
Development of Novel Class of Phenylpyrazolo[3,4-d]pyrimidine-Based Analogs with Potent Anticancer Activity and Multitarget Enzyme Inhibition Supported by Docking Studies
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
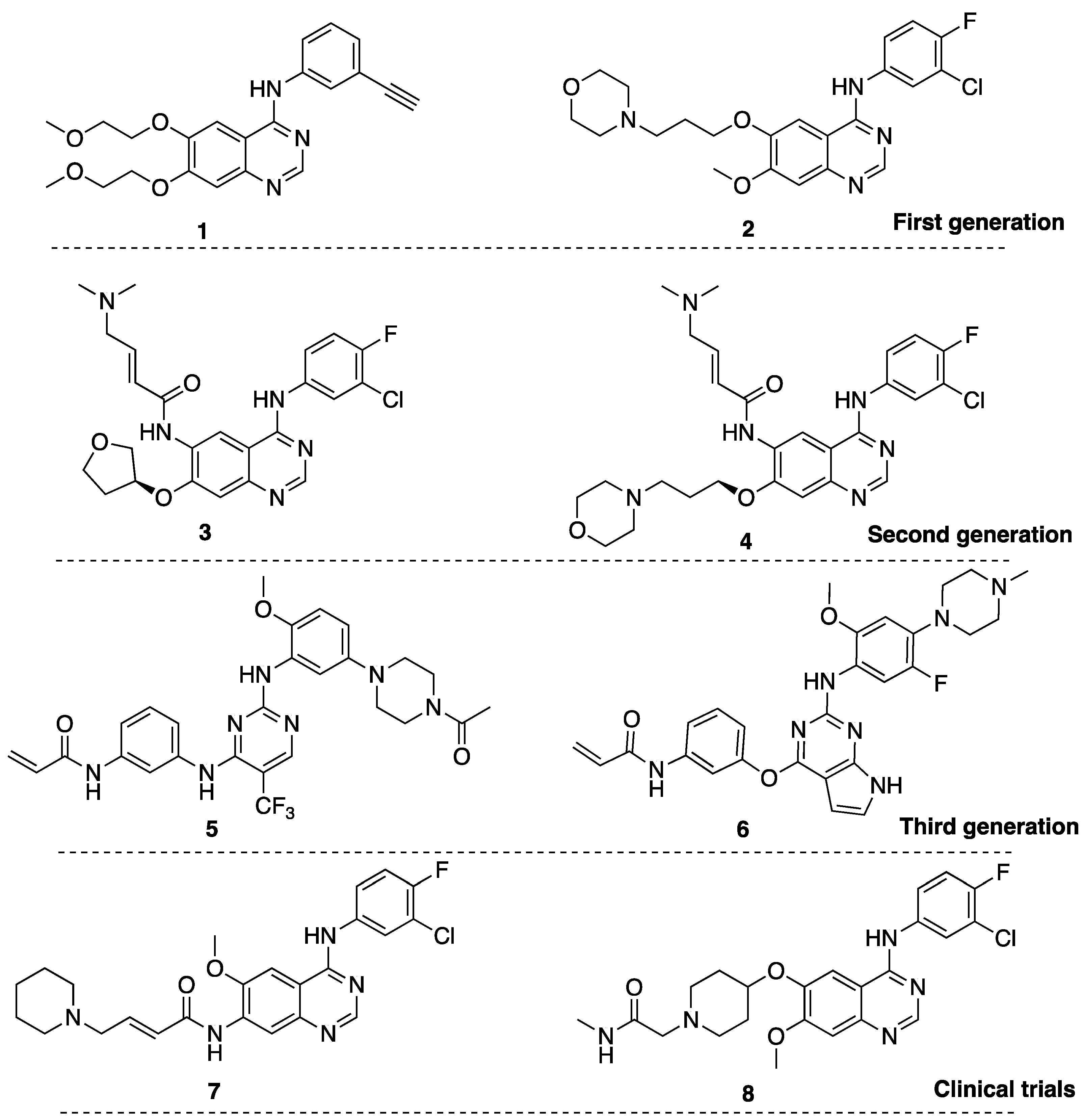
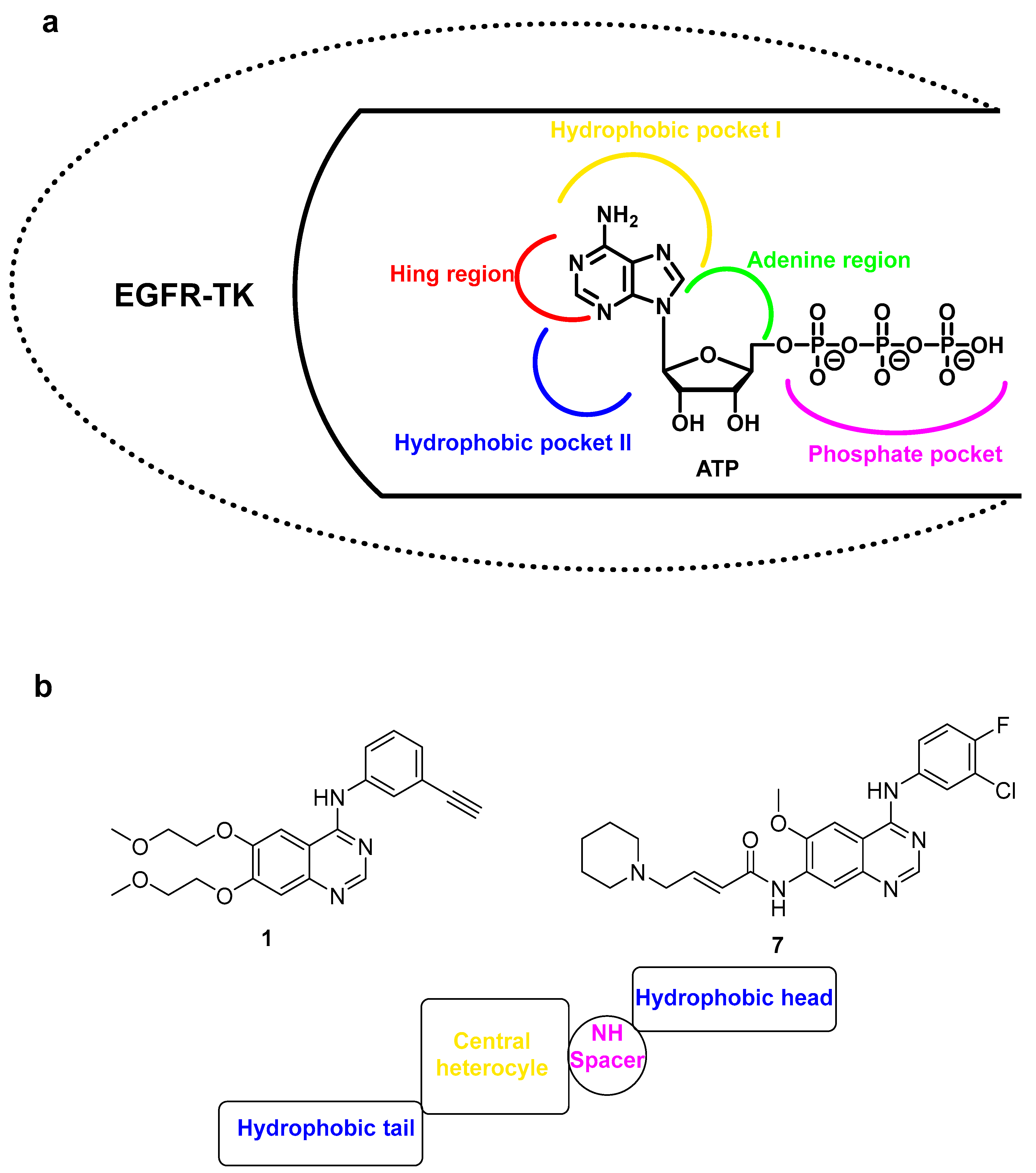
2.1. Structure-Based Scaffold and Compound Design
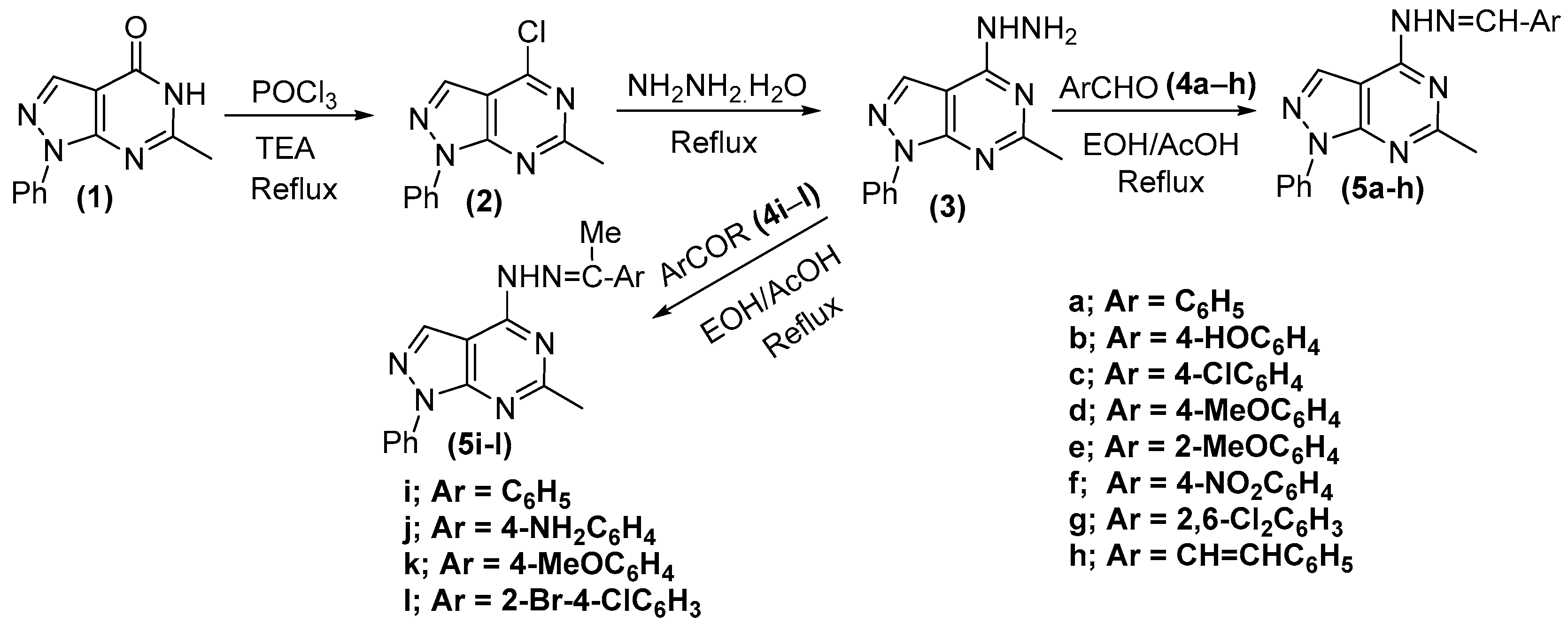
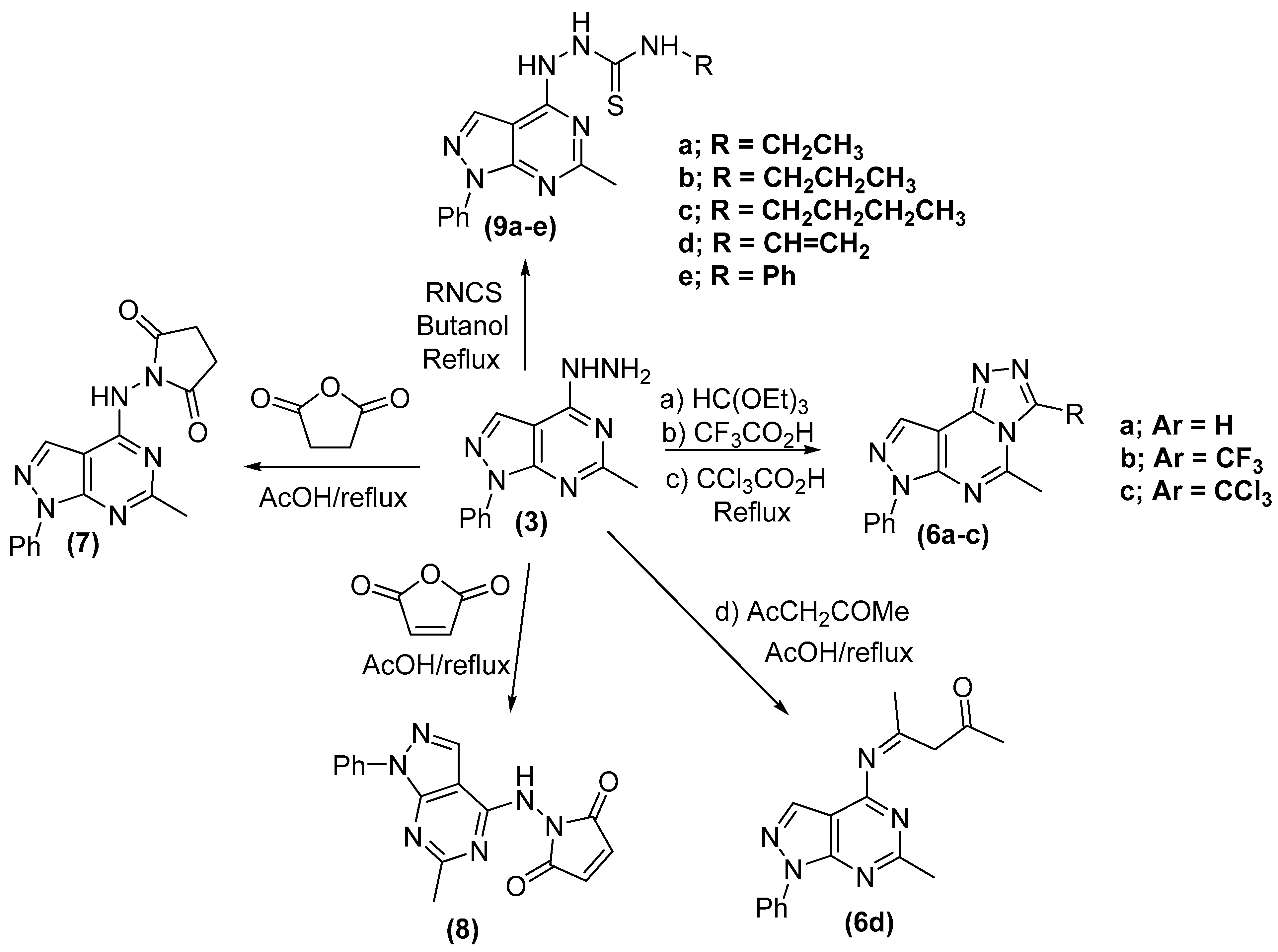
2.2. Chemistry
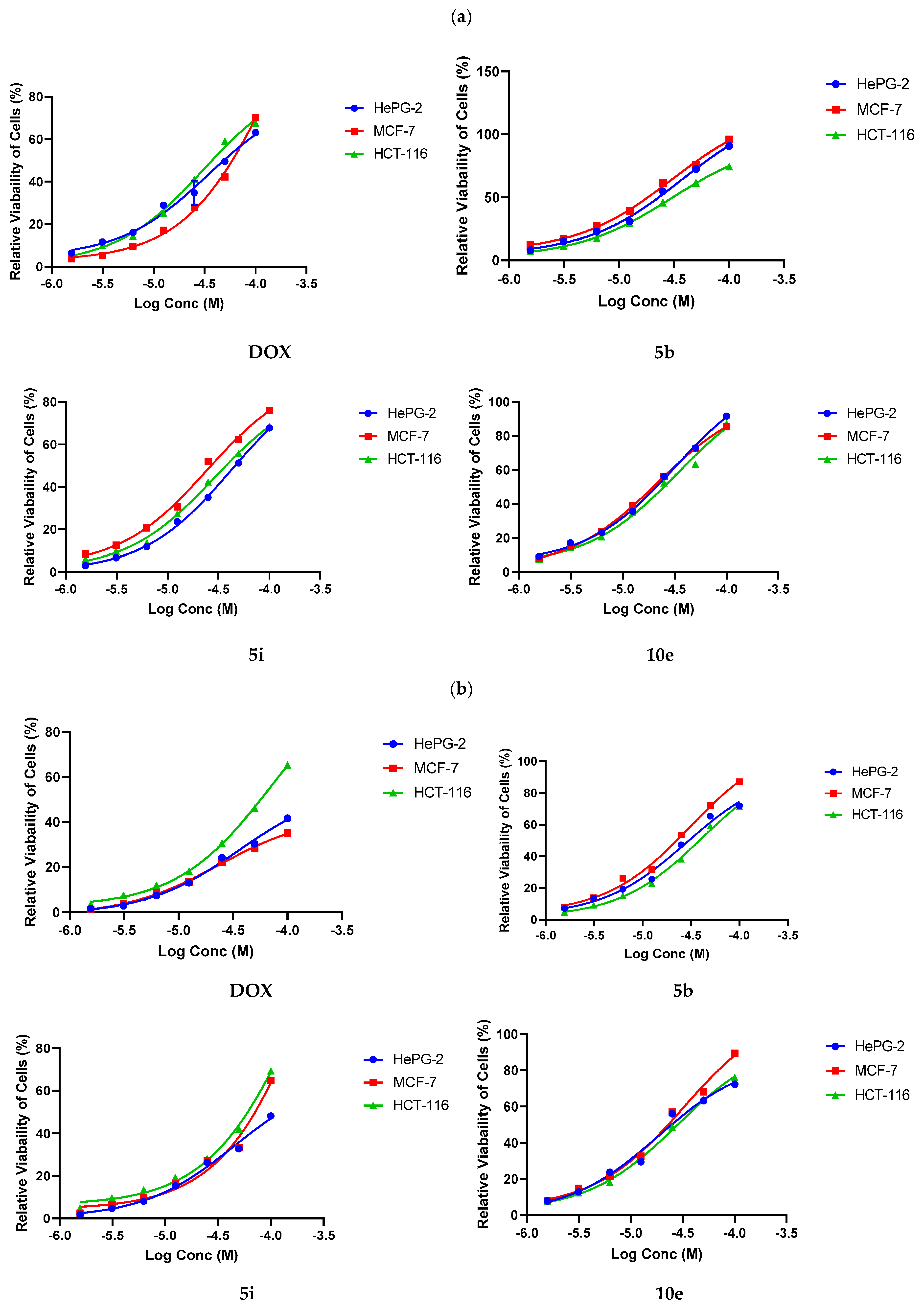
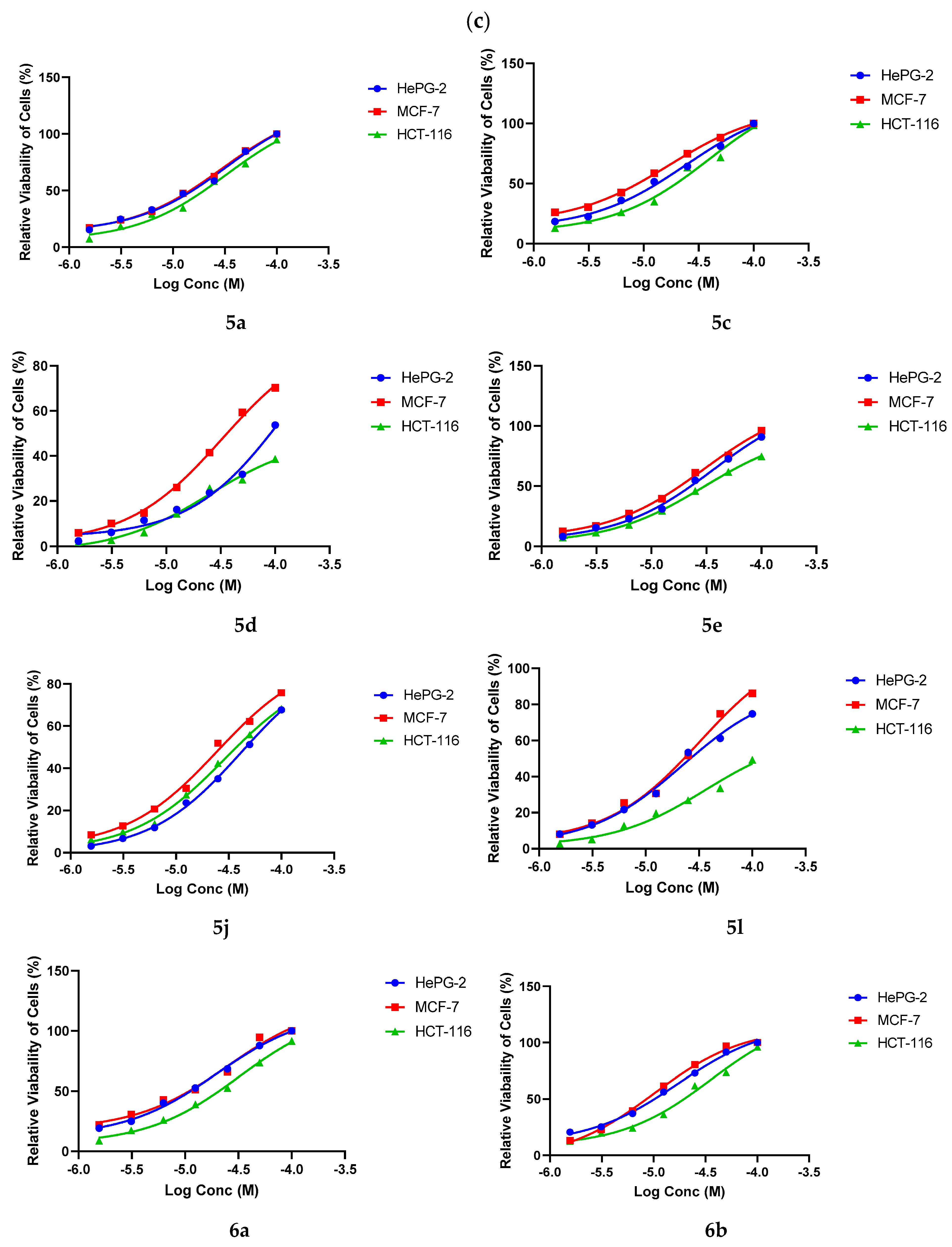
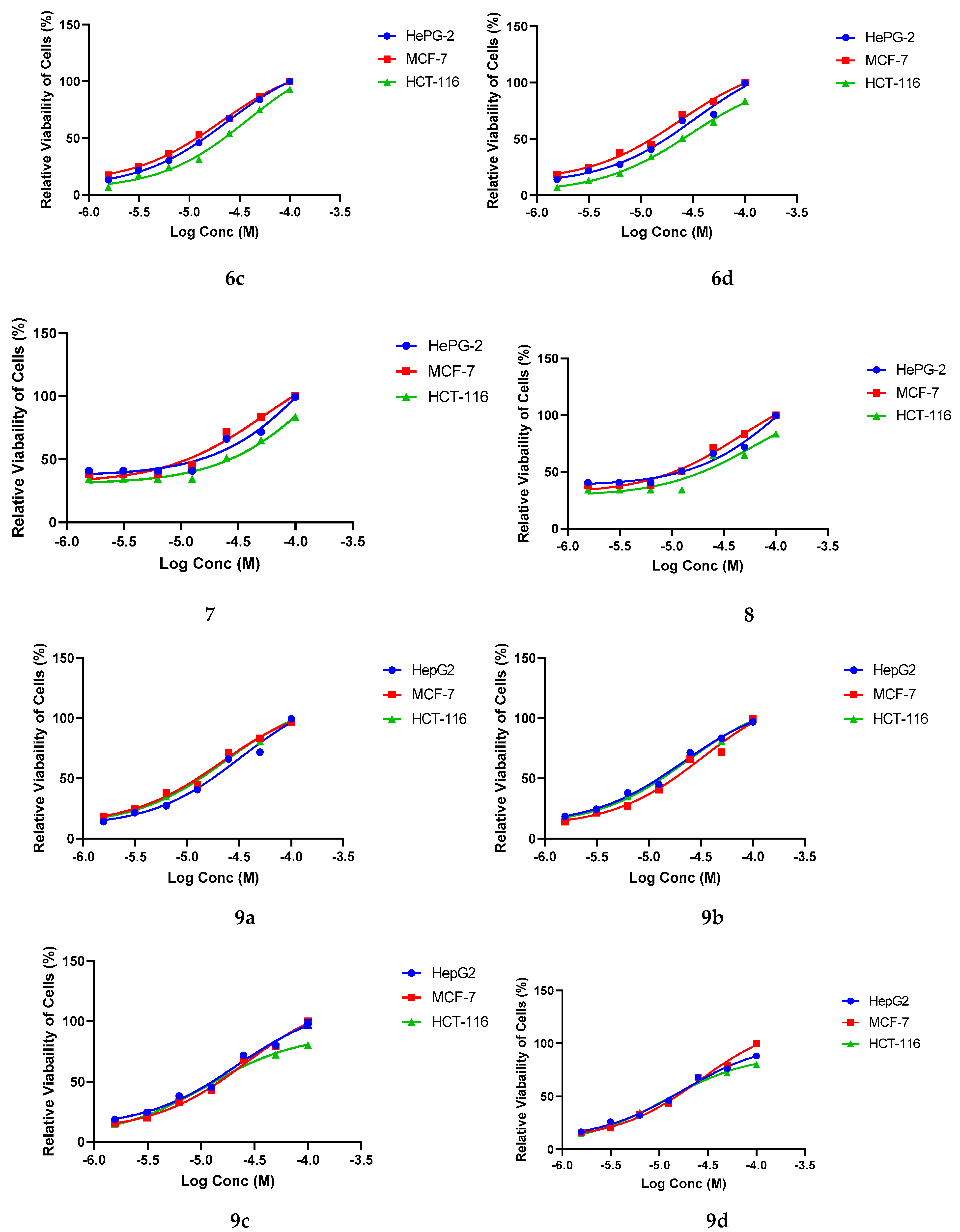
2.3. Anticancer Assay
2.4. In Vitro Cancer Related Targets Inhibition Analysis
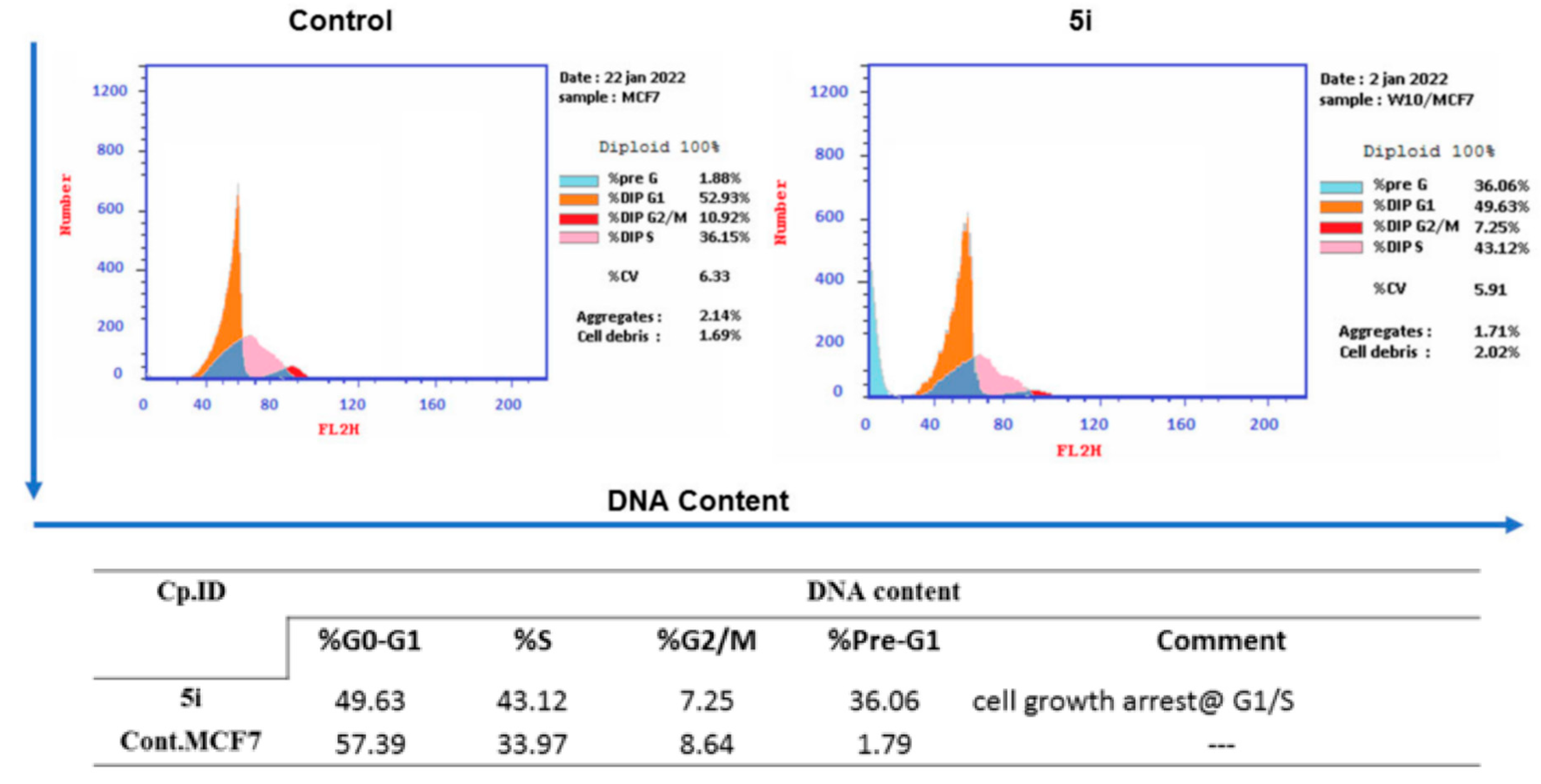
2.5. Cell Cycle Analysis
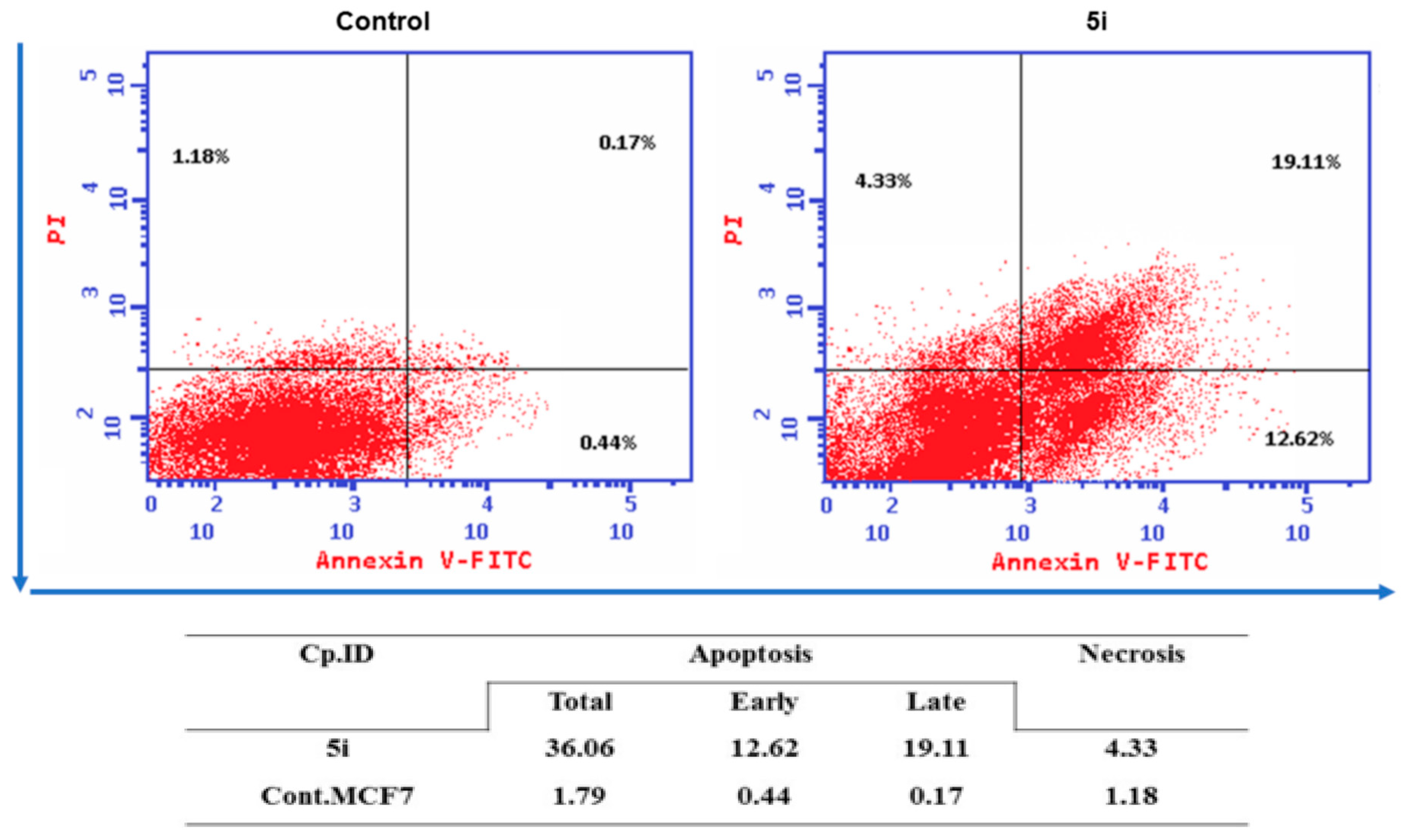
2.6. Annexin V-FITC Apoptosis Assay
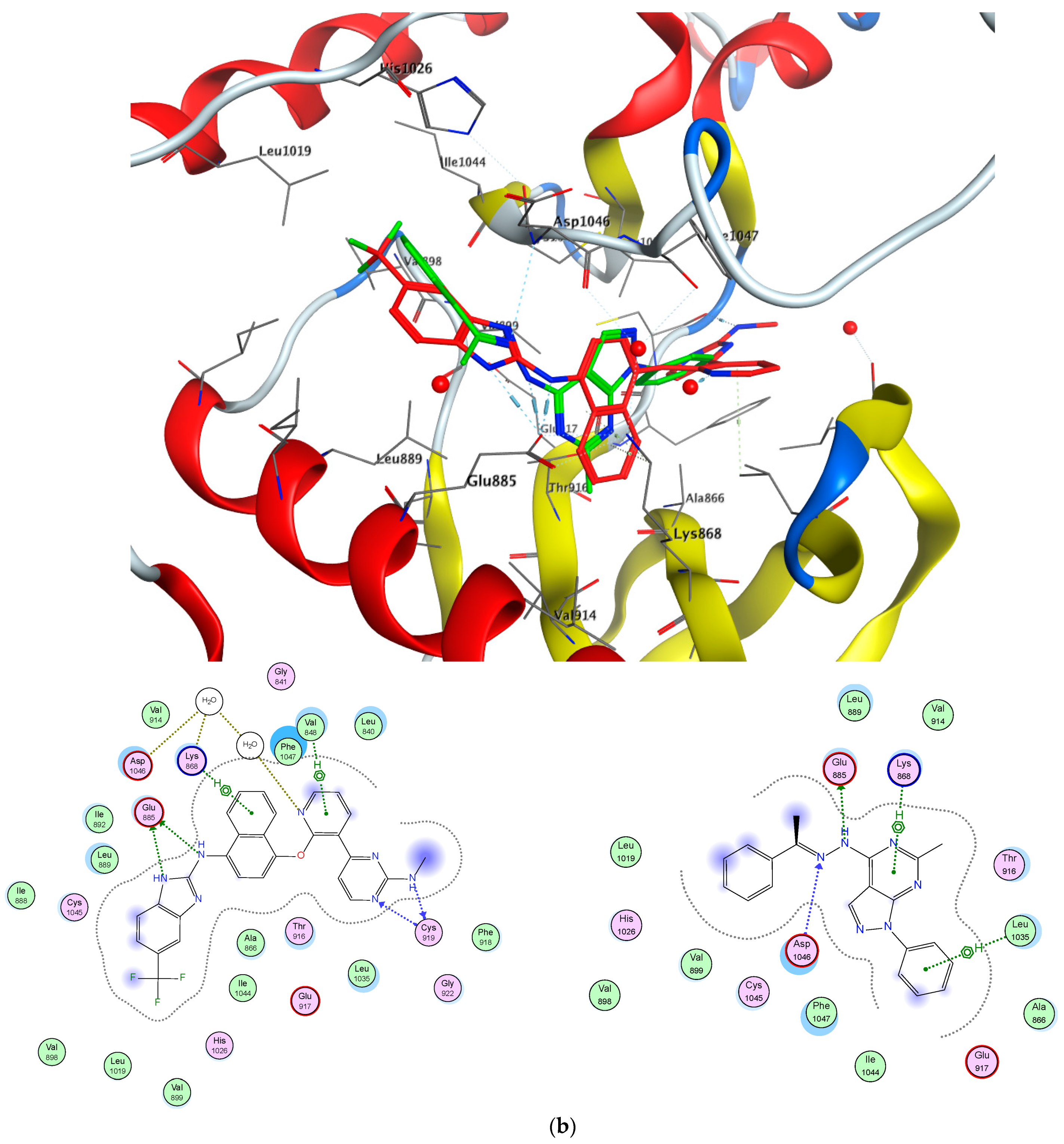
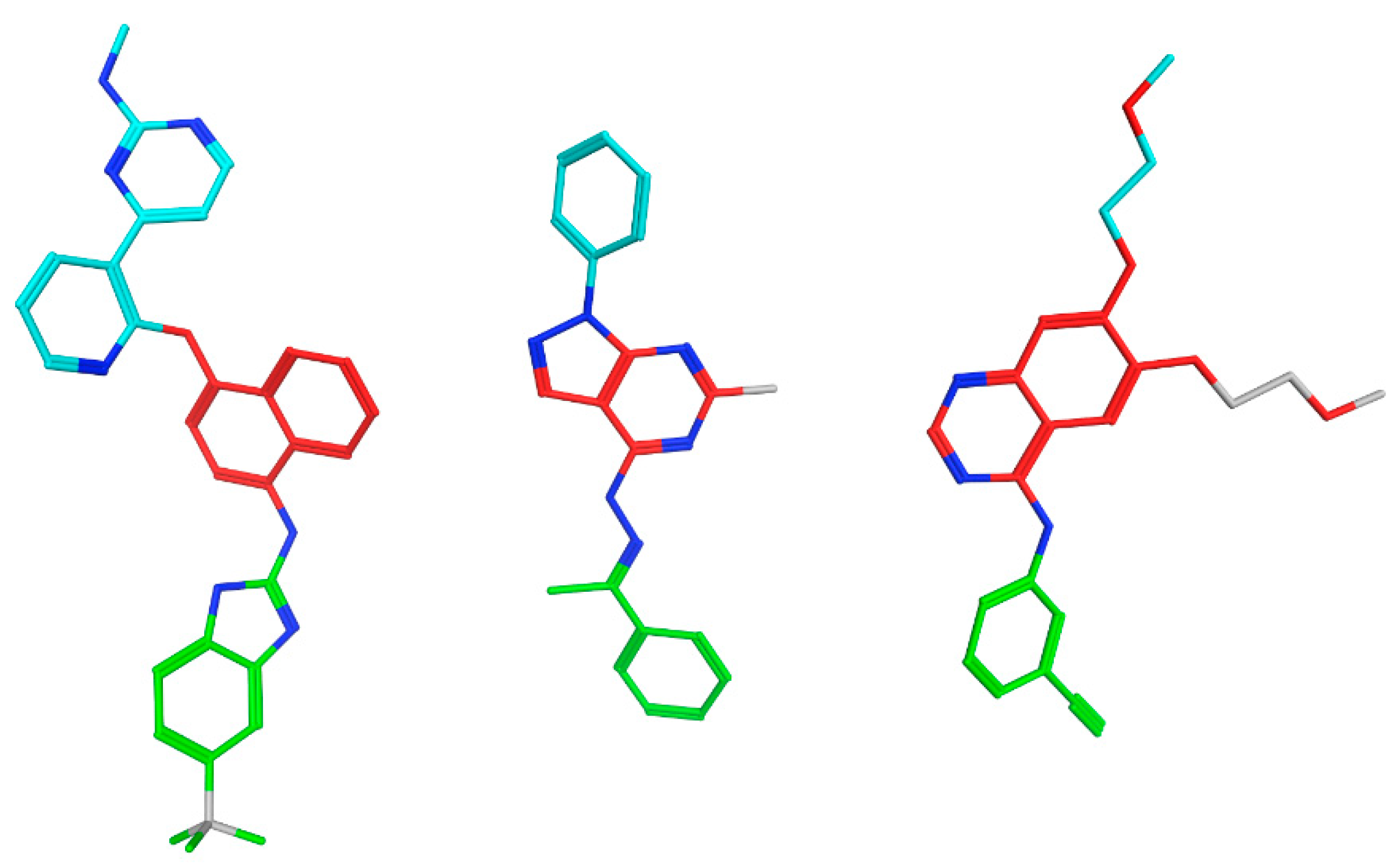
2.7. EGFR/VGFR2 Target Docking Simulations
2.8. Prediction of Drug-Likeness and ADME Properties
3. Conclusions
4. Experimental Section
4.1. Chemistry
4.1.1. General Procedures
6-Methyl-1-phenyl-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (1)
4-Chloro-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (2)
4-Hydrazinyl-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (3)
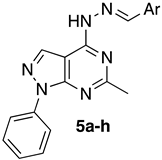
General Procedure for the Synthesis of 4-(2-Arylidenehydrazinyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5a–l)
- 4-(2-Benzylidenehydrazinyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5a)
- 4-((2-(6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)hydrazineylidene)methyl)phenol (5b)
- 4-(2-(4-Chlorobenzylidene)hydrazinyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5c)
- (4-(2-(4-Methoxybenzylidene)hydrazineyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5d)
- 4-(2-(2-Methoxybenzylidene)hydrazineyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5e)
- 6-Methyl-4-(2-(4-nitrobenzylidene)hydrazineyl)-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5f)
- 4-(2-(2,6-Dichlorobenzylidene)hydrazineyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5g)
- 6-Methyl-1-phenyl-4-(2-(-3-phenylallylidene)hydrazineyl)-1H-pyrazolo[3,4-d]pyrimidine (5h)
- 6-Methyl-1-phenyl-4-(2-(1-phenylethylidene)hydrazinyl)-1H-pyrazolo[3,4-d]pyrimidine (5i)
- 4-(1-(2-(6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)hydrazineylidene)ethyl)aniline (5j)
- 4-(2-(1-(4-Methoxyphenyl)ethylidene)hydrazinyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine (5k)
- 4-(2-(1-(2-Bromo-4-chlorophenyl)ethylidene)hydrazineyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d] pyrimidine (5l)
General Procedure for the Synthesis of Pyrazolopyrimidine Derivatives (6a–d)
- 5-Methyl-7-phenyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[4,3-c]pyrimidine (6a)
- 5-Methyl-7-phenyl-3-(trifluoromethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[4,3-c]pyrimidine (6b)
- 5-Methyl-7-phenyl-3-(trichloromethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[4,3-c]pyrimidine (6c)
- 4-((6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)imino)pentan-2-one (6d)
General Procedure for the Synthesis of Pyrrole-2,5-Dione Derivatives (7,8)
- 1-((6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)pyrrolidine-2,5-dione (7)
- 1-((6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)-1H-pyrrole-2,5-dione (8)
- General Procedure for the Synthesis of N-alkyl/aryl-2-(6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)hydrazinecarbothioamides (9a–e)
- N-Ethyl-2-(6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)hydrazinecarbothioamide (9a)
- 2-(6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-propylhydrazinecarbothioamide (9b)
- N-Butyl-2-(6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)hydrazinecarbothioamide (9c)
- 2-(6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-vinylhydrazine-1-carbothioamide (9d)
- 2-(6-Methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-phenylhydrazinecarbothioamide (9e)
4.2. Biological Evaluation
4.2.1. Cytotoxicity Evaluation Using Viability Assay
4.2.2. In Vitro Enzyme Inhibitory Assays (against EGFRwt, EGFRT790M, VEGFR-2, and Topo II)
4.2.3. Quantification of Apoptosis by Flow Cytometry
4.2.4. Cell Cycle Analysis
4.3. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sener, S.F.; Grey, N. The global burden of cancer. J. Surg. Oncol. 2005, 92, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Salminen, E.; Izewska, J.; Andreo, P. IAEA’s role in the global management of cancer-focus on upgrading radiotherapy services. Acta Oncol. 2005, 44, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Zucchinetti, C.; Buda, G.; Orciuolo, E. Tumor dormancy as an alternative step in the development of chemoresistance and metastasis—Clinical implications. Cell. Oncol. 2020, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- El Azab, I.H.; Bakr, R.B.; Elkanzi, N.A.A. Facile One-Pot Multicomponent Synthesis of Pyrazolo-Thiazole Substituted Pyridines with Potential Anti-Proliferative Activity: Synthesis, In Vitro and In Silico Studies. Molecules 2021, 26, 3103. [Google Scholar] [CrossRef]
- Omar, A.M.; Bajorath, J.; Ihmaid, S.; Mohamed, H.M.; El-Agrody, A.M.; Mora, A.; El-Araby, M.E.; Ahmed, H.E.A. Novel molecular discovery of promising amidine-based thiazole analogues as potent dual Matrix Metalloproteinase-2 and 9 inhibitors: Anticancer activity data with prominent cell cycle arrest and DNA fragmentation analysis effects. Bioorganic Chem. 2020, 101, 103992. [Google Scholar] [CrossRef]
- Garraway, L.A.; Janne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Papavassiliou, A.G. Seeing the future of cancer-associated transcription factor drug targets. JAMA 2011, 305, 2349–2350. [Google Scholar] [CrossRef] [PubMed]
- Chohan, T.A.; Qayyum, A.; Rehman, K.; Tariq, M.; Akash, M.S.H. An insight into the emerging role of cyclin-dependent kinase inhibitors as potential therapeutic agents for the treatment of advanced cancers. Biomed. Pharmacother. 2018, 107, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Xiang, M.; Frank, D.A. Distinct roles of STAT3 and STAT5 in the pathogenesis and targeted therapy of breast cancer. Mol. Cell. Endocrinol. 2014, 382, 616–621. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Wu, X.; Xiong, Z.; Yang, T.; Fu, Z.; Liu, X.; Tan, X.; Zhong, F.; Wan, X.; et al. Deep Learning Enhancing Kinome-Wide Polypharmacology Profiling: Model Construction and Experiment Validation. J. Med. Chem. 2020, 63, 8723–8737. [Google Scholar] [CrossRef] [PubMed]
- Nassar, I.F.; Abdel Aal, M.T.; El-Sayed, W.A.; Shahin, M.A.E.; Elsakka, E.G.E.; Mokhtar, M.M.; Hegazy, M.; Hagras, M.; Mandour, A.A.; Ismail, N.S.M. Discovery of pyrazolo[3,4-d]pyrimidine and pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives as novel CDK2 inhibitors: Synthesis, biological and molecular modeling investigations. RSC Adv. 2022, 12, 14865–14882. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, M.A.; Elkanzi, N.A.A.; Nayl, A.A.; Musa, A.; Alotaibi, N.H.; Arafa, W.A.A.; Gomha, S.M.; Bakr, R.B. Targeting tumor cells with pyrazolo[3,4-d]pyrimidine scaffold: A literature review on synthetic approaches, structure activity relationship, structural and target-based mechanisms. Arab. J. Chem. 2022, 15, 103781. [Google Scholar] [CrossRef]
- Almehmadi, S.J.; Alsaedi, A.M.R.; Harras, M.F.; Farghaly, T.A. Synthesis of a new series of pyrazolo[1,5-a]pyrimidines as CDK2 inhibitors and anti-leukemia. Bioorganic Chem. 2021, 117, 105431. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, M.; Kumar, R. Medicinal attributes of pyrazolo[3,4-d]pyrimidines: A review. Bioorganic Med. Chem. 2013, 21, 5657–5668. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Radi, M.; Musumeci, F.; Brullo, C.; Botta, M. Biologically Driven Synthesis of Pyrazolo[3,4-d]pyrimidines As Protein Kinase Inhibitors: An Old Scaffold As a New Tool for Medicinal Chemistry and Chemical Biology Studies. Chem. Rev. 2014, 114, 7189–7238. [Google Scholar] [CrossRef]
- Husseiny, E.M. Synthesis, cytotoxicity of some pyrazoles and pyrazolo[1,5-a]pyrimidines bearing benzothiazole moiety and investigation of their mechanism of action. Bioorganic Chem. 2020, 102, 104053. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Bakr, R.B. Pyrimidine and fused pyrimidine derivatives as promising protein kinase inhibitors for cancer treatment. Med. Chem. Res. 2021, 30, 31–49. [Google Scholar] [CrossRef]
- Cherukupalli, S.; Chandrasekaran, B.; Aleti, R.R.; Sayyad, N.; Hampannavar, G.A.; Merugu, S.R.; Rachamalla, H.R.; Banerjee, R.; Karpoormath, R. Synthesis of 4,6-disubstituted pyrazolo[3,4-d]pyrimidine analogues: Cyclin-dependent kinase 2 (CDK2) inhibition, molecular docking and anticancer evaluation. J. Mol. Struct. 2019, 1176, 538–551. [Google Scholar] [CrossRef]
- Rahmouni, A.; Souiei, S.; Belkacem, M.A.; Romdhane, A.; Bouajila, J.; Ben Jannet, H. Synthesis and biological evaluation of novel pyrazolopyrimidines derivatives as anticancer and anti-5-lipoxygenase agents. Bioorganic Chem. 2016, 66, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J. The Role of VEGF and EGFR Inhibition: Implications for Combining Anti–VEGF and Anti–EGFR Agents. Mol. Cancer Res. 2007, 5, 203–220. [Google Scholar] [CrossRef]
- Le, X.; Nilsson, M.; Goldman, J.; Reck, M.; Nakagawa, K.; Kato, T.; Ares, L.P.; Frimodt-Moller, B.; Wolff, K.; Visseren-Grul, C.; et al. Dual EGFR-VEGF Pathway Inhibition: A Promising Strategy for Patients With EGFR-Mutant NSCLC. J. Thorac. Oncol. 2021, 16, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Alblewi, F.F.; Okasha, R.M.; Eskandrani, A.A.; Afifi, T.H.; Mohamed, H.M.; Halawa, A.H.; Fouda, A.M.; Al-Dies, A.M.; Mora, A.; El-Agrody, A.M. Design and Synthesis of Novel Heterocyclic-Based 4H-benzo[h]chromene Moieties: Targeting Antitumor Caspase 3/7 Activities and Cell Cycle Analysis. Molecules 2019, 24, 1060. [Google Scholar] [CrossRef] [PubMed]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.J.M. Design, synthesis and cytotoxic evaluation of novel chalcone derivatives bearing triazolo [4, 3-a]-quinoxaline moieties as potent anticancer agents with dual EGFR kinase and tubulin polymerization inhibitory effects. Molecules 2018, 23, 48. [Google Scholar] [CrossRef] [PubMed]
- Ihmaid, S.; Ahmed, H.E.A.; Zayed, M.F. The design and development of potent small molecules as anticancer agents targeting EGFR TK and tubulin polymerization. Int. J. Mol. Sci. 2018, 19, 408. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.M.; Ihmaid, S.; Habib, E.-S.E.; Althagfan, S.S.; Ahmed, S.; Abulkhair, H.S.; Ahmed, H.E. The Rational Design, Synthesis, and Antimicrobial Investigation of 2-Amino-4-Methylthiazole Analogues Inhibitors of GlcN-6-P Synthase. Bioorganic Chem. 2020, 99, 103781. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T. Targeting multiple kinase pathways: A change in paradigm. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1973–1978. [Google Scholar] [CrossRef]
- Herbst, R.S.; Johnson, D.H.; Mininberg, E.; Carbone, D.P.; Henderson, T.; Kim, E.S.; Blumenschein, G., Jr.; Lee, J.J.; Liu, D.D.; Truong, M.T.; et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 2544–2555. [Google Scholar]
- Fox, J.L.; MacFarlane, M. Targeting cell death signalling in cancer: Minimising ‘Collateral damage’. Br. J. Cancer 2016, 115, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.B.; Gasparini, G.; Harris, A.L. Angiogenesis: Pathological, prognostic, and growth-factor pathways and their link to trial design and anticancer drugs. Lancet. Oncol. 2001, 2, 278–289. [Google Scholar] [CrossRef]
- Ellis, L.M. Epidermal growth factor receptor in tumor angiogenesis. Hematol./Oncol. Clin. North Am. 2004, 18, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.E.; El-Nassag, M.A.; Hassan, A.H.; Mohamed, H.M.; Halawa, A.H.; Okasha, R.M.; Ihmaid, S.; El-Gilil, S.M.A.; Khattab, E.S.; Fouda, A.M.; et al. Developing lipophilic aromatic halogenated fused systems with specific ring orientations, leading to potent anticancer analogs and targeting the c-Src Kinase enzyme. J. Mol. Struct. 2019, 1186, 212–223. [Google Scholar] [CrossRef]
- Ahmed, H.E.A.; El-Nassag, M.A.A.; Hassan, A.H.; Okasha, R.M.; Ihmaid, S.; Fouda, A.M.; Afifi, T.H.; Aljuhani, A.; El-Agrody, A.M. Introducing novel potent anticancer agents of 1H-benzo [f] chromene scaffolds, targeting c-Src kinase enzyme with MDA-MB-231 cell line anti-invasion effect. J. Enzym. Inhib. Med. Chem. 2018, 33, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.E.A.; Ihmaid, S.K.; Omar, A.M.; Shehata, A.M.; Rateb, H.S.; Zayed, M.F.; Ahmed, S.; Elaasser, M.M. Design, synthesis, molecular docking of new lipophilic acetamide derivatives affording potential anticancer and antimicrobial agents. Bioorganic Chem. 2018, 76, 332–342. [Google Scholar] [CrossRef] [PubMed]
- El-Moghazy, S.M.; George, R.F.; Osman, E.E.A.; Elbatrawy, A.A.; Kissova, M.; Colombo, A.; Crespan, E.; Maga, G. Novel pyrazolo[3,4-d]pyrimidines as dual Src-Abl inhibitors active against mutant form of Abl and the leukemia K-562 cell line. Eur. J. Med. Chem. 2016, 123, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.; Dawson, J.C.; Dowling, R.; Houston, D.R.; Weiss, J.T.; Munro, A.F.; Muir, M.; Harrington, L.; Webster, S.P.; Frame, M.C.; et al. Rapid Discovery and Structure–Activity Relationships of Pyrazolopyrimidines That Potently Suppress Breast Cancer Cell Growth via SRC Kinase Inhibition with Exceptional Selectivity over ABL Kinase. J. Med. Chem. 2016, 59, 4697–4710. [Google Scholar] [CrossRef]
- Traxler, P.; Bold, G.; Frei, J.; Lang, M.; Lydon, N.; Mett, H.; Buchdunger, E.; Meyer, T.; Mueller, M.; Furet, P. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines. J. Med. Chem. 1997, 40, 3601–3616. [Google Scholar] [CrossRef]
- Ducray, R.; Ballard, P.; Barlaam, B.C.; Hickinson, M.D.; Kettle, J.G.; Ogilvie, D.J.; Trigwell, C.B. Novel 3-alkoxy-1H-pyrazolo[3,4-d]pyrimidines as EGFR and erbB2 receptor tyrosine kinase inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 959–962. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Alkhoja, O.A.; Mohamed, W.R. Design, synthesis and antitumor activity of novel pyrazolo[3,4-d]pyrimidine derivatives as EGFR-TK inhibitors. Bioorganic Chem. 2016, 66, 88–96. [Google Scholar] [CrossRef]
- Elshaier, Y.A.M.M.; Shaaban, M.A.; Abd El Hamid, M.K.; Abdelrahman, M.H.; Abou-Salim, M.A.; Elgazwi, S.M.; Halaweish, F. Design and synthesis of pyrazolo[3,4-d]pyrimidines: Nitric oxide releasing compounds targeting hepatocellular carcinoma. Bioorganic Med. Chem. 2017, 25, 2956–2970. [Google Scholar] [CrossRef]
- Cohen, S. Epidermal growth factor. Vitr. Cell. Dev. Biol. 1987, 23, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Nasser, A.A.; Eissa, I.H.; Oun, M.R.; El-Zahabi, M.A.; Taghour, M.S.; Belal, A.; Saleh, A.M.; Mehany, A.B.; Luesch, H.; Mostafa, A.E. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M. Org. Biomol. Chem. 2020, 18, 7608–7634. [Google Scholar] [CrossRef] [PubMed]
- Traxler, P.; Furet, P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Nandekar, P.P.; Sangamwar, A.; Pérez-Sánchez, H.; Agarwal, S.M. Structure guided design and binding analysis of EGFR inhibiting analogues of erlotinib and AEE788 using ensemble docking, molecular dynamics and MM-GBSA. RSC Adv. 2016, 6, 65725–65735. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Rezki, N.; Almehmadi, M.A.; Ihmaid, S.; Shehata, A.M.; Omar, A.M.; Ahmed, H.E.; Aouad, M.R. Novel scaffold hopping of potent benzothiazole and isatin analogues linked to 1, 2, 3-triazole fragment that mimic quinazoline epidermal growth factor receptor inhibitors: Synthesis, antitumor and mechanistic analyses. Bioorganic Chem. 2020, 103, 104133. [Google Scholar] [CrossRef] [PubMed]
- Aouad, M.R.; Soliman, M.A.; Alharbi, M.O.; Bardaweel, S.K.; Sahu, P.K.; Ali, A.A.; Messali, M.; Rezki, N.; Al-Soud, Y.A. Design, synthesis and anticancer screening of novel benzothiazole-piperazine-1, 2, 3-triazole hybrids. Molecules 2018, 23, 2788. [Google Scholar] [CrossRef] [PubMed]
- Aouad, M.R.; Almehmadi, M.A.; Rezki, N.; Al-blewi, F.F.; Messali, M.; Ali, I. Design, click synthesis, anticancer screening and docking studies of novel benzothiazole-1, 2, 3-triazoles appended with some bioactive benzofused heterocycles. J. Mol. Struct. 2019, 1188, 153–164. [Google Scholar] [CrossRef]
- Sainsbury, J.R.; Farndon, J.R.; Needham, G.K.; Malcolm, A.J.; Harris, A.L. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet 1987, 1, 1398–1402. [Google Scholar] [PubMed]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol./Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Musa, A.; Ihmaid, S.K.; Hughes, D.L.; Said, M.A.; Abulkhair, H.S.; El-Ghorab, A.H.; Abdelgawad, M.A.; Shalaby, K.; Shaker, M.E.; Alharbi, K.S.; et al. The anticancer and EGFR-TK/CDK-9 dual inhibitory potentials of new synthetic pyranopyrazole and pyrazolone derivatives: X-ray crystallography, in vitro, and in silico mechanistic investigations. J. Biomol. Struct. Dyn. 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ihmaid, S.K.; Alraqa, S.Y.; Aouad, M.R.; Aljuhani, A.; Elbadawy, H.M.; Salama, S.A.; Rezki, N.; Ahmed, H.E.A. Design of molecular hybrids of phthalimide-triazole agents with potent selective MCF-7/HepG2 cytotoxicity: Synthesis, EGFR inhibitory effect, and metabolic stability. Bioorganic Chem. 2021, 111, 104835. [Google Scholar] [CrossRef]
- Othman, D.I.A.; Hamdi, A.; Tawfik, S.S.; Elgazar, A.A.; Mostafa, A.S. Identification of new benzimidazole-triazole hybrids as anticancer agents: Multi-target recognition, in vitro and in silico studies. J. Enzym. Inhib. Med. Chem. 2023, 38, 2166037. [Google Scholar] [CrossRef] [PubMed]
- Elgaafary, M.; Fouda, A.M.; Mohamed, H.M.; Hamed, A.; El-Mawgoud, H.K.A.; Jin, L.; Ulrich, J.; Simmet, T.; Syrovets, T.; El-Agrody, A.M. Synthesis of β-Enaminonitrile-Linked 8-Methoxy-1H-Benzo[f]Chromene Moieties and Analysis of Their Antitumor Mechanisms. Front. Chem. 2021, 9, 759148. [Google Scholar] [CrossRef]
- Ismail, N.S.M.; Ali, E.M.H.; Ibrahim, D.A.; Serya, R.A.T.; Abou El Ella, D.A. Pyrazolo[3,4-d]pyrimidine based scaffold derivatives targeting kinases as anticancer agents. Future J. Pharm. Sci. 2016, 2, 20–30. [Google Scholar] [CrossRef]
- Kanipandian, N.; Li, D.; Kannan, S. Induction of intrinsic apoptotic signaling pathway in A549 lung cancer cells using silver nanoparticles from Gossypium hirsutum and evaluation of in vivo toxicity. Biotechnol. Rep. 2019, 23, e00339. [Google Scholar] [CrossRef]
- Wilkins, R.C.; Kutzner, B.; Truong, M.; Sanchez-Dardon, J.; McLean, J. Analysis of radiation-induced apoptosis in human lymphocytes: Flow cytometry using Annexin V and propidium iodide versus the neutral comet assay. Cytom. J. Int. Soc. Anal. Cytol. 2002, 48, 14–19. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Gaber, A.A.; Sobhy, M.; Turky, A.; Abdulwahab, H.G.; Al-Karmalawy, A.A.; Elhendawy, M.A.; Radwan, M.M.; Elkaeed, E.B.; Ibrahim, I.M.; Elzahabi, H.S.A.; et al. Discovery of new 1H-pyrazolo[3,4-d]pyrimidine derivatives as anticancer agents targeting EGFR(WT) and EGFR(T790M). J. Enzym. Inhib. Med. Chem. 2022, 37, 2283–2303. [Google Scholar] [CrossRef]
- Allouche, F.; Chabchoub, F.; Carta, F.; Supuran, C.T. Synthesis of aminocyanopyrazoles via a multi-component reaction and anti-carbonic anhydrase inhibitory activity of their sulfamide derivatives against cytosolic and transmembrane isoforms. J. Enzym. Inhib. Med. Chem. 2013, 28, 343–349. [Google Scholar] [CrossRef] [PubMed]
- He, H.Y.; Zhao, J.N.; Jia, R.; Zhao, Y.L.; Yang, S.Y.; Yu, L.T.; Yang, L. Novel pyrazolo[3,4-d]pyrimidine derivatives as potential antitumor agents: Exploratory synthesis, preliminary structure-activity relationships, and in vitro biological evaluation. Molecules 2011, 16, 10685–10694. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Demirci, F.; Başer, K.H.C. Bioassay Techniques for Drug Development By Atta-ur-Rahman, M. Iqbal Choudhary (HEJRIC, University of Karachi, Pakistan), William J. Thomsen (Areana Pharmaceuticals, San Diego, CA). Harwood Academic Publishers, Amsterdam, The Netherlands. 2001. xii + 223 pp. 15.5 × 23.5 cm. $79.00. ISBN 90-5823-051-1. J. Nat. Prod. 2002, 65, 1086–1087. [Google Scholar]
- El-Mawgoud, H.K.A.; Fouda, A.M.; El-Nassag, M.A.A.; Elhenawy, A.A.; Alshahrani, M.Y.; El-Agrody, A.M. Discovery of novel rigid analogs of 2-naphthol with potent anticancer activity through multi-target topoisomerase I & II and tyrosine kinase receptor EGFR & VEGFR-2 inhibition mechanism. Chem.-Biol. Interact. 2022, 355, 109838. [Google Scholar]
- Van Engeland, M.; Nieland, L.J.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Schipany, K.; Hengstschlager, M. Merging high-quality biochemical fractionation with a refined flow cytometry approach to monitor nucleocytoplasmic protein expression throughout the unperturbed mammalian cell cycle. Nat. Protoc. 2013, 8, 602–626. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) Chemical Computing Group, Quebec, Canada. 2012. Available online: http://www.chemcomp.com (accessed on 30 February 2013).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Ar | HePG-2 | MCF-7 | HCT-116 | WI-38 |
|---|---|---|---|---|---|
| DOX | 4.50 ± 0.2 | 4.17 ± 0.2 | 5.23 ± 0.3 | 10.63 ± 0.47 | |
 | |||||
| 5a | C6H5 | 15.10 ± 1.2 | 21.13 ± 1.5 | 11.69 ± 0.9 | |
| 5b | 4-OH-C6H5 | 9.12 ± 0.7 | 10.48 ± 0.7 | 5.65 ± 0.3 | 35 ± 0.3 |
| 5c | 4-Cl-C6H5 | 83.71 ± 4.4 | 66.61 ± 3.8 | 39.54 ± 2.6 | - |
| 5g | 2,6-Cl-C6H5 | >100 | >100 | >100 | - |
| 5d | 4-OMe-C6H5 | 18.34 ± 1.5 | 19.36 ± 1.3 | 9.87 ± 0.7 | - |
| 5e | 2-OMe-C6H5 | 13.79 ± 1.0 | 16.07 ± 1.1 | 8.15 ± 0.5 | - |
| 5f | 4-NO2-C6H5 | >100 | >100 | 89.68 ± 4.8 | - |
| 5h | 4-CH=CH-C6H5 | >100 | >100 | 76.84 ± 4.1 | - |
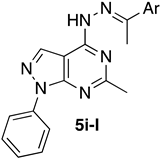 | |||||
| 5i | C6H5 | 6.38 ± 0.1 | 3.81 ± 0.3 | 4.38 ± 0.2 | >100 |
| 5j | 4-NH2-C6H5 | 44.31 ± 2.9 | 36.04 ± 2.4 | 21.50 ± 1.5 | - |
| 5k | 4-OMe-C6H5 | 91.63 ± 5.1 | 88.29 ± 4.7 | 78.05 ± 4.2 | - |
| 5l | 2-Br-4-Cl-C6H5 | 17.08 ± 1.3 | 29.15 ± 1.8 | 14.59 ± 1.1 | - |
 | |||||
| 6a | H | 37.50 ± 2.7 | 49.65 ± 3.1 | 34.60 ± 2.2 | - |
| 6b | CF3 | 25.16 ± 2.0 | 39.10 ± 2.5 | 26.06 ± 1.8 | - |
| 6c | CCl3 | 56.18 ± 3.5 | 54.19 ± 3.4 | 37.18 ± 2.4 | - |
 | |||||
| 6d | 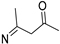 | 89.22 ± 4.7 | 84.58 ± 4.6 | 93.43 ± 5.2 | - |
| 7 | 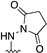 | 87.21 ± 4.5 | 81.02 ± 4.6 | 73.50 ± 3.8 | - |
| 8 | 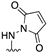 | 78.53 ± 4.3 | 79.46 ± 4.4 | 66.30 ± 3.5 | - |
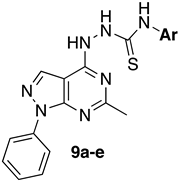 | |||||
| 9a | -CH2CH3 | 5.97 ± 0.3 | 7.61 ± 0.4 | 2.96 ± 0.1 | - |
| 9b | -CH2CH2CH3 | 23.88 ± 1.8 | 34.18 ± 2.0 | 17.59 ± 1.3 | - |
| 9c | -CH2CH2CH2CH3 | 20.34 ± 1.6 | 31.42 ± 1.9 | 16.62 ± 1.2 | - |
| 9d | -=CH2 | 32.61 ± 2.4 | 35.19 ± 2.2 | 19.72 ± 1.4 | - |
| 9e | Phenyl | 7.80 ± 0.4 | 8.99 ± 0.5 | 6.87 ± 0.5 | 66 ± 0.23 |
| Cell Line | Time | 5b | 5i | 9e | Dox |
|---|---|---|---|---|---|
| HePG-2 | 24 h | 9.12 ± 0.7 | 6.38 ± 0.1 | 7.80 ± 0.4 | 4.50 ± 0.2 |
| 48 h | 7.60 ± 0.7 | 3.34 ± 0.2 | 8.28 ± 0.8 | 3.24 ± 0.2 | |
| 72 h | 5.16 ± 0.4 | 2.42 ± 0.2 | 5.93 ± 0.5 | 1.07 ± 0.1 | |
| MCF-7 | 24 h | 10.48 ± 0.7 | 3.81 ± 0.3 | 8.99 ± 0.5 | 4.17 ± 0.2 |
| 48 h | 9.82 ± 0.9 | 3.34 ± 0.2 | 8.16 ± 0.8 | 2.82 ± 0.1 | |
| 72 h | 5.16 ± 0.4 | 1.24 ± 0.3 | 7.45 ± 0.7 | 0.69 ± 0.1 | |
| HCT116 | 24 h | 5.65 ± 0.3 | 4.38 ± 0.2 | 6.87 ± 0.5 | 5.23 ± 0.3 |
| 48 h | 9.82 ± 0.9 | 4.01 ± 0.3 | 6.77 ± 0.4 | 4.07 ± 0.3 | |
| 72 h | 4.09 ± 0.3 | 2.76 ± 0.3 | 5.55 ± 0.4 | 2.79 ± 0.2 |
| Compound | IC50 (µM) | |||
|---|---|---|---|---|
| EGFRWT | EGFRT790M | VGFR2 | Topo-II | |
| 5b | 0.78 ± 0.02 | 1.93 ± 0.08 | 15.7 ± 0.32 | 108.3 ± 2.20 |
| 5i | 0.37 ± 0.18 | 0.56 ± 0.04 | 7.60 ± 0.15 | 24.7 ± 0.51 |
| 9e | 0.77 ± 0.018 | - | 16.24 ± 0.36 | 24.6 ± 0.54 |
| Etoposide | - | - | - | 33.6 ± 1.16 |
| Sorafenib | - | - | 0.073 ± 0.002 | - |
| Erlotinib | 0.13 ± 0.003 | 0.035 ± 0.001 | - | - |
| ID | Molecular Properties | Druggability | Toxicity Risks | %ABS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TPSA | Mwt | Solubility | ClogP | Drug-likeness | Drug-Score | M | T | I | R | ||
| 5b | 88.22 | 344 | −4.09 | 4.29 | 3.97 | 0.66 | Safe | 78.56 | |||
| 5i | 67.99 | 342 | −4.74 | 4.57 | 4.26 | 0.58 | Safe | 85.54 | |||
| Sorafenib | 92.35 | 464 | −6.69 | 4.14 | −4.2 | 0.2 | Safe | 77.13 | |||
| Erlotinib | 74.73 | 393 | −3.53 | 3.07 | −6.73 | 0.38 | Safe | 83.21 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aljohani, A.K.B.; El Zaloa, W.A.Z.; Alswah, M.; Seleem, M.A.; Elsebaei, M.M.; Bayoumi, A.H.; El-Morsy, A.M.; Almaghrabi, M.; Awaji, A.A.; Hammad, A.; et al. Development of Novel Class of Phenylpyrazolo[3,4-d]pyrimidine-Based Analogs with Potent Anticancer Activity and Multitarget Enzyme Inhibition Supported by Docking Studies. Int. J. Mol. Sci. 2023, 24, 15026. https://doi.org/10.3390/ijms241915026
Aljohani AKB, El Zaloa WAZ, Alswah M, Seleem MA, Elsebaei MM, Bayoumi AH, El-Morsy AM, Almaghrabi M, Awaji AA, Hammad A, et al. Development of Novel Class of Phenylpyrazolo[3,4-d]pyrimidine-Based Analogs with Potent Anticancer Activity and Multitarget Enzyme Inhibition Supported by Docking Studies. International Journal of Molecular Sciences. 2023; 24(19):15026. https://doi.org/10.3390/ijms241915026
Chicago/Turabian StyleAljohani, Ahmed K. B., Waheed Ali Zaki El Zaloa, Mohamed Alswah, Mohamed A. Seleem, Mohamed M. Elsebaei, Ashraf H. Bayoumi, Ahmed M. El-Morsy, Mohammed Almaghrabi, Aeshah A. Awaji, Ali Hammad, and et al. 2023. "Development of Novel Class of Phenylpyrazolo[3,4-d]pyrimidine-Based Analogs with Potent Anticancer Activity and Multitarget Enzyme Inhibition Supported by Docking Studies" International Journal of Molecular Sciences 24, no. 19: 15026. https://doi.org/10.3390/ijms241915026
APA StyleAljohani, A. K. B., El Zaloa, W. A. Z., Alswah, M., Seleem, M. A., Elsebaei, M. M., Bayoumi, A. H., El-Morsy, A. M., Almaghrabi, M., Awaji, A. A., Hammad, A., Alsulaimany, M., & Ahmed, H. E. A. (2023). Development of Novel Class of Phenylpyrazolo[3,4-d]pyrimidine-Based Analogs with Potent Anticancer Activity and Multitarget Enzyme Inhibition Supported by Docking Studies. International Journal of Molecular Sciences, 24(19), 15026. https://doi.org/10.3390/ijms241915026