and Functional Subcluster Analysis
4.4. GO and KEGG Pathway Enrichment Analyses
4.5. GSEA of GO and KEGG Pathway
4.6. Clinical HT Biomarker Identification Via Comparison of Gene Signatures in the CMap Database
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Caturegli, P.; De Remigis, A.; Rose, N.R. Hashimoto thyroiditis: Clinical and diagnostic criteria. Autoimmun. Rev. 2014, 13, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Abbasgholizadeh, P.; Naseri, A.; Nasiri, E.; Sadra, V. Is Hashimoto thyroiditis associated with increasing risk of thyroid malignancies? A systematic review and meta-analysis. Thyroid. Res. 2021, 14, 26. [Google Scholar] [CrossRef]
- Kim, H.G.; Kim, E.-K.; Han, K.H.; Kim, H.; Kwak, J.Y. Pathologic Spectrum of Lymphocytic Infiltration and Recurrence of Papillary Thyroid Carcinoma. Yonsei Med. J. 2014, 55, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Bogusławska, J.; Godlewska, M.; Gajda, E.; Piekiełko-Witkowska, A. Cellular and molecular basis of thyroid autoimmunity. Eur. Thyroid J. 2022, 11, e210024. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.R.; Gadgil, N.M.; Yadav, S.; Kalgutkar, A.D. Importance of combined approach of investigations for detection of asymptomatic Hashimoto Thyroiditis in early stage. J. Lab. Physicians 2018, 10, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Di Domenicantonio, A.; Fallahi, P. Autoimmune thyroid disorders. Autoimmun. Rev. 2015, 14, 174–180. [Google Scholar] [CrossRef]
- Zaletel, K.; Gaberšček, S. Hashimoto’s Thyroiditis: From Genes to the Disease. Curr. Genom. 2011, 12, 576–588. [Google Scholar] [CrossRef]
- Davidson, A.; Diamond, B. Autoimmune diseases. N. Engl. J. Med. 2001, 345, 340–350. [Google Scholar] [CrossRef]
- Goodnow, C.C. Balancing immunity and tolerance: Deleting and tuning lymphocyte repertoires. Proc. Natl. Acad. Sci. USA 1996, 93, 2264–2271. [Google Scholar] [CrossRef]
- Davidson, A.; Diamond, B. General Features of Autoimmune Disease. In The Autoimmune Diseases; Academic Press: Cambridge, MA, USA, 2020; pp. 17–44. [Google Scholar] [CrossRef]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of Autoantibodies before the Clinical Onset of Systemic Lupus Erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef]
- Marrack, P.; Kappler, J.W.; Kotzin, B.L. Autoimmune disease: Why and where it occurs. Nat. Med. 2001, 7, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, M.J.; Gough, S.C.L. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genom. 2007, 8, 453–465. [Google Scholar] [CrossRef]
- Shin, D.-H.; Baek, I.-C.; Kim, H.J.; Choi, E.-J.; Ahn, M.; Jung, M.H.; Suh, B.-K.; Cho, W.K.; Kim, T.-G. HLA alleles, especially amino-acid signatures of HLA-DPB1, might contribute to the molecular pathogenesis of early-onset autoimmune thyroid disease. PLoS ONE 2019, 14, e0216941. [Google Scholar] [CrossRef] [PubMed]
- Hewagama, A.; Richardson, B. The genetics and epigenetics of autoimmune diseases. J. Autoimmun. 2009, 33, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.M.; Rahal, E.A. The role of viral infections in the development of autoimmune diseases. Crit. Rev. Microbiol. 2019, 45, 394–412. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular Mimicry as a Mechanism of Autoimmune Disease. Clin. Rev. Allergy Immunol. 2011, 42, 102–111. [Google Scholar] [CrossRef]
- Piccinini, A.M.; Midwood, K.S. DAMPening Inflammation by Modulating TLR Signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef]
- Di Crescenzo, V.; D’Antonio, A.; Tonacchera, M.; Carlomagno, C.; Vitale, M. Human herpes virus associated with Hashimoto’s thyroiditis. Le Infez. Med. 2013, 21, 224–228. [Google Scholar]
- Janegova, A.; Janega, P.; Rychly, B.; Kuracinova, K.; Babal, P. Rola infekcji wirusem Epstein-Barr’a w rozwoju autoimmunologicznych chorób tarczycy. Endokrynol. Polska 2015, 66, 132–136. [Google Scholar] [CrossRef]
- Antonelli, A.; Ferri, C.; Pampana, A.; Fallahi, P.; Nesti, C.; Pasquini, M.; Marchi, S.; Ferrannini, E. Thyroid disorders in chronic hepatitis C. Am. J. Med. 2004, 117, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, X.-L.; Xie, J.-P.; Shao, J.-G.; Lu, Y.-H.; Zhang, S.; Qin, G. Thyroid Disturbance in Patients with Chronic Hepatitis C Infection: A Systematic Review and Meta-analysis. J. Gastrointest. Liver Dis. 2016, 25, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Trevino, V.; Falciani, F.; Barrera-Saldaña, H.A. DNA Microarrays: A Powerful Genomic Tool for Biomedical and Clinical Research. Mol. Med. 2007, 13, 527–541. [Google Scholar] [CrossRef]
- Qiu, K.; Li, K.; Zeng, T.; Liao, Y.; Min, J.; Zhang, N.; Peng, M.; Kong, W.; Chen, L.-L. Integrative Analyses of Genes Associated with Hashimoto’s Thyroiditis. J. Immunol. Res. 2021, 2021, 8263829. [Google Scholar] [CrossRef]
- Jacobson, D.L.; Gange, S.J.; Rose, N.R.; Graham, N.M. Epidemiology and Estimated Population Burden of Selected Autoimmune Diseases in the United States. Clin. Immunol. Immunopathol. 1997, 84, 223–243. [Google Scholar] [CrossRef]
- Münz, C.; Lünemann, J.D.; Getts, M.T.; Miller, S.D. Antiviral immune responses: Triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 2009, 9, 246–258. [Google Scholar] [CrossRef]
- Han, F.; Lin, L.; Warby, S.C.; Faraco, J.; Li, J.; Dong, S.X.; An, P.; Zhao, L.; Wang, L.H.; Li, Q.Y.; et al. Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in china. Ann. Neurol. 2011, 70, 410–417. [Google Scholar] [CrossRef]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2020, 33, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Yazdanpanah, N.; Rezaei, N. Autoimmune complications of COVID-19. J. Med. Virol. 2021, 94, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Duntas, L.H.; Jonklaas, J. COVID-19 and Thyroid Diseases: A Bidirectional Impact. J. Endocr. Soc. 2021, 5, bvab076. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; O’Callaghan, K.; Sinclair, H.; Hawke, K.; Love, A.; Hajkowicz, K.; Stewart, A.G. Risk factors, treatment and outcomes of subacute thyroiditis secondary to COVID-19: A systematic review. Intern. Med. J. 2021, 52, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.A.U.; Farooq, H.; Ali, M.M.; Dar, Q.A.; Hussain, A. The Association of Subacute Thyroiditis with COVID-19: A Systematic Review. SN Compr. Clin. Med. 2021, 3, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Niewold, T.B. Interferon Alpha as a Primary Pathogenic Factor in Human Lupus. J. Interf. Cytokine Res. 2011, 31, 887–892. [Google Scholar] [CrossRef]
- Buscema, M.; Todd, I.; Deuss, U.; Hammond, L.; Mirakian, R.; Pujol-Borrell, R.; Bottazzo, G.F. Influence of Tumor Necrosis Factor-α on the Modulation by Interferon-γ of HLA Class II Molecules in Human Thyroid Cells and Its Effect on Interferon-γ Binding. J. Clin. Endocrinol. Metab. 1989, 69, 433–439. [Google Scholar] [CrossRef]
- Weider, T.; Richardson, S.J.; Morgan, N.G.; Paulsen, T.H.; Dahl-Jørgensen, K.; Hammerstad, S.S. Upregulation of HLA class I and antiviral tissue responses in Hashimoto’s thyroiditis. Thyroid 2020, 30, 432–442. [Google Scholar] [CrossRef]
- Li, S. Regulation of Ribosomal Proteins on Viral Infection. Cells 2019, 8, 508. [Google Scholar] [CrossRef]
- Wu, C.; Zhao, Y.; Lin, Y.; Yang, X.; Yan, M.; Min, Y.; Pan, Z.; Xia, S.; Shao, Q. Bioinformatics analysis of differentially expressed gene profiles associated with systemic lupus erythematosus. Mol. Med. Rep. 2017, 17, 3591–3598. [Google Scholar] [CrossRef]
- Jiménez-Vacas, J.M.; Herrero-Aguayo, V.; Montero-Hidalgo, A.J.; Gómez-Gómez, E.; Fuentes-Fayos, A.C.; León-González, A.J.; Sáez-Martínez, P.; Alors-Pérez, E.; Pedraza-Arévalo, S.; González-Serrano, T. Dysregulation of the splicing machinery is directly associated to aggressiveness of prostate cancer. EBioMedicine 2020, 51, 102547. [Google Scholar] [CrossRef] [PubMed]
- Wojcechowskyj, J.A.; Didigu, C.A.; Lee, J.Y.; Parrish, N.F.; Sinha, R.; Hahn, B.H.; Bushman, F.D.; Jensen, S.T.; Seeholzer, S.H.; Doms, R.W. Quantitative Phosphoproteomics Reveals Extensive Cellular Reprogramming during HIV-1 Entry. Cell Host Microbe 2013, 13, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, U.; Benoit-Pilven, C.; Lacroix, V.; Navratil, V.; Naffakh, N. Advances in Analyzing Virus-Induced Alterations of Host Cell Splicing. Trends Microbiol. 2018, 27, 268–281. [Google Scholar] [CrossRef]
- Liao, K.-C.; Garcia-Blanco, M. Role of Alternative Splicing in Regulating Host Response to Viral Infection. Cells 2021, 10, 1720. [Google Scholar] [CrossRef] [PubMed]
- Akamizu, T.; Amino, N. Hashimoto’s Thyroiditis. Endotext [Internet]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK285557/ (accessed on 17 July 2017).
- Strieder, T.G.A. Prediction of Progression to Overt Hypothyroidism or Hyperthyroidism in Female Relatives of Patients With Autoimmune Thyroid Disease Using the Thyroid Events Amsterdam (THEA) Score. Arch. Intern. Med. 2008, 168, 1657–1663. [Google Scholar] [CrossRef]
- Subhi, O.; Schulten, H.-J.; Bagatian, N.; Al-Dayini, R.; Karim, S.; Bakhashab, S.; Alotibi, R.; Al-Ahmadi, A.; Ata, M.; Elaimi, A.; et al. Genetic relationship between Hashimoto‘s thyroiditis and papillary thyroid carcinoma with coexisting Hashimoto‘s thyroiditis. PLoS ONE 2020, 15, e0234566. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin/Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2–27. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Bonnot, T.; Gillard, M.; Nagel, D. A Simple Protocol for Informative Visualization of Enriched Gene Ontology Terms. Bio-Protocol 2019, 9. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. R Package Version 2019, 1. Available online: https://bioconductor.org/packages/release/bioc/html/enrichplot.html (accessed on 17 July 2017).
- Musa, A.; Ghoraie, L.S.; Zhang, S.-D.; Galzko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Briefings Bioinform. 2017, 19, 506–523. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | log2(Fold Change) | −Log10(P) | |
|---|---|---|---|---|
| Up regulated | KIF5B | Kinesin family member 5B | 2.092 | 9.687 |
| SLC30A7 | Solute carrier family 30, member 7 | 1.613 | 8.584 | |
| RHNO1 | RAD9-HUS1-RAD1 interacting nuclear orphan 1 | 1.948 | 8.386 | |
| PGK1 | phosphoglycerate kinase 1 | 1.628 | 8.321 | |
| ATP5EP2 | ATP synthase, H+ transporting, mitochondrial F1 complex, epsilon subunit pseudogene 2 | 1.865 | 8.107 | |
| PIGS | Phosphatidylinositol glycan anchor biosynthesis class S | 2.408 | 8.092 | |
| CFL1 | Cofilin 1 | 1.441 | 7.967 | |
| ACER3 | Alkaline ceramidase 3 | 1.833 | 7.798 | |
| PARP3 | Poly(ADP-ribose) polymerase family member 3 | 2.09 | 7.783 | |
| ATXN7L1 | Ataxin 7 like 1 | 2.526 | 7.756 | |
| TMA7 | Translation machinery associated 7 homolog | 2.999 | 7.713 | |
| PTMA | Prothymosin, alpha | 1.341 | 7.685 | |
| HLA-DMB | Major histocompatibility complex, Class II, DM beta | 2.011 | 7.663 | |
| SAMD9L | Sterile alpha motif domain containing 9 like | 3.262 | 7.617 | |
| UNC93B1 | Unc-93 homolog B1 (C. elegans) | 1.973 | 7.581 | |
| Downregulated | PTH | Parathyroid hormone | −7.228 | 14.79 |
| CKM | Creatine kinase, M-type | −6.094 | 10.002 | |
| MYL2 | Myosin light chain 2 | −7.113 | 9.68 | |
| MYH2 | Myosin heavy chain 2 | −5.091 | 9.511 | |
| ATP2A1 | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1 | −4.317 | 9.467 | |
| AKR1C3 | Aldo-keto reductase family 1, member C3 | −3.636 | 9.222 | |
| GPT2 | Glutamic–pyruvic transaminase 2 | −1.961 | 8.731 | |
| CHGA | Chromogranin A | −1.615 | 8.614 | |
| MYBPC1 | Myosin binding protein C, slow type | −6.775 | 8.608 | |
| IGBP1 | Immunoglobulin (CD79A) binding protein 1 | −1.63 | 8.189 | |
| SAMD8 | Sterile alpha motif domain containing 8 | −1.652 | 8.067 | |
| SOD1 | Superoxide dismutase 1, soluble | −1.722 | 7.862 | |
| AKR1C1 | Aldo-keto reductase family 1, member C1 | −3.674 | 7.796 | |
| FKBP3 | FK506 binding protein 3 | −1.872 | 7.688 | |
| TMEM159 | Transmembrane protein 159 | −1.87 | 7.661 |
| ID | Description | Enrichment Score | NES | p-Value |
|---|---|---|---|---|
| GO:0002250 | Adaptive immune response | 0.658 | 2.637 | 0.00011 |
| GO:0050870 | Positive regulation of T-cell activation | 0.674 | 2.564 | 0.00012 |
| GO:0046651 | Lymphocyte proliferation | 0.641 | 2.502 | 0.00011 |
| GO:0046649 | Lymphocyte activation | 0.608 | 2.494 | 0.00010 |
| GO:0007159 | Leukocyte cell–cell adhesion | 0.625 | 2.483 | 0.00011 |
| GO:0019724 | B-cell mediated immunity | 0.684 | 2.475 | 0.00013 |
| GO:1990868 | Response to chemokine | 0.706 | 2.445 | 0.00013 |
| GO:0002366 | Leukocyte activation involved in immune response | 0.619 | 2.407 | 0.00011 |
| GO:0002263 | Cell activation involved in immune response | 0.615 | 2.389 | 0.00011 |
| GO:0070661 | Leukocyte proliferation | 0.606 | 2.387 | 0.00011 |
| GO:0050865 | Regulation of cell activation | 0.584 | 2.377 | 0.00011 |
| GO:0002252 | Immune effector process | 0.580 | 2.359 | 0.00011 |
| GO:0002684 | Positive regulation of immune system process | 0.570 | 2.347 | 0.00010 |
| GO:0050776 | Regulation of immune response | 0.559 | 2.304 | 0.00010 |
| GO:0019882 | Antigen processing and presentation | 0.658 | 2.286 | 0.00013 |
| ID | Description | Enrichment Score | NES | p-Value |
|---|---|---|---|---|
| hsa04061 | Viral protein interaction with cytokine and cytokine receptor | 0.753 | 2.683 | 0.00014 |
| hsa04672 | Intestinal immune network for IgA production | 0.843 | 2.672 | 0.00015 |
| hsa04640 | Hematopoietic cell lineage | 0.742 | 2.643 | 0.00014 |
| hsa05322 | Systemic lupus erythematosus | 0.785 | 2.524 | 0.00015 |
| hsa05320 | Autoimmune thyroid disease | 0.797 | 2.499 | 0.00015 |
| hsa05340 | Primary immunodeficiency | 0.814 | 2.480 | 0.00016 |
| hsa04514 | Cell-adhesion molecules | 0.647 | 2.449 | 0.00013 |
| hsa05330 | Allograft rejection | 0.821 | 2.427 | 0.00016 |
| hsa05310 | Asthma | 0.837 | 2.426 | 0.00016 |
| hsa04658 | Th1- and Th2-cell differentiation | 0.678 | 2.405 | 0.00014 |
| hsa05332 | Graft-versus-host disease | 0.817 | 2.385 | 0.00016 |
| hsa05140 | Leishmaniasis | 0.693 | 2.379 | 0.00014 |
| hsa05150 | Staphylococcus aureus infection | 0.672 | 2.365 | 0.00014 |
| hsa04662 | B-cell receptor signaling pathway | 0.675 | 2.361 | 0.00014 |
| hsa04062 | Chemokine signaling pathway | 0.607 | 2.360 | 0.00013 |
| hsa04940 | Type I diabetes mellitus | 0.775 | 2.351 | 0.00016 |
| hsa05323 | Rheumatoid arthritis | 0.663 | 2.341 | 0.00014 |
| hsa04659 | Th17-cell differentiation | 0.637 | 2.313 | 0.00014 |
| hsa05169 | Epstein–Barr virus infection | 0.587 | 2.291 | 0.00013 |
| hsa04650 | Natural-killer-cell-mediated cytotoxicity | 0.622 | 2.274 | 0.00014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, D.-W.; Choi, M.-S.; Kim, S.-M. Bioinformatics and Connectivity Map Analysis Suggest Viral Infection as a Critical Causative Factor of Hashimoto’s Thyroiditis. Int. J. Mol. Sci. 2023, 24, 1157. https://doi.org/10.3390/ijms24021157
Lim D-W, Choi M-S, Kim S-M. Bioinformatics and Connectivity Map Analysis Suggest Viral Infection as a Critical Causative Factor of Hashimoto’s Thyroiditis. International Journal of Molecular Sciences. 2023; 24(2):1157. https://doi.org/10.3390/ijms24021157
Chicago/Turabian StyleLim, Dong-Woo, Min-Seo Choi, and Seok-Mo Kim. 2023. "Bioinformatics and Connectivity Map Analysis Suggest Viral Infection as a Critical Causative Factor of Hashimoto’s Thyroiditis" International Journal of Molecular Sciences 24, no. 2: 1157. https://doi.org/10.3390/ijms24021157
APA StyleLim, D.-W., Choi, M.-S., & Kim, S.-M. (2023). Bioinformatics and Connectivity Map Analysis Suggest Viral Infection as a Critical Causative Factor of Hashimoto’s Thyroiditis. International Journal of Molecular Sciences, 24(2), 1157. https://doi.org/10.3390/ijms24021157