
Broadband EPR Spectroscopy of the Triplet State: Multi-Frequency Analysis of Copper Acetate Monohydrate


Abstract
:1. Introduction
2. Results and Discussion
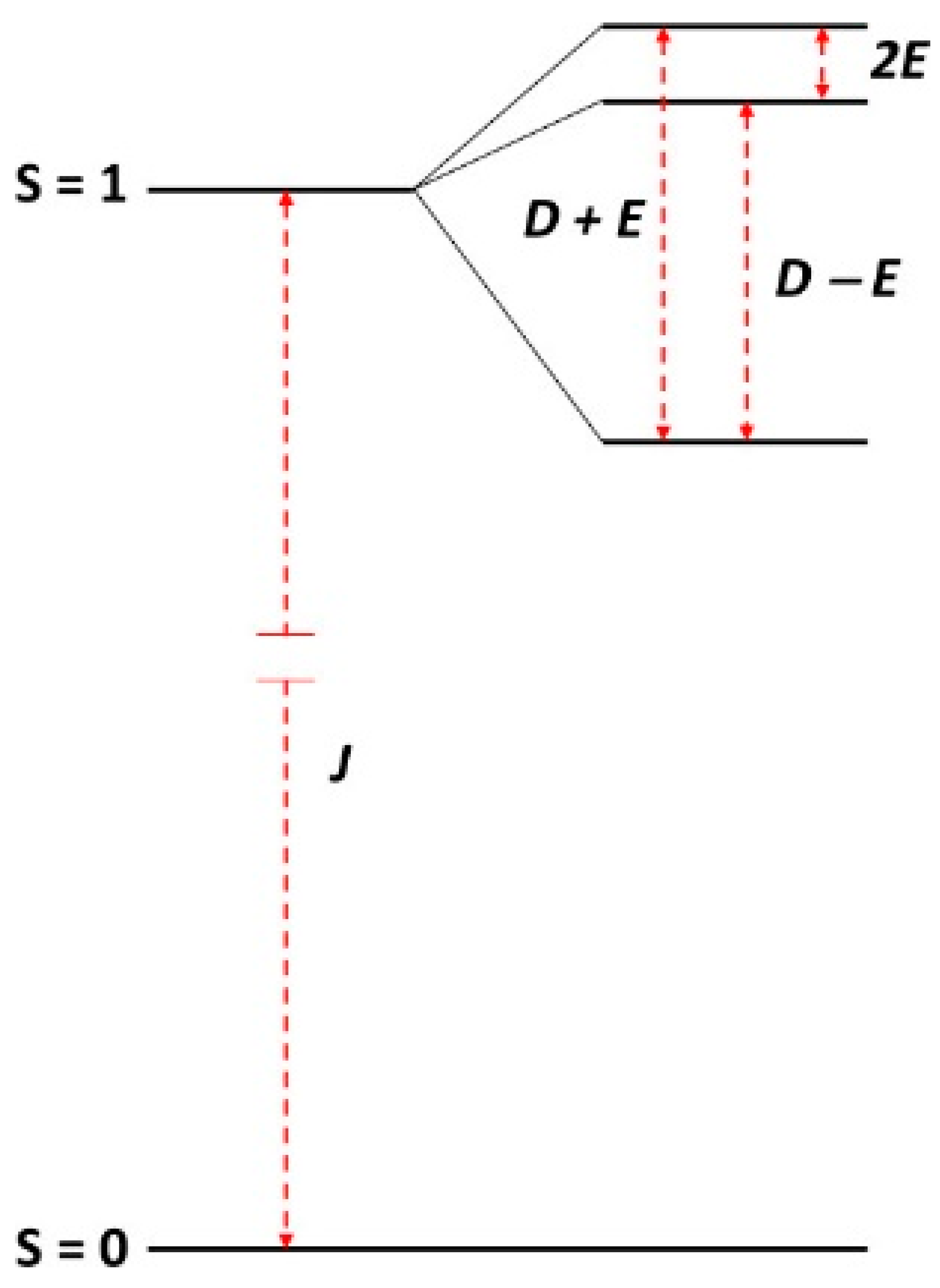
2.1. Description of the Model
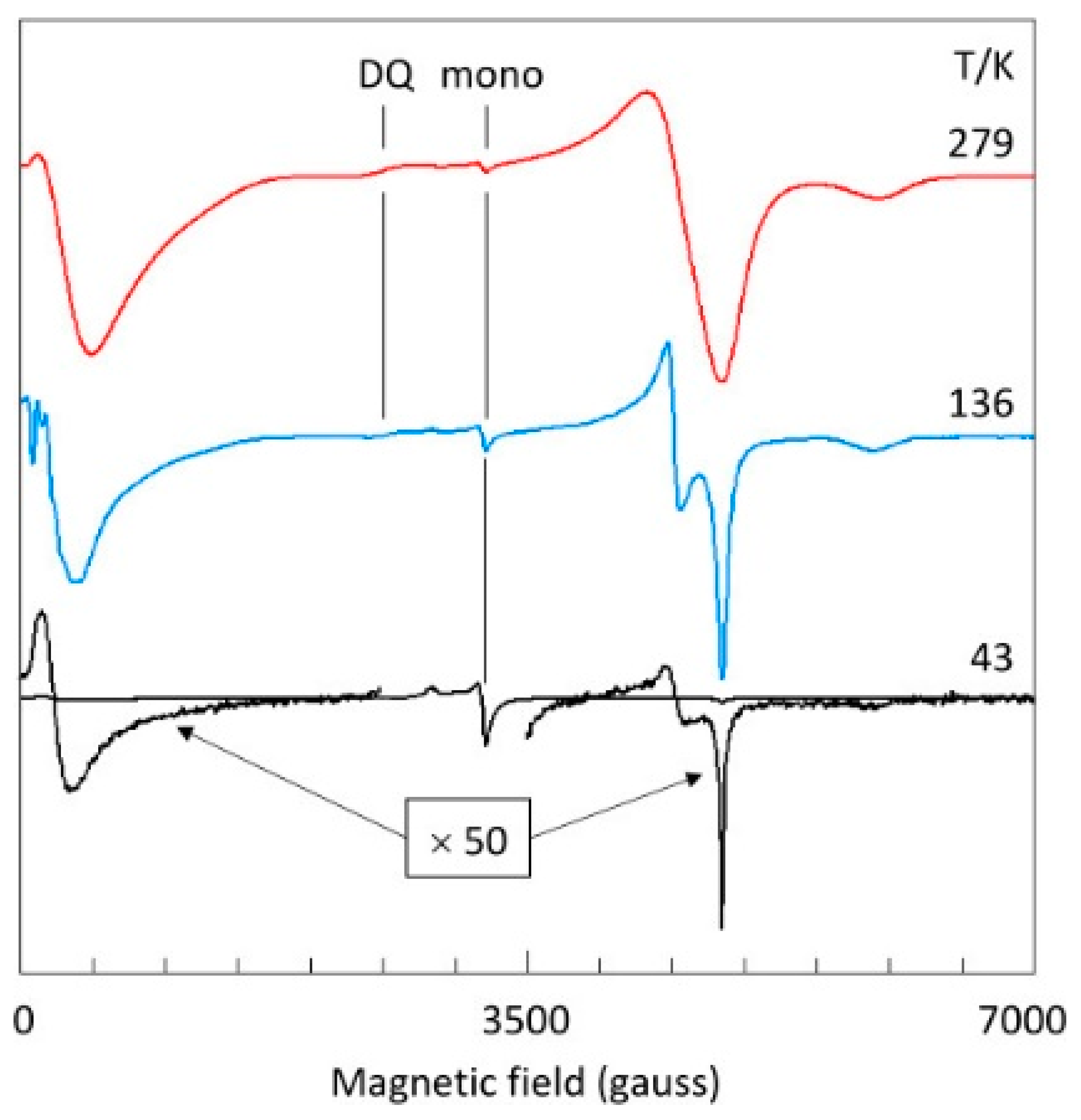
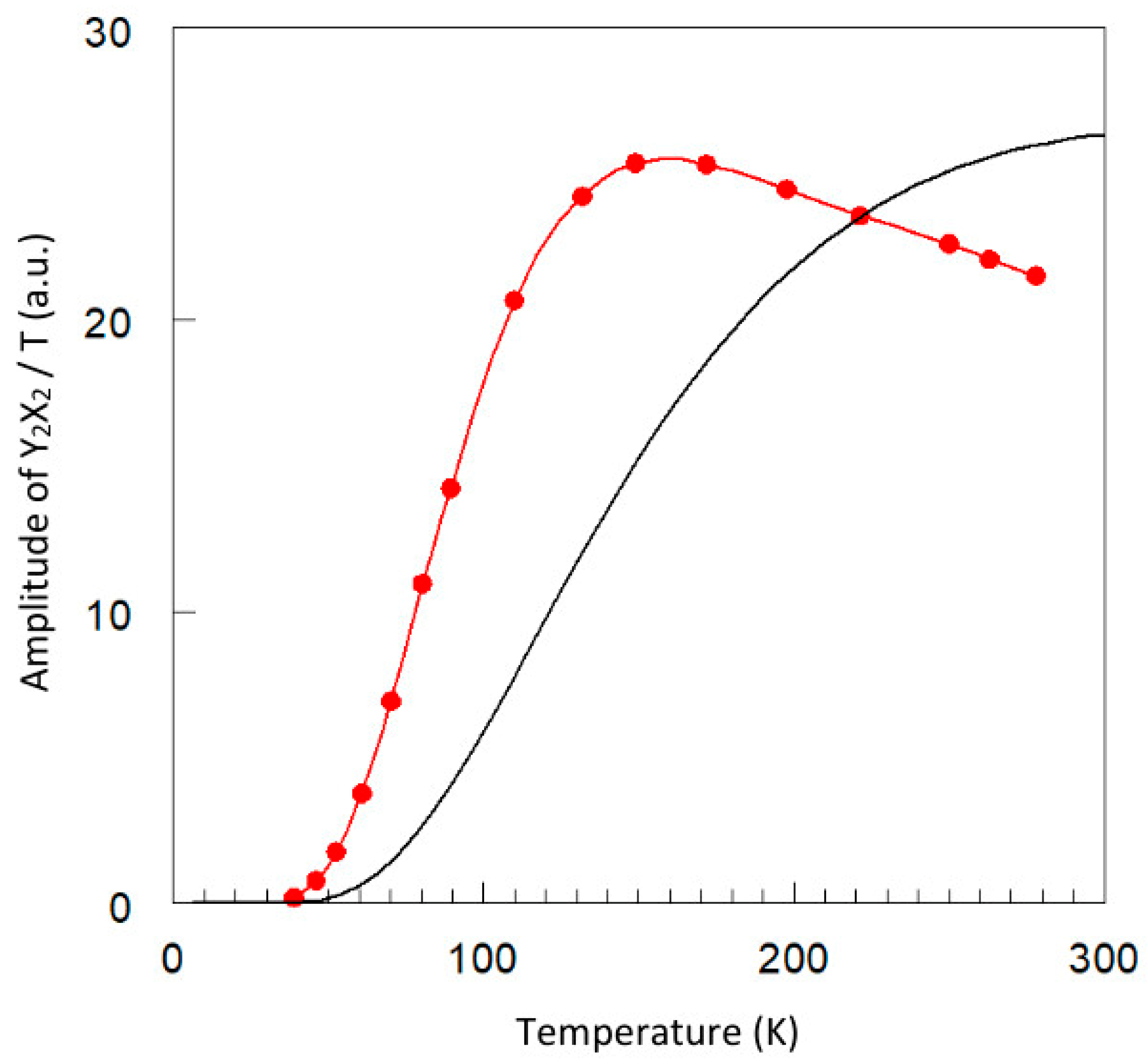
2.2. Temperature Dependence of Copper Acetate X-Band EPR
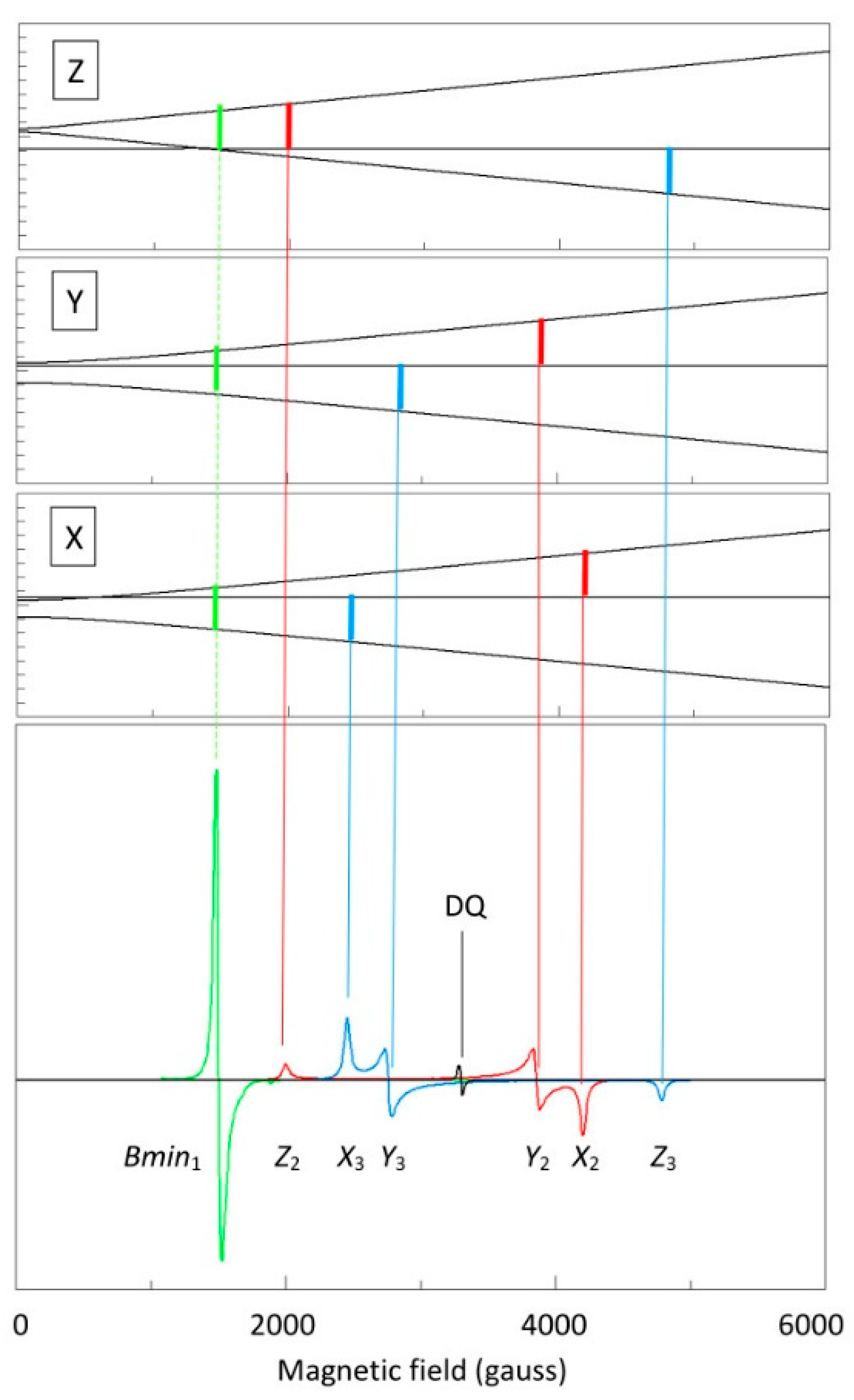
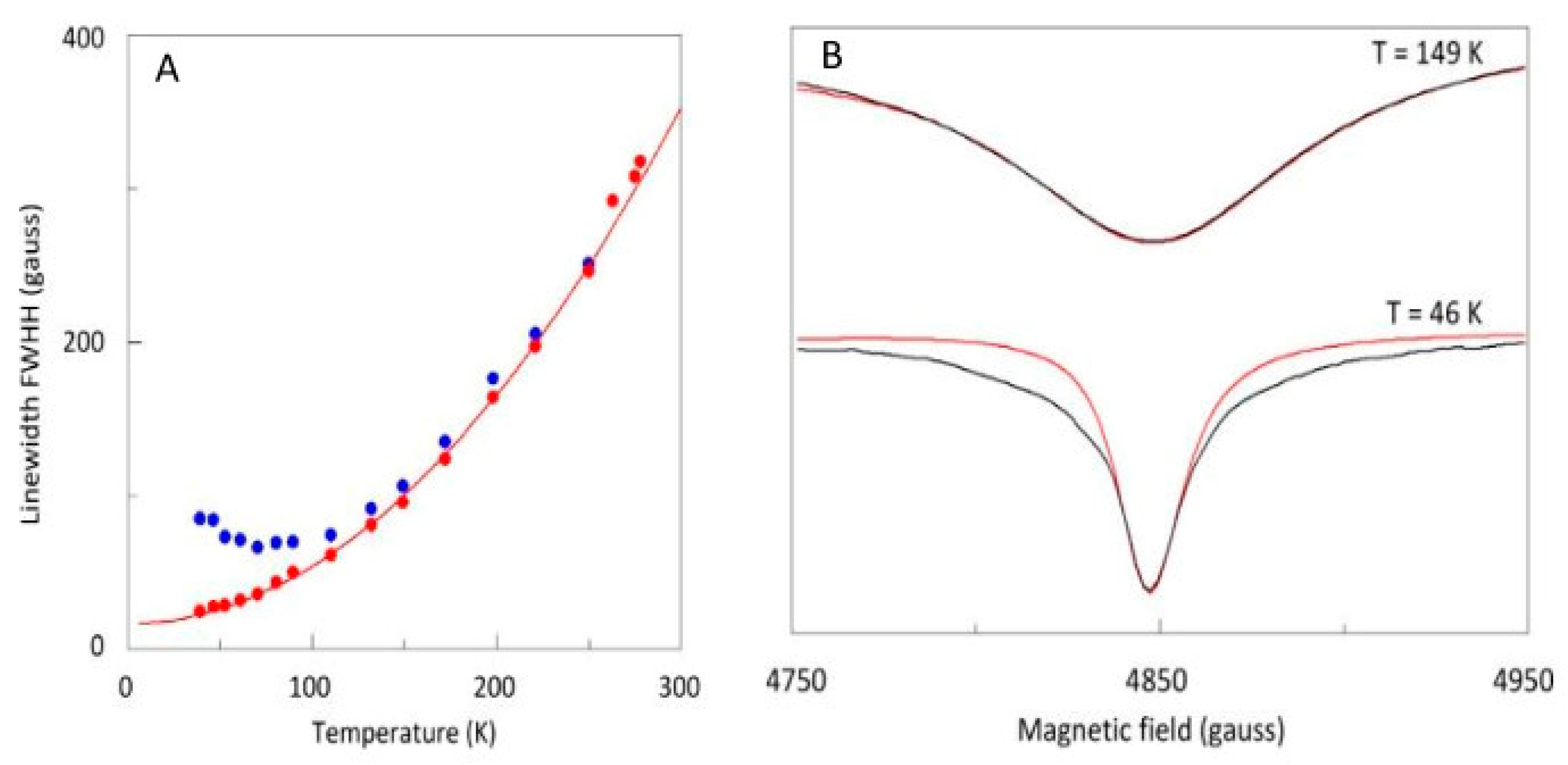
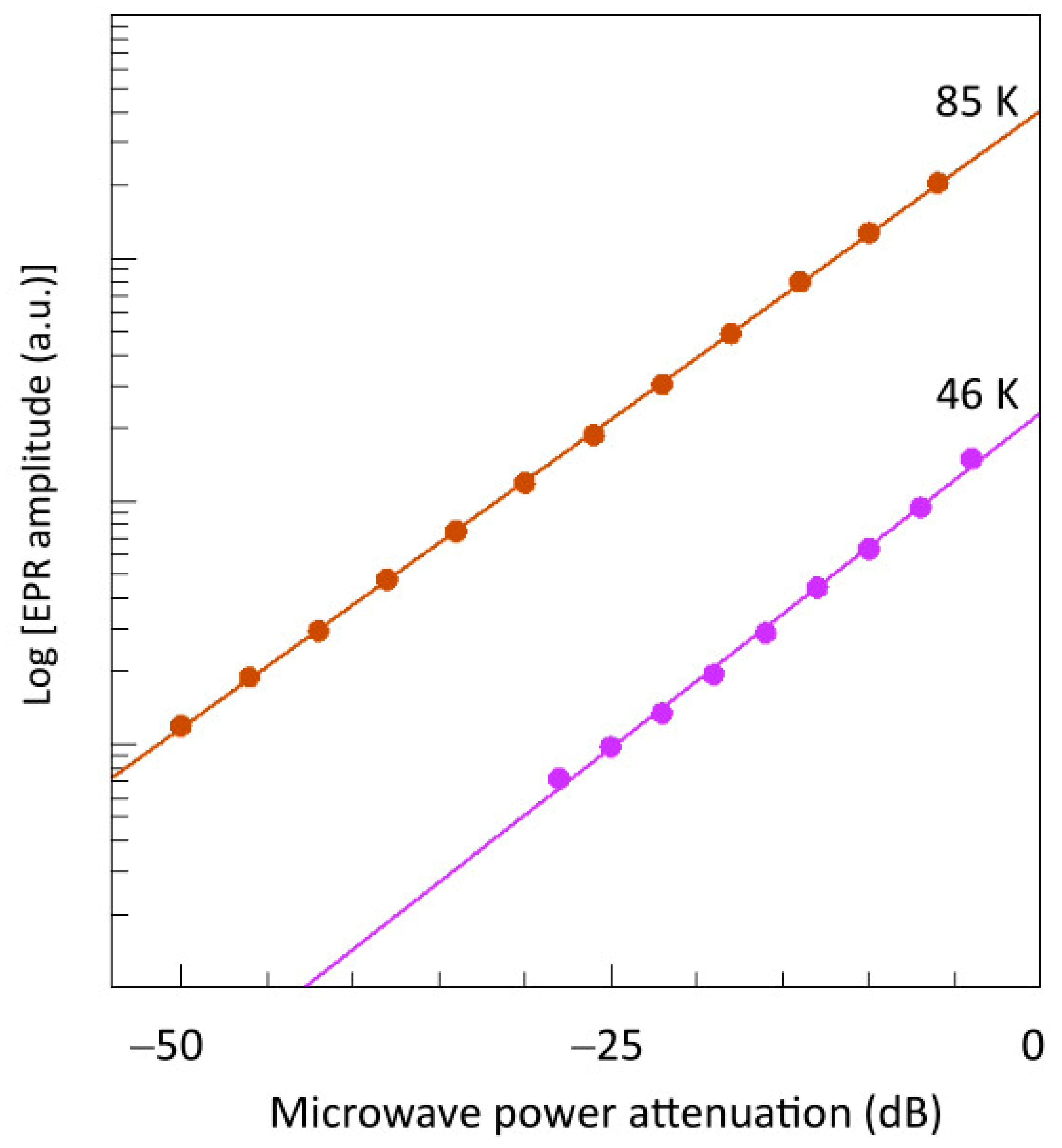
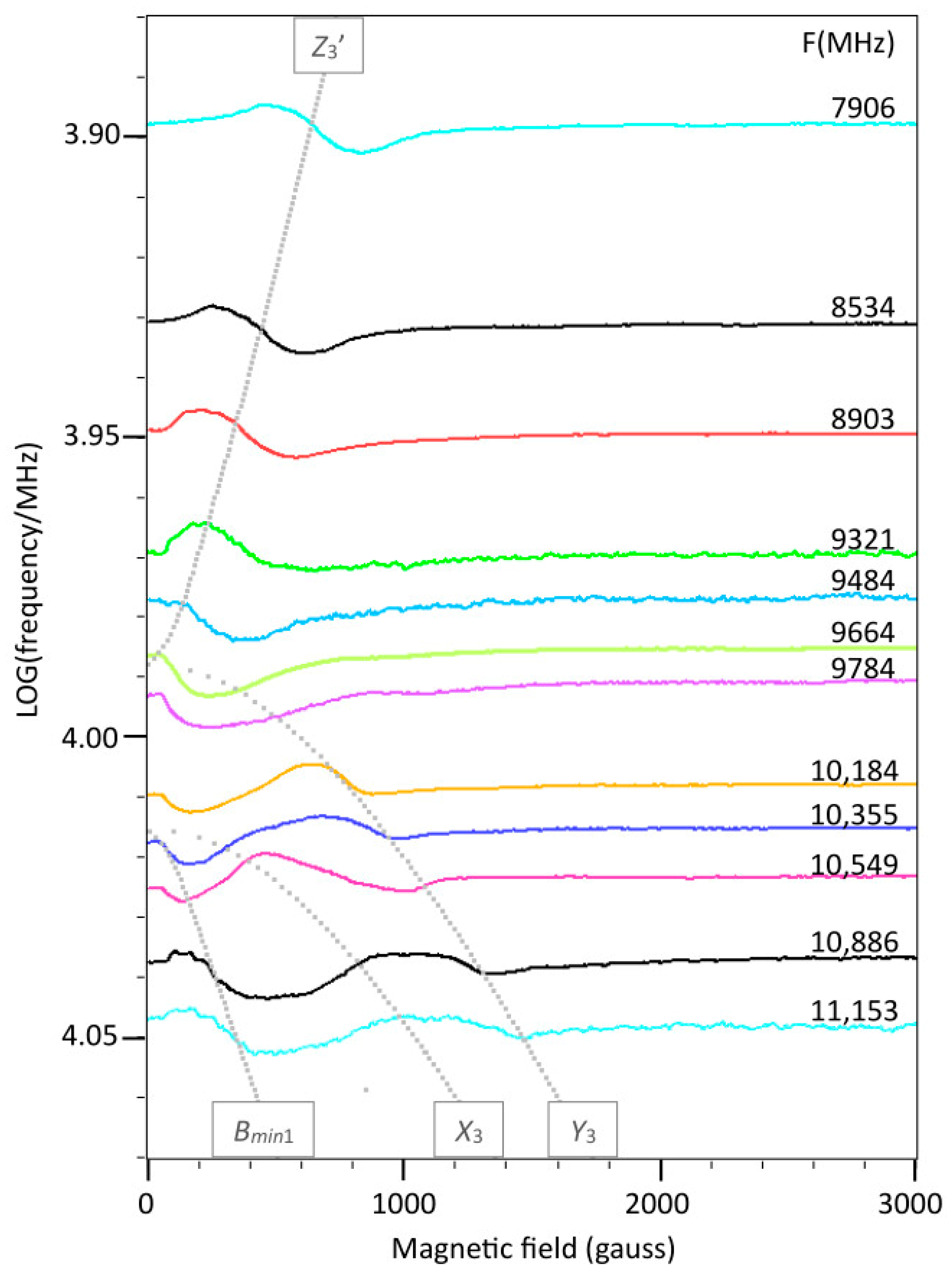
2.3. Broadband EPR of Copper Acetate
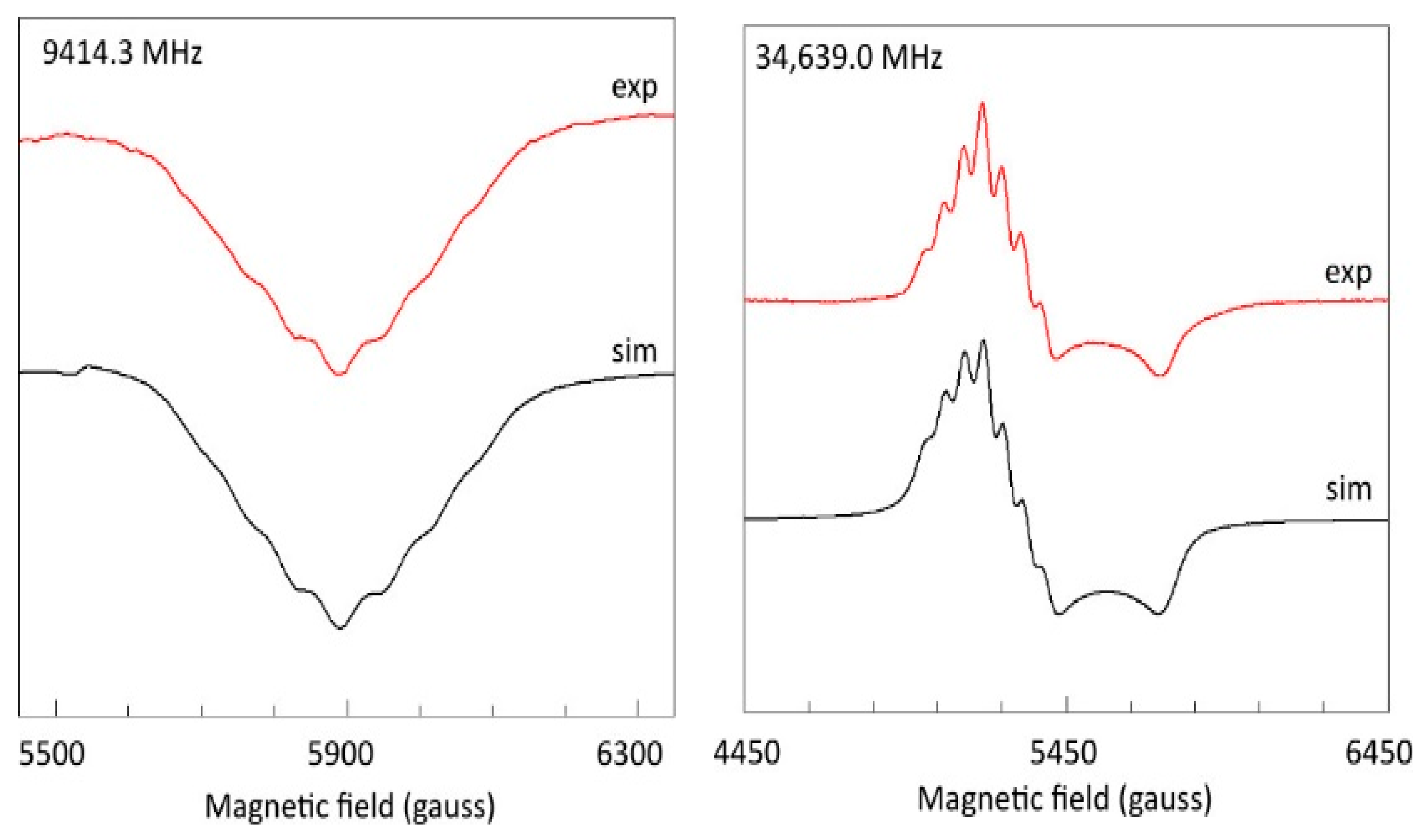
2.4. Spectral Simulation
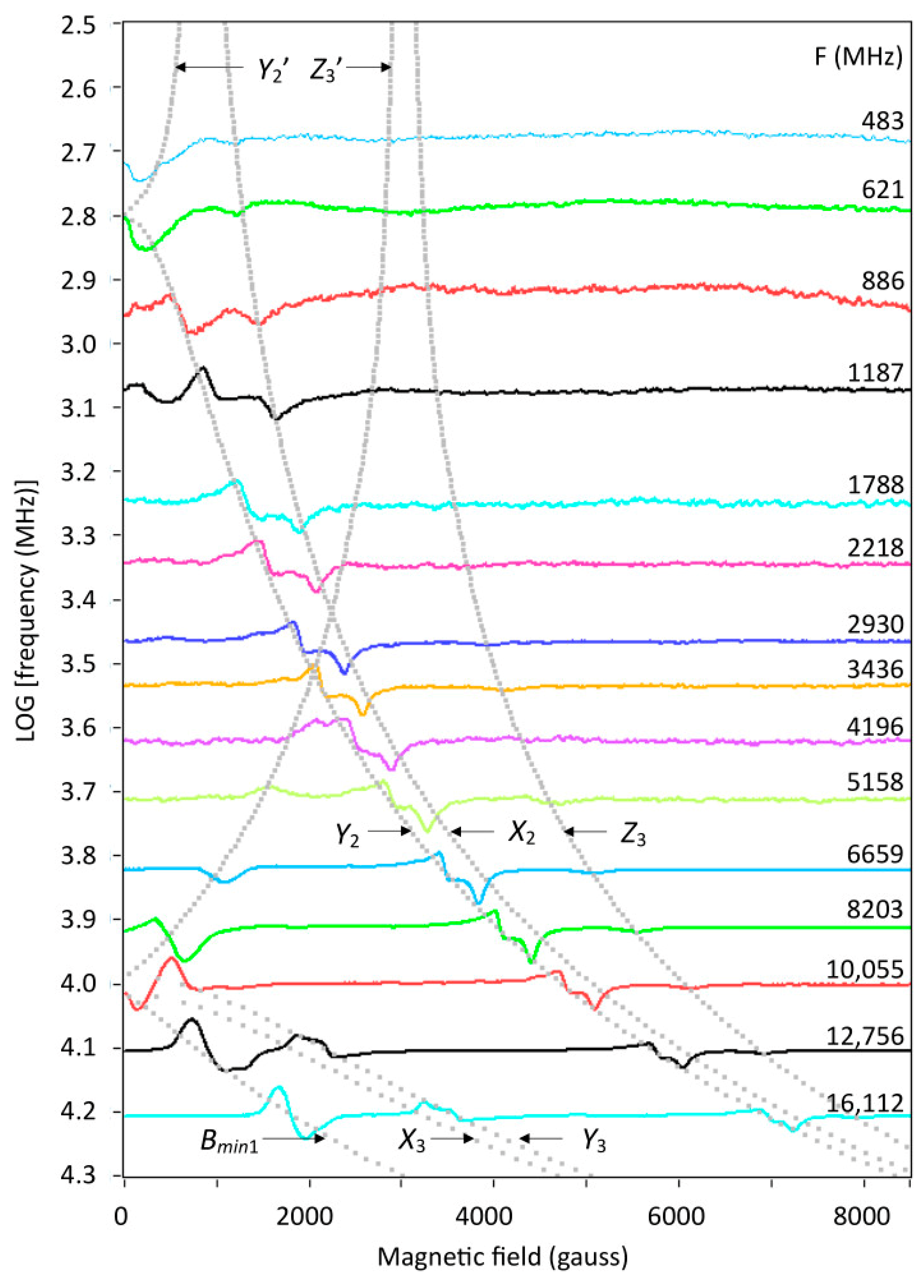
2.5. Multi-Fequency Spectra Close to Zero Field
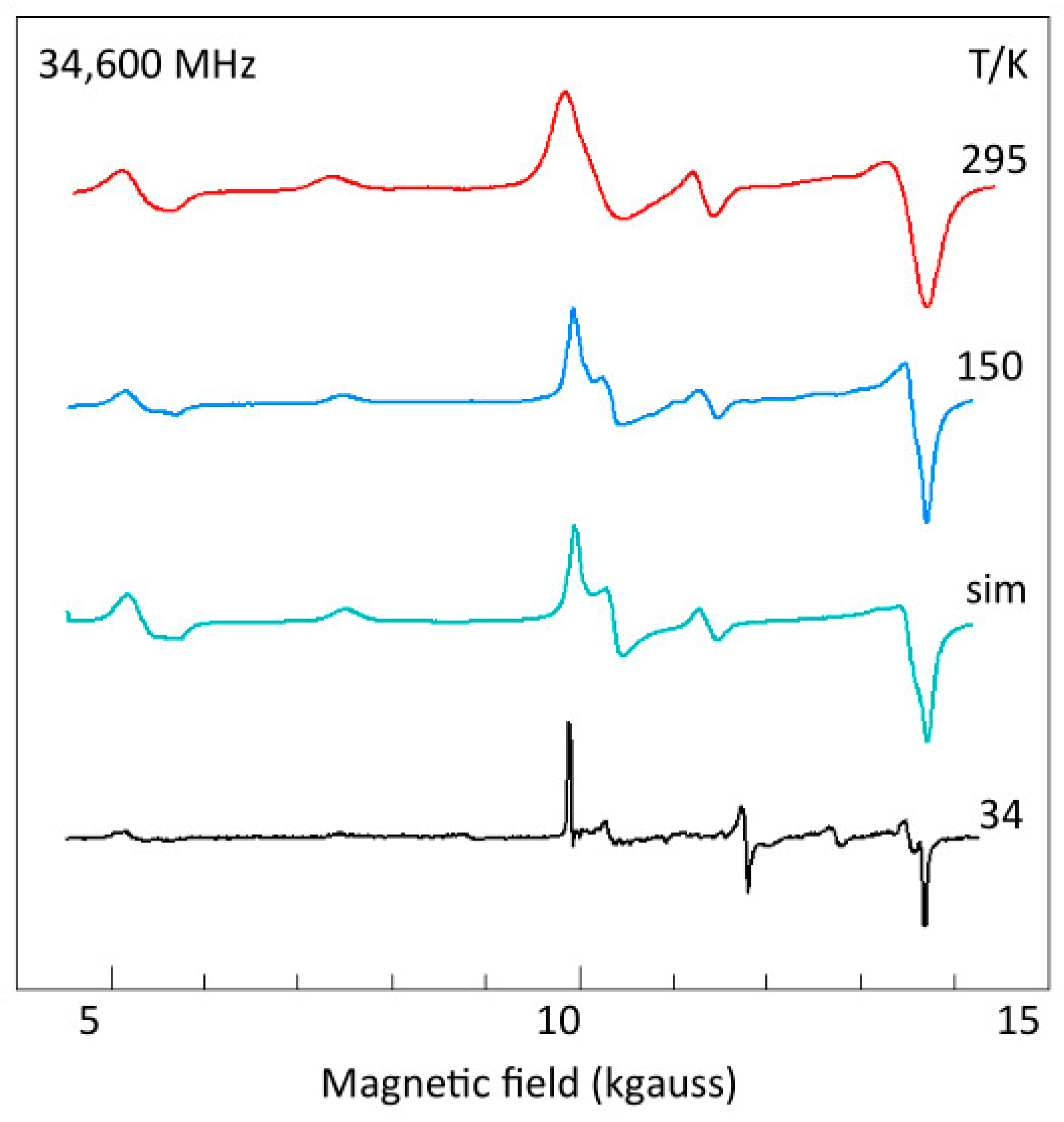
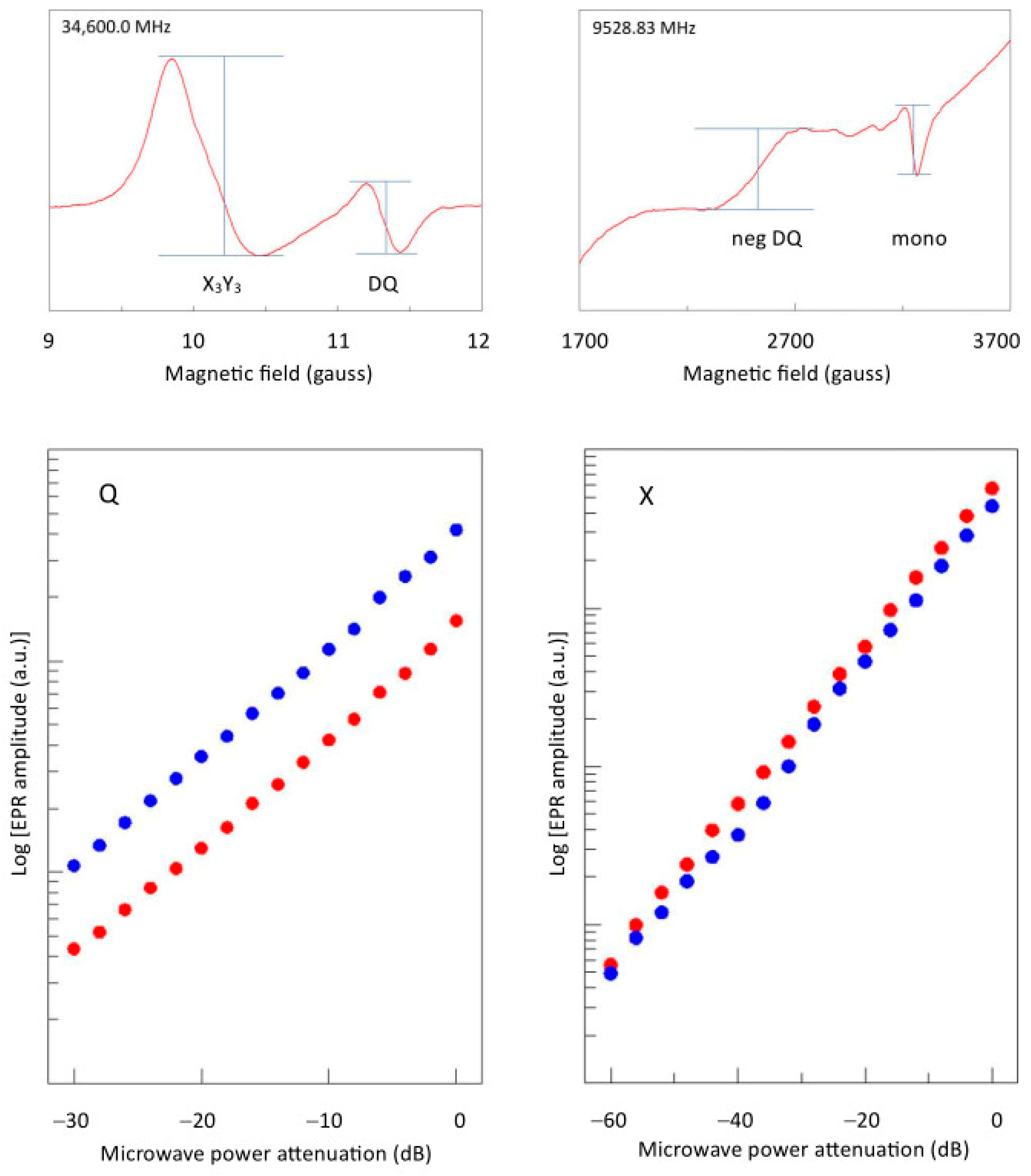
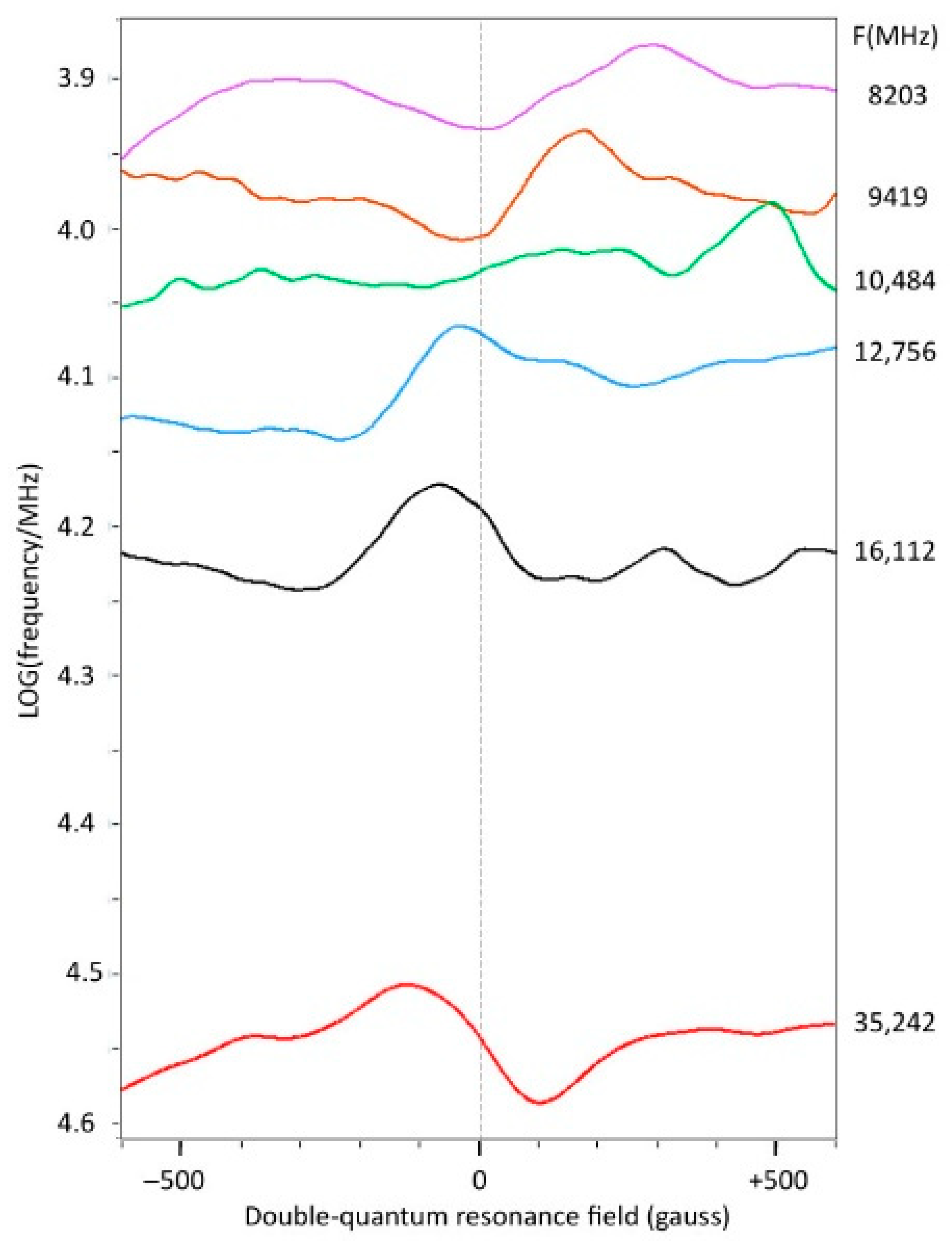
2.6. The Double-Quantum Line in Broadband EPR
2.7. The Copper Hyperfine Interaction
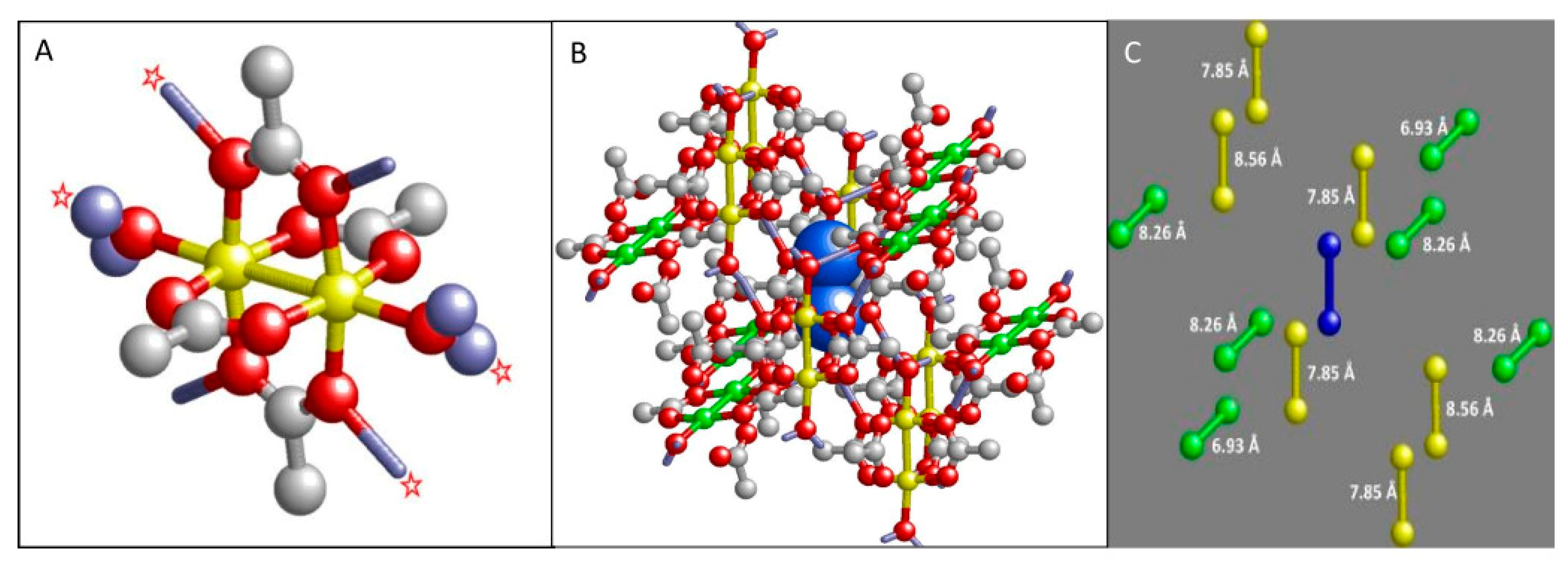
3. Materials and Methods
4. Conclusions
- Low-field broadband EPR spectroscopy allows for comprehensive study of S = 1 systems, in particular when the axial zero-field splitting parameter D is comparable to the X-band quantum or less;
- All regimes D > hν, D ≈ hν, and D < hν can be covered;
- All three zero-field transitions at hν = 2E, D − E, and D + E, can be sampled;
- Global analysis of broadband S = 1 EPR gives spin Hamiltonian parameters with error estimates;
- The EPR of copper acetate monohydrate is rhombic (gx ≠ gy; E ≠ 0) consistent with deformation by hydrogen bonds in a plane;
- Broadband EPR allows for unequivocal line shape and linewidth analysis, e.g., in the present case of copper acetate monohydrate a homogeneous Lorentzian line shape from T1 relaxation is found with no, or negligible, inhomogeneous contributions from g strain, D strain, and/or unresolved superhyperfine splittings, but possibly with finite contributions from E strain;
- Contributions to the linewidth from dipolar interactions are negligible under the assumption of the point-dipole model, however, they may be significant under a spatial dipole model;
- Spin-lattice relaxation includes a thus far rarely encountered inverted Orbach mechanism, which–contrary to regular Orbach relaxation–is virtually temperature-independent;
- At temperatures below 150 K, the X2 resonance continues to sharpen but the Y2 resonance re-broadens; the latter phenomenon is not understood;
- Broadband EPR of copper acetate monohydrate encompasses a linear double-quantum transition, with a complex line shape dependency on frequency, which calls for future experimental and theoretical studies.
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weil, J.A.; Bolton, J.R. Electron Paramagnetic Resonance; Elementary Theory and Practical Applications, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Hyde, J.; Froncisz, W. The role of microwave frequency in EPR spectroscopy of copper complexes. Ann. Rev. Biophys. Bioeng. 1982, 11, 391–417. [Google Scholar] [CrossRef] [PubMed]
- Hagen, W.R.; Albracht, S.P.J. Analysis of strain-induced EPR-line shapes and anisotropic spin-lattice relaxation in a [2Fe-2S] ferredoxin. Biochim. Biophys. Acta 1982, 702, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Hagen, W.R. Biomolecular EPR Spectroscopy; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Hagen, W.R. High-frequency EPr of transition ion complexes and metalloproteins. Coord. Chem. Rev. 1999, 190–192, 209–229. [Google Scholar] [CrossRef]
- Bennati, M.; Prisner, T.F. New developments in high field electron paramagnetic resonance with applications in structural biology. Rep. Prog. Phys. 2005, 68, 411–448. [Google Scholar] [CrossRef]
- Krzystek, J.; Zvyagin, S.A.; Ozarowski, A.; Trofimenko, S.; Telser, J. Tunable-frequency high-field electron paramagnetic resonance. J. Magn. Reson. 2006, 178, 174–183. [Google Scholar] [CrossRef]
- Möbium, K.; Savitsky, A. High-field/high-frequency EPR spectroscopy in protein research: Principles and examples. Appl. Magn. Reason. 2023, 54, 207–287. [Google Scholar]
- Hagen, W.R. Broadband transmission EPR spectroscopy. PLoS ONE 2013, 8, e59874. [Google Scholar] [CrossRef]
- Hagen, W.R. Broadband tunable electron paramagnetic resonance spectroscopy of dilute metal complexes. J. Phys. Chem. A 2019, 123, 6986–6995. [Google Scholar] [CrossRef]
- Hagen, W.R. Very low-frequency broadband electron paramagnetic resonance spectroscopy of metalloproteins. J. Phys. Chem. A 2021, 125, 3208–3218. [Google Scholar] [CrossRef]
- Hagen, W.R. Low-frequency EPR of ferrimyoglobin fluoride and ferrimyoglobin cyanide: A case study on the applicability of broadband analysis to high-spin hemoproteins and to HALS hemoproteins. J. Biol. Inorg. Chem. 2022, 27, 497–507. [Google Scholar] [CrossRef]
- Hagen, W.R. Conversion of a single-frequency X-band EPR spectrometer into a broadband multi-frequency 0.1–18 GHz instrument for analysis of complex molecular spin Hamiltonians. Molecules 2023, 28, 5281. [Google Scholar] [CrossRef] [PubMed]
- Hagen, W.R.; Louro, R.O. A comparative multi-frequency EPR study of dipolar interaction in tetra-heme cytochromes. Int. J. Mol. Sci. 2023, 24, 12713. [Google Scholar] [CrossRef] [PubMed]
- Abragam, A.; Bleaney, B. Electron Paramagnetic Resonance of Transition Ions; Clarendon Press: Oxford, UK, 1970. [Google Scholar]
- Troup, G.J.; Hutton, D.R. Paramagnetic resonance of Fe3+ in kyanite. Br. J. Appl. Phys. 1964, 15, 1493–1499. [Google Scholar] [CrossRef]
- Wasserman, E.; Snyder, L.C.; Yager, W.A. ESR of the triplet states of randomly oriented molecules. J. Chem. Phys. 1964, 41, 1763–1772. [Google Scholar] [CrossRef]
- Krzystek, J.; Fiedler, A.T.; Sokol, J.J.; Ozarowski, A.; Zvyagin, S.A.; Brunold, T.C.; Long, J.R.; Brunel, L.-C.; Telser, J. Pseudooctahedral complexes of vanadium(III): Electronic structure investigations by magnetic and electronic spectroscopy. Inorg. Chem. 2004, 43, 5645–5658. [Google Scholar] [CrossRef] [PubMed]
- Kottis, P.; Lefebre, R. Calculation of the electron spin resonance line shape of randomly oriented molecules in a triplet state. II. Correlation of the spectrum with the zero-field splittings. Introduction of an orientation-dependent linewidth. J. Chem. Phys. 1964, 41, 379–393. [Google Scholar] [CrossRef]
- Kottis, P.; Lefebre, R. Calculation of the electron spin resonance line shape of randomly oriented molecules in a triplet state. I. The ∆m = 2 transition with a constant linewidth. J. Chem. Phys. 1963, 39, 393–403. [Google Scholar] [CrossRef]
- de Groot, M.S.; van der Waals, J.H. Paramagnetic resonance in phosphorescent aromatic hydrocarbons II: Determination of the zero-field splitting from solution spectra. Mol. Phys. 1960, 3, 190–200. [Google Scholar] [CrossRef]
- van Dam, P.J.; Klaassen, A.A.K.; Reijerse, E.J.; Hagen, W.R. Application of high frequency EPR to integer spin systems: Unusual behaviour of the double-quantum line. J. Magn. Reson. 1998, 130, 140–144. [Google Scholar] [CrossRef]
- de Groot, M.S.; van der Waals, J.H. Paramagnetic resonance in phosphorescent aromatic hydrocarbons III: Conformational isomerism in benzene and triptycene. Mol. Phys. 1963, 6, 545–562. [Google Scholar] [CrossRef]
- Collison, D.; Helliwell, M.; Jones, V.M.; Mabbs, F.E.; McInnes, E.J.L.; Riedi, P.C.; Smith, G.M.; Pritchard, R.G.; Cross, W.I. Single and double quantum transitions in the multi-frequency continuous wave electron paramagnetic resonance (cwEPR) of three six co-ordinated nickel(II) complexes: [Ni(EtL)2(Me5dien)] and [Ni(5-methylpyrazole)6]X2, X = (ClO4)− or (BF4)−. The single crystal X-ray structure at room temperature of [Ni(5-methylpyrazole)6](ClO4)2. J. Chem. Soc. Faraday Trans. 1998, 94, 3015–3025. [Google Scholar]
- Bleaney, B.; Bowers, K.D. Anomalous paramagnetism of copper acetate. Proc. R. Soc. A 1952, 214, 451–456. [Google Scholar] [CrossRef]
- Jezierska, J.; Głowiak, T.; Ożarowski, A.; Yablokov, Y.V.; Rzączyńska, Z. Crystal structure, EPR and magnetic susceptibility studies of tetrakis[μ-(β-alanine)-O,O’]dichlorodicopper(II) dichloro monohydrate. Inorg. Chim. Acta 1998, 275–276, 28–36. [Google Scholar]
- Barquín, M.; González Garmendia, M.J.; Pacheco, S.; Pinilla, E.; Quintela, S.; Seco, J.M.; Torres, M.R. Synthesis, crystal structure, magnetic properties and EPR spectra of copper(II) acetate derivatives: Tetra-(μ-acetato)bis(2-methylaminopyridine)copper(II) and catena-poly(aquadiacetato-μ-3-aminomethylpyridine)copper(II). Sheets formed by chains connected through hydrogen-bonds. Inorg. Chim. Acta 2004, 357, 3230–3236. [Google Scholar]
- Lancaster, F.W.; Gordy, W. Paramagnetic resonance absorption of microwaves. J. Chem. Phys. 1951, 19, 1181–1191. [Google Scholar] [CrossRef]
- Kumagai, H.; Aba, H.; Shimada, J. Anomalous magnetic resonance absorption of Cu++. Phys. Rev. 1952, 87, 385–387. [Google Scholar] [CrossRef]
- Abe, H.; Shimada, J. Anomalous magnetic resonance absorption of copper acetate at 40 kMc/sec. Phys. Rev. 1953, 90, 316. [Google Scholar] [CrossRef]
- Elmali, A. The magnetic super-exchange coupling on copper(II) acetate monohydrate and a redetermination of the crystal structure. Turk. J. Phys. 2000, 24, 667–672. [Google Scholar]
- van Niekerk, J.N.; Schoening, F.R.L. A new type of copper complex as found in the crystal structure of cupric acetate, Cu2(CH3COO)4.2H2O. Acta Cryst. 1953, 6, 227–232. [Google Scholar] [CrossRef]
- Brown, G.M.; Chidambaram, R. Dinuclear copper(II) acetate monohydrate: A redetermination of the structure by neutron-diffraction analysis. Acta Cryst. B 1973, 29, 2393–2403. [Google Scholar] [CrossRef]
- de Meester, P.; Fletcher, S.; Skapski, A.C. Refined crystal structure of tetra-μ-acetato-bisaquodicopper(II). J. Chem. Soc. Dalton Trans. 1973, 2575–2578. [Google Scholar] [CrossRef]
- Kyuzou, M.; Mori, W.; Tanake, J. Electronic structure and spectra of cupric acetate mon-hydrate revisited. Inorg. Chim. Acta 2010, 363, 930–934. [Google Scholar]
- Bertolotti, F.; Forni, A.; Gervasio, G.; Marabello, D.; Diana, E. Experimental and theoretical charge density of hydrated copper acetate. Polyhedron 2012, 42, 118–127. [Google Scholar] [CrossRef]
- Shee, N.K.; Verma, R.; Kumar, D.; Datta, D. On copper-copper bond in hydrated copper acetate. Comput. Theor. Chem. 2015, 1061, 1–5. [Google Scholar] [CrossRef]
- Orbach, R. Spin-lattice relaxation in rare-earth salts. Proc. R. Soc. Lond. A Math. Phys. Sci. 1961, 264, 458–484. [Google Scholar]
- Harris, E.A.; Yngvesson, K.S. Spin-lattice relaxation in some iridium salts I. Relaxation of the isolated (IrCl6)2− complex. J. Phys. C Solid State Phys. 1968, 1, 990–1010. [Google Scholar]
- Harris, E.A.; Yngvesson, K.S. Spin-lattice relaxation in some iridium salts II. Relaxation of nearest-neighbour exchange-coupled pairs. J. Phys. C Solid State Phys. 1968, 1, 1011–1023. [Google Scholar] [CrossRef]
- Kokoszka, G.F.; Allen, H.C.; Gordon, G. Electron paramagnetic resonance spectra of zinc-doped copper acetate monohydrate. J. Chem. Phys. 1965, 42, 3693–3697. [Google Scholar] [CrossRef]
- Göppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen. Ann. Phys. 1931, 9, 273–294. [Google Scholar] [CrossRef]
- Sorokin, P.P.; Gelles, I.L.; Smith, W.V. Multiple quantum transitions in paramagnetic resonance. Phys. Rev. 1958, 112, 1513–1515. [Google Scholar] [CrossRef]
- Hughes, V.W.; Geiger, J.S. Two-quantum transitions in the microwave zeeman spectrum of atomic oxygen. Phys. Rev. 1955, 99, 1842–1845. [Google Scholar] [CrossRef]
- McDonald, C.C. Multiple-quantum transitions in EPR spectra of atomic oxygen. J. Chem. Phys. 1963, 39, 3159–3160. [Google Scholar] [CrossRef]
- Carrington, A.; Levy, D.H.; Miller, T.A.; Hyde, J.S. Double quantum transitions in gas-phase electron resonance. J. Chem. Phys. 1967, 47, 4859–4860. [Google Scholar] [CrossRef]
- Katayama, M. Double quantum transition in electron spin resonance of gamma-irradiated acetyl-d,l-alanine. Phys. Rev. 1962, 126, 1440–1442. [Google Scholar] [CrossRef]
- de Groot, M.S.; van der Waals, J.H. The two-quantum transition in the electron resonance spectrum of phosphorescent aromatic hydrocarbons. Physica 1963, 29, 1128–1132. [Google Scholar] [CrossRef]
- Grivet, J.P.; Mispelter, J. Electron spin resonance of triplet molecules: The hyperfine structure of the double quantum transition. Mol. Phys. 1974, 27, 15–32. [Google Scholar] [CrossRef]
- Orton, J.W.; Auzins, P.; Wertz, J.E. Double-quantum electron spin resonance transitions of nickel in magnesium oxide. Phys Rev. Lett. 1960, 4, 128–129. [Google Scholar] [CrossRef]
- Orton, J.W.; Auzins, P.; Wertz, J.E. Estimate of the nuclear moment of Ni61 from electron spin resonance. Phys. Rev. 1960, 119, 1681–1682. [Google Scholar] [CrossRef]
- Smith, S.R.P.; Dravnieks, F.; Wertz, J.E. Electron-paramagnetic-resonance line shape of Ni2+ in MgO. Phys. Rev. 1969, 178, 471–480. [Google Scholar] [CrossRef]
- Holmberg, G.E.; Slifkin, L.M.; Hempel, J.C. Double-quantum EPR transition in AgCl:Ni. J. Chem. Phys. 1979, 71, 2712–2715. [Google Scholar] [CrossRef]
- Lupei, V. EPR of cubic Pr3+ in fluorite. J. Phys. C Solid State Phys. 1979, 12, L45–L48. [Google Scholar] [CrossRef]
- Backs, D.; Stösser, R.; Lieberenz, M. Multiple-quantum transitions and optical excitation processes in the EPR of Mn2+ in ZnS. Phys. Status Solidi B 1985, 131, 291–297. [Google Scholar] [CrossRef]
- Ke, S.C.; Tohver, H.T. Double quantum transitions of Mn2+ in CaO. J. Phys. Condens. Matter 1994, 6, 8331–8334. [Google Scholar] [CrossRef]
- Reedijk, J.; Nieuwenhuijse, B. Interpretation of E.P.R.-spectra of powdered octahedral nickel(II) complexes with nitrogen-donor ligands. Recl. Trav. Chim. Pays-Bas 1972, 91, 533–551. [Google Scholar] [CrossRef]
- Mabbs, F.E.; Collison, D. Electron Paramagnetic Resonance of D Transition Metal Complexes; Elsevier: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Pardi, L.A.; Hassan, A.K.; Hulsbergen, F.B.; Reedijk, J.; Spek, A.L.; Brunel, L.-C. Direct determination of the single-ion anisotropy in a one-dimensional magnetic system by high-field EPR spectroscopy; synthesis, EPR, and X-ray structure of NixZn1−x(C2O4)(dmiz)2 [x = 0.07]. Inorg. Chem. 2000, 39, 159–164. [Google Scholar] [CrossRef]
- Desrochers, P.J.; Telser, J.; Zvyagin, S.A.; Ozarowski, A.; Krzystek, J.; Vicic, D.A. Electronic structure of four-coordinate C3V nickel(II) scorpionate complexes: Investigation by high-frequency and -field electron paramagnetic resonance and electron absorption spectroscopies. Inorg. Chem. 2006, 45, 8930–8941. [Google Scholar] [CrossRef]
- Schweinfurth, D.; Krzystek, J.; Schapiro, I.; Demeshko, S.; Klein, J.; Telser, J.; Ozarowski, A.; Su, C.-Y.; Meyer, F.; Atanasov, M.; et al. Electronic structures of octahedral Ni(II) complexes with “click” derived triazole ligands: A combined structural, magnetometric, spectroscopic, and theoretical study. Inorg. Chem. 2013, 52, 6880–6892. [Google Scholar] [CrossRef]
- Suaud, N.; Rogez, G.; Rebilly, J.-N.; Bouammali, M.-A.; Guihéry, N.; Barra, A.-L.; Mallah, T. Playing with magnetic anisotropy in hexacoordinated mononuclear Ni(II) complexes, an interplay between symmetry and geometry. Appl. Magn. Reson. 2020, 51, 1215–1231. [Google Scholar] [CrossRef]
- Wojnar, M.K.; Laorenza, D.W.; Schaller, R.D.; Freedman, D.E. Nickel(II) metal complexes as optically addressable qubit candidates. J. Am. Chem. Soc. 2020, 142, 14826–14830. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.J.; Shepit, M.; van Lierop, J.; Krzystek, J.; Telser, J.; Herbert, D. Characterization of the ligand field in pseudo-octahedral Ni(II) complexes of pincer-type amido ligands: Magnetism, redox behaviour, electronic absorption and high-frequency and -field EPR spectroscopy. Eur. J. Inorg. Chem. 2023, e202300446. [Google Scholar] [CrossRef]
- Yang, E.-C.; Kirman, C.; Lawrence, J.; Zakharov, L.N.; Rheingold, A.L.; Hill, S.; Hendrickson, D.N. Single-molecule magnets: High-field electron paramagnetic resonance evaluation of the single-ion zero-field interaction in a ZnII3NiII complex. Inorg. Chem. 2005, 44, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Ajibola, A.A.; Obaleye, J.A.; Sieroń, L.; Maniukiewicz, W.; Wojciechowska, A.; Ozarowski, A. Structural, spectroscopic insights, and antimicrobial properties of mononuclear and dinuclear metal(II) carboxylate derivatives with metronidazole. Polyhedron 2021, 194, 114931. [Google Scholar] [CrossRef]
- Dall’Olio, A.; Dascola, G.; Varacca, V. EPR evidence of exchange-coupled ion pairs and of isolated ions in some chlorosubsitutued cupric actates. Phys. Status Solidi (B) 1967, 22, 365–370. [Google Scholar] [CrossRef]
- Wasson, J.R.; Shyr, C.-I.; Trapp, C. The spectral and magnetic properties of copper(II) cyanoacetate. Inorg. Chem. 1968, 7, 469–473. [Google Scholar] [CrossRef]
- Sharrock, P.; Melník, M. Copper(II) acetates: From dimer to monomer. Can. J. Chem. 1985, 63, 52–56. [Google Scholar] [CrossRef]
- Parades-García, V.; Santana, R.C.; Madrid, R.; Vega, A.; Spodine, E.; Venegas-Yazigi, D. Unusual conformation of a dinuclear paddle wheel copper(II) complex. Synthesis, structural characterization and EPR studies. Inorg. Chem. 2013, 52, 8369–8377. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, I.A.; Nikiforova, M.E.; Koshenskova, K.A.; Kiskin, M.A.; Nelyubina, Y.V.; Primakov, P.V.; Fedin, M.V.; Becker, O.B.; Shender, V.O.; Malyants, I.K.; et al. Binuclear complexes of Cu(II) and Mg(II) with 2-furancarboxylic acid: Synthesis, structure, EPR spectroscopy, and results of in vitro biological activity against Mycolicibacterium smegmatis and SCOV3. Russ. J. Coord. Chem. 2021, 47, 881–890. [Google Scholar] [CrossRef]
- Lundin, A.; Aasa, R. A simple device to maintain temperatures in the range 4.2–100 K for EPR measurements. J. Magn. Reson. 1972, 8, 70–73. [Google Scholar] [CrossRef]
- Albracht, S.P.J. A low-cost cooling device for EPR measurements at 35 GHz down to 4.8 °K. J. Magn. Reson. 1974, 13, 299–303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| |D| = 0.335 (0.002) cm−1 | |E| = 0.0105 (0.0003) cm−1 | E/D < 0 |
| gz = 2.365 (0.008) | gy = 2.055 (0.010) | gx = 2.077 (0.005) |
| Az = 64 gauss | Ay = 1 gauss | Ax = 1 gauss (dummy value) |
| Wz = 85 gauss | Wy = 85 gauss | Wx = 70 gauss |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagen, W.R. Broadband EPR Spectroscopy of the Triplet State: Multi-Frequency Analysis of Copper Acetate Monohydrate. Int. J. Mol. Sci. 2023, 24, 14793. https://doi.org/10.3390/ijms241914793
Hagen WR. Broadband EPR Spectroscopy of the Triplet State: Multi-Frequency Analysis of Copper Acetate Monohydrate. International Journal of Molecular Sciences. 2023; 24(19):14793. https://doi.org/10.3390/ijms241914793
Chicago/Turabian StyleHagen, Wilfred R. 2023. "Broadband EPR Spectroscopy of the Triplet State: Multi-Frequency Analysis of Copper Acetate Monohydrate" International Journal of Molecular Sciences 24, no. 19: 14793. https://doi.org/10.3390/ijms241914793
APA StyleHagen, W. R. (2023). Broadband EPR Spectroscopy of the Triplet State: Multi-Frequency Analysis of Copper Acetate Monohydrate. International Journal of Molecular Sciences, 24(19), 14793. https://doi.org/10.3390/ijms241914793