
Deciphering the Mysterious Relationship between the Cross-Pathogenetic Mechanisms of Neurodegenerative and Oncological Diseases



Abstract
1. Introduction
2. Oxidative Stress and Mitochondrial Dysfunction in Oncological and Neurodegenerative Diseases
2.1. Concept of Oxidative Stress and Sources of Free Radicals
2.2. Free Radical Theory of the Occurrence of a Pathological Condition in a Cell
2.3. Dysfunction of the Cell Antioxidant Defense System
2.4. ROS-Mediated Signaling Pathways in Oncology and Neurodegeneration
2.5. Potential Neuroprotective and Antitumor Therapeutic Candidates Targeting ROS
3. General Aspects of the Epigenetic Regulation of Neurodegenerative Diseases and Cancer Pathogenesis
3.1. Histone Deacetylases as Major Epigenetic Regulators: Structure and Function
3.2. Changes in the Intensity of Histone Acetylation during Oncogenesis
3.3. Role of Histone Deacetylases in the Pathogenesis of Neurodegenerative Disorders
3.4. Advances in the Development of Histone Deacetylase Inhibitors in the Treatment of Cancer and Neurodegenerative Diseases
4. Alterations in the Bioenergetic Metabolism of Cells during Oncogenesis and Neurodegeneration
4.1. Determination of the Main Metabolic Processes of the Cell and Energy Metabolism
4.2. Molecular Subtleties of Tumor Cell Metabolism: Dysregulation of Aerobic Glycolysis and the Warburg Effect
4.3. Correction of Anomalies in Oxidative Phosphorylation in Mitochondria as a Promising Therapeutic Approach in the Development of Neuroprotective Drugs
4.4. Determination of the Main Metabolic Processes of the Cell and Energy Metabolism
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- Katsnelson, A.; De Strooper, B.; Zoghbi, H.Y. Neurodegeneration: From cellular concepts to clinical applications. Sci. Transl. Med. 2016, 8, 364ps18. [Google Scholar] [CrossRef]
- Lanni, C.; Masi, M.; Racchi, M.; Govoni, S. Cancer and Alzheimer’s disease inverse relationship: An age-associated diverging derailment of shared pathways. Mol. Psychiatry 2021, 26, 280–295. [Google Scholar] [CrossRef]
- French, P.W. Unfolded p53 in non-neuronal cells supports bacterial etiology of Alzheimer’s disease. Neural Regen. Res. 2022, 17, 2619–2622. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Yang, H. Balancing Apoptosis and Autophagy for Parkinson’s Disease Therapy: Targeting BCL-2. ACS Chem. Neurosci. 2019, 10, 792–802. [Google Scholar] [CrossRef]
- Callens, M.; Kraskovskaya, N.; Derevtsova, K.; Annaert, W.; Bultynck, G.; Bezprozvanny, I.; Vervliet, T. The role of Bcl-2 proteins in modulating neuronal Ca(2+) signaling in health and in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118997. [Google Scholar] [CrossRef]
- Pemberton, J.M.; Pogmore, J.P.; Andrews, D.W. Neuronal cell life, death, and axonal degeneration as regulated by the BCL-2 family proteins. Cell Death Differ. 2021, 28, 108–122. [Google Scholar] [CrossRef]
- Fairlie, W.D.; Lee, E.F. Co-Operativity between MYC and BCL-2 Pro-Survival Proteins in Cancer. Int. J. Mol. Sci. 2021, 22, 2841. [Google Scholar] [CrossRef]
- Opferman, J.T. Attacking cancer’s Achilles heel: Antagonism of anti-apoptotic BCL-2 family members. FEBS J. 2016, 283, 2661–2675. [Google Scholar] [CrossRef]
- Jin, J.; Xiong, Y.; Cen, B. Bcl-2 and Bcl-xL mediate resistance to receptor tyrosine kinase-targeted therapy in lung and gastric cancer. Anticancer Drugs 2017, 28, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Thomalla, D.; Beckmann, L.; Grimm, C.; Oliverio, M.; Meder, L.; Herling, C.D.; Nieper, P.; Feldmann, T.; Merkel, O.; Lorsy, E.; et al. Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies. Blood 2022, 140, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; ME, L.L. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Santos, R.; Ruiz de Almodovar, C.; Bulteau, A.L.; Gomes, C.M. Neurodegeneration, neurogenesis, and oxidative stress. Oxid. Med. Cell. Longev. 2013, 2013, 730581. [Google Scholar] [CrossRef]
- Xu, S.C.; Chen, Y.B.; Lin, H.; Pi, H.F.; Zhang, N.X.; Zhao, C.C.; Shuai, L.; Zhong, M.; Yu, Z.P.; Zhou, Z.; et al. Damage to mtDNA in liver injury of patients with extrahepatic cholestasis: The protective effects of mitochondrial transcription factor A. Free Radic. Biol. Med. 2012, 52, 1543–1551. [Google Scholar] [CrossRef]
- Lin, J.C.; Wang, X.Z.; Shen, T.; Zhang, J.Y. iTRAQ-based quantitative analysis reveals the mechanism underlying the changes in physiological activity in a glutamate racemase mutant strain of Streptococcus mutans UA159. Mol. Biol. Rep. 2020, 47, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Goldsamt, A.; Damayanti, N.P.; De Nigris, F.; Pili, R. Epigenetic Dysregulation in Advanced Kidney Cancer: Opportunities for Therapeutic Interventions. Cancer J. 2020, 26, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Farrell, K.; Navabpour, S.; Narayanan, S.N.; Jarome, T.J. Epigenetic Mechanisms in Memory and Cognitive Decline Associated with Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guo, S.; Zhang, X.; Tang, S.; Shao, W.; Han, X.; Wang, L.; Du, Y. Inverse relationship between cancer and Alzheimer’s disease: A systemic review meta-analysis. Neurol. Sci. 2015, 36, 1987–1994. [Google Scholar] [CrossRef]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef]
- Gao, X.; Ning, Y. Cancer and Parkinson’s disease: The odd couple. Drugs Today 2011, 47, 215–222. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.; Liu, J.; Aleksandrova, Y.; Klochkov, S.; Fan, R. Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment. Cancers 2021, 13, 6062. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Lu, W.; Shi, Y.; Wang, R.; Su, D.; Tang, M.; Liu, Y.; Li, Z. Antioxidant Activity and Healthy Benefits of Natural Pigments in Fruits: A Review. Int. J. Mol. Sci. 2021, 22, 4945. [Google Scholar] [CrossRef]
- Kang, S.W.; Lee, S.; Lee, E.K. ROS and energy metabolism in cancer cells: Alliance for fast growth. Arch. Pharm. Res. 2015, 38, 338–345. [Google Scholar] [CrossRef]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Huang, Z.; Goo, N.; Bae, H.J.; Jeong, Y.; Park, H.J.; Cai, M.; Cho, K.; Jung, S.Y.; et al. Theracurmin Ameliorates Cognitive Dysfunctions in 5XFAD Mice by Improving Synaptic Function and Mitigating Oxidative Stress. Biomol. Ther. 2019, 27, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Peng, A.; Gao, Y.; Zhuang, X.; Lin, Y.; He, W.; Wang, Y.; Chen, W.; Chen, T.; Huang, X.; Yang, R.; et al. Bazhu Decoction, a Traditional Chinese Medical Formula, Ameliorates Cognitive Deficits in the 5xFAD Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 1391. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Kim, D.H.; Jeon, W.K.; Han, J.S. 4-Hydroxynonenal Immunoreactivity Is Increased in the Frontal Cortex of 5XFAD Transgenic Mice. Biomedicines 2020, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lim, D.K.; Suh, Y.H.; Chang, K.A. Long-Term Treatment of Cuban Policosanol Attenuates Abnormal Oxidative Stress and Inflammatory Response via Amyloid Plaques Reduction in 5xFAD Mice. Antioxidants 2021, 10, 1321. [Google Scholar] [CrossRef]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.S. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, W.; Wang, X.; Ge, W. Impact of mitochondrial aldehyde dehydrogenase 2 on cognitive impairment in the AD model mouse. Acta Biochim. Biophys. Sin. 2021, 53, 837–847. [Google Scholar] [CrossRef]
- Foroumandi, E.; Javan, R.; Moayed, L.; Fahimi, H.; Kheirabadi, F.; Neamatshahi, M.; Shogofteh, F.; Zarghi, A. The effects of fenugreek seed extract supplementation in patients with Alzheimer’s disease: A randomized, double-blind, placebo-controlled trial. Phytother. Res. 2023, 37, 285–294. [Google Scholar] [CrossRef]
- Khalil, A.; Berrougui, H.; Pawelec, G.; Fulop, T. Impairment of the ABCA1 and SR-BI-mediated cholesterol efflux pathways and HDL anti-inflammatory activity in Alzheimer’s disease. Mech. Ageing Dev. 2012, 133, 20–29. [Google Scholar] [CrossRef]
- Ton, A.M.M.; Campagnaro, B.P.; Alves, G.A.; Aires, R.; Coco, L.Z.; Arpini, C.M.; Guerra, E.O.T.; Campos-Toimil, M.; Meyrelles, S.S.; Pereira, T.M.C.; et al. Oxidative Stress and Dementia in Alzheimer’s Patients: Effects of Synbiotic Supplementation. Oxid. Med. Cell. Longev. 2020, 2020, 2638703. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Tirle, T.; Lopez-Nogueroles, M.; Vento, M.; Baquero, M.; Chafer-Pericas, C. Oxidative Damage of DNA as Early Marker of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 6136. [Google Scholar] [CrossRef]
- Hasina, Z.; Wang, N.; Wang, C.C. Developmental Neuropathology and Neurodegeneration of Down Syndrome: Current Knowledge in Humans. Front. Cell Dev. Biol. 2022, 10, 877711. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. Brain lipid peroxidation and alzheimer disease: Synergy between the Butterfield and Mattson laboratories. Ageing Res. Rev. 2020, 64, 101049. [Google Scholar] [CrossRef] [PubMed]
- Leu, T.; Schutzhold, V.; Fandrey, J.; Ferenz, K.B. When the Brain Yearns for Oxygen. Neurosignals 2019, 27, 50–61. [Google Scholar] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Guan, L.; Mao, Z.; Yang, S.; Wu, G.; Chen, Y.; Yin, L.; Qi, Y.; Han, L.; Xu, L. Dioscin alleviates Alzheimer’s disease through regulating RAGE/NOX4 mediated oxidative stress and inflammation. Biomed. Pharmacother. 2022, 152, 113248. [Google Scholar] [CrossRef]
- Meng, M.; Zhang, L.; Ai, D.; Wu, H.; Peng, W. beta-Asarone Ameliorates beta-Amyloid-Induced Neurotoxicity in PC12 Cells by Activating P13K/Akt/Nrf2 Signaling Pathway. Front. Pharmacol. 2021, 12, 659955. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Schrag, M.; Mueller, C.; Zabel, M.; Crofton, A.; Kirsch, W.M.; Ghribi, O.; Squitti, R.; Perry, G. Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Neurobiol. Dis. 2013, 59, 100–110. [Google Scholar] [CrossRef]
- Zabel, M.; Nackenoff, A.; Kirsch, W.M.; Harrison, F.E.; Perry, G.; Schrag, M. Markers of oxidative damage to lipids, nucleic acids and proteins and antioxidant enzymes activities in Alzheimer’s disease brain: A meta-analysis in human pathological specimens. Free Radic. Biol. Med. 2018, 115, 351–360. [Google Scholar] [CrossRef]
- Trares, K.; Chen, L.J.; Schottker, B. Association of F(2)-isoprostane levels with Alzheimer’s disease in observational studies: A systematic review and meta-analysis. Ageing Res. Rev. 2022, 74, 101552. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Alvarez, L.; Baquero, M.; Ferrer, I.; Garcia, L.; Hervas-Marin, D.; Chafer-Pericas, C. Plasma isoprostanoids assessment as Alzheimer’s disease progression biomarkers. J. Neurochem. 2021, 157, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T.; Nakagawa, K.; Takekoshi, H.; Higuchi, O.; Kato, S.; Kondo, M.; Kimura, F.; Miyazawa, T. Ingestion of Chlorella reduced the oxidation of erythrocyte membrane lipids in senior Japanese subjects. J. Oleo Sci. 2013, 62, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Streck, E.L.; Czapski, G.A.; Goncalves da Silva, C. Neurodegeneration, mitochondrial dysfunction, and oxidative stress. Oxid. Med. Cell. Longev. 2013, 2013, 826046. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otin, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Smith, M.A.; Avila, J.; Nunomura, A.; Siedlak, S.L.; Zhu, X.; Perry, G.; Sayre, L.M. In Alzheimer’s disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. J. Neurochem. 2000, 75, 1234–1241. [Google Scholar] [CrossRef]
- Takagane, K.; Nojima, J.; Mitsuhashi, H.; Suo, S.; Yanagihara, D.; Takaiwa, F.; Urano, Y.; Noguchi, N.; Ishiura, S. Abeta induces oxidative stress in senescence-accelerated (SAMP8) mice. Biosci. Biotechnol. Biochem. 2015, 79, 912–918. [Google Scholar] [CrossRef]
- Akude, E.; Zherebitskaya, E.; Roy Chowdhury, S.K.; Girling, K.; Fernyhough, P. 4-Hydroxy-2-nonenal induces mitochondrial dysfunction and aberrant axonal outgrowth in adult sensory neurons that mimics features of diabetic neuropathy. Neurotox. Res. 2010, 17, 28–38. [Google Scholar] [CrossRef]
- Jiang, D.; Men, L.; Wang, J.; Zhang, Y.; Chickenyen, S.; Wang, Y.; Zhou, F. Redox reactions of copper complexes formed with different beta-amyloid peptides and their neuropathological [correction of neuropathalogical] relevance. Biochemistry 2007, 46, 9270–9282. [Google Scholar] [CrossRef]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.; Ferreira, R.S.; Santos, A.C.D. Overview of cisplatin-induced neurotoxicity and ototoxicity, and the protective agents. Food Chem. Toxicol. 2020, 136, 111079. [Google Scholar] [CrossRef] [PubMed]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Sadati Zarrini, A.; Moslemi, D.; Parsian, H.; Vessal, M.; Mosapour, A.; Shirkhani Kelagari, Z. The status of antioxidants, malondialdehyde and some trace elements in serum of patients with breast cancer. Casp. J. Intern. Med. 2016, 7, 31–36. [Google Scholar]
- do Val Carneiro, J.L.; Nixdorf, S.L.; Mantovani, M.S.; da Silva do Amaral Herrera, A.C.; Aoki, M.N.; Amarante, M.K.; Fabris, B.A.; Pelegrinelli Fungaro, M.H.; Ehara Watanabe, M.A. Plasma malondialdehyde levels and CXCR4 expression in peripheral blood cells of breast cancer patients. J. Cancer Res. Clin. Oncol. 2009, 135, 997–1004. [Google Scholar] [CrossRef]
- Gonenc, A.; Ozkan, Y.; Torun, M.; Simsek, B. Plasma malondialdehyde (MDA) levels in breast and lung cancer patients. J. Clin. Pharm. Ther. 2001, 26, 141–144. [Google Scholar] [CrossRef]
- Arif, M.; Rashid, A.; Majeed, A.; Qaiser, F.; Razak, S. Evaluation of correlation between expression of P53 and Malondialdehyde levels in prostate cancer patients. J. Pak. Med. Assoc. 2018, 68, 1373–1377. [Google Scholar]
- Dillioglugil, M.O.; Mekik, H.; Muezzinoglu, B.; Ozkan, T.A.; Demir, C.G.; Dillioglugil, O. Blood and tissue nitric oxide and malondialdehyde are prognostic indicators of localized prostate cancer. Int. Urol. Nephrol. 2012, 44, 1691–1696. [Google Scholar] [CrossRef]
- Drozdz-Afelt, J.M.; Koim-Puchowska, B.B.; Kaminski, P. Analysis of oxidative stress indicators in Polish patients with prostate cancer. Environ. Sci. Pollut. Res. Int. 2022, 29, 4632–4640. [Google Scholar] [CrossRef]
- Lepara, Z.; Lepara, O.; Fajkic, A.; Rebic, D.; Alic, J.; Spahovic, H. Serum malondialdehyde (MDA) level as a potential biomarker of cancer progression for patients with bladder cancer. Rom. J. Intern. Med. 2020, 58, 146–152. [Google Scholar] [CrossRef]
- Gecit, I.; Eryilmaz, R.; Kavak, S.; Meral, I.; Demir, H.; Pirincci, N.; Gunes, M.; Taken, K. The Prolidase Activity, Oxidative Stress, and Nitric Oxide Levels of Bladder Tissues with or Without Tumor in Patients with Bladder Cancer. J. Membr. Biol. 2017, 250, 455–459. [Google Scholar] [CrossRef]
- Firdausa, A.Y.; Ahimsa, S.S.; Ahmada, R.A.; Sukmawati, N.F.; Ernawati, D.S.; Parmadiati, A.E.; Soebadi, B.; Radithia, D.; Winias, S.; Mahdani, F.Y.; et al. Malondialdehyde Level and Tissue Apoptosis Count as an Early-Detection Marker of Oral Potentially Malignant Disorders. Eur. J. Dent. 2023, 17, 155–160. [Google Scholar] [CrossRef]
- Marakala, V.; Malathi, M.; Shivashankara, A.R. Lipid peroxidation and antioxidant vitamin status in oral cavity and oropharyngeal cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 5763–5765. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pande, D.; Negi, R.; Khanna, S.; Khanna, R.; Khanna, H.D. Vascular endothelial growth factor levels in relation to oxidative damage and antioxidant status in patients with breast cancer. J. Breast Cancer 2011, 14, 181–184. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Hosokawa, K.; Tamura, T.; Kanno, H.; Urabe, M.; Honjo, H. Urinary 8-hydroxy-2’-deoxyguanosine (8-OHdG) levels in women with or without gynecologic cancer. J. Obstet. Gynaecol. Res. 1996, 22, 359–363. [Google Scholar] [CrossRef]
- Pylvas, M.; Puistola, U.; Laatio, L.; Kauppila, S.; Karihtala, P. Elevated serum 8-OHdG is associated with poor prognosis in epithelial ovarian cancer. Anticancer Res. 2011, 31, 1411–1415. [Google Scholar] [PubMed]
- Xu, X.; Wang, Y.; Guo, W.; Zhou, Y.; Lv, C.; Chen, X.; Liu, K. The significance of the alteration of 8-OHdG in serous ovarian carcinoma. J. Ovarian Res. 2013, 6, 74. [Google Scholar] [CrossRef]
- Plachetka, A.; Adamek, B.; Strzelczyk, J.K.; Krakowczyk, L.; Migula, P.; Nowak, P.; Wiczkowski, A. 8-hydroxy-2’-deoxyguanosine in colorectal adenocarcinoma—Is it a result of oxidative stress? Med. Sci. Monit. 2013, 19, 690–695. [Google Scholar]
- Sato, T.; Takeda, H.; Otake, S.; Yokozawa, J.; Nishise, S.; Fujishima, S.; Orii, T.; Fukui, T.; Takano, J.; Sasaki, Y.; et al. Increased plasma levels of 8-hydroxydeoxyguanosine are associated with development of colorectal tumors. J. Clin. Biochem. Nutr. 2010, 47, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Rozalski, R.; Gackowski, D.; Siomek-Gorecka, A.; Starczak, M.; Modrzejewska, M.; Banaszkiewicz, Z.; Olinski, R. Urinary 5-hydroxymethyluracil and 8-oxo-7,8-dihydroguanine as potential biomarkers in patients with colorectal cancer. Biomarkers 2015, 20, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Sedlic, F.; Seiwerth, F.; Sepac, A.; Sikiric, S.; Cindric, M.; Milavic, M.; Batelja Vuletic, L.; Jakopovic, M.; Seiwerth, S. Mitochondrial ROS Induce Partial Dedifferentiation of Human Mesothelioma via Upregulation of NANOG. Antioxidants 2020, 9, 606. [Google Scholar] [CrossRef]
- Migliario, M.; Pittarella, P.; Fanuli, M.; Rizzi, M.; Reno, F. Laser-induced osteoblast proliferation is mediated by ROS production. Lasers Med. Sci. 2014, 29, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Chang, C.H.; Pauklin, S. ROS and TGFbeta: From pancreatic tumour growth to metastasis. J. Exp. Clin. Cancer Res. 2021, 40, 152. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Demirci-Cekic, S.; Ozkan, G.; Avan, A.N.; Uzunboy, S.; Capanoglu, E.; Apak, R. Biomarkers of Oxidative Stress and Antioxidant Defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef]
- Grazioli, V.; Schiavo, R.; Casari, E.; Marzatico, F.; Rodriguez y Baena, R.; Gaetani, P. Antioxidant enzymatic activities and lipid peroxidation in cultured human chondrocytes from vertebral plate cartilage. FEBS Lett. 1998, 431, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protein Cell 2010, 1, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Flohe, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Averill-Bates, D.A. The antioxidant glutathione. Vitam. Horm. 2023, 121, 109–141. [Google Scholar]
- Alanazi, A.M.; Mostafa, G.A.; Al-Badr, A.A. Glutathione. Profiles Drug Subst. Excip. Relat. Methodol. 2015, 40, 43–158. [Google Scholar] [PubMed]
- Owen, J.B.; Butterfield, D.A. Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol. 2010, 648, 269–277. [Google Scholar] [PubMed]
- Poljsak, B.; Milisav, I. The Role of Antioxidants in Cancer, Friends or Foes? Curr. Pharm. Des. 2018, 24, 5234–5244. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Patel, S.; Dey, T.; Pandey, U.; Mishra, S.P. A study of oxidative stress in cervical cancer- an institutional study. Biochem. Biophys. Rep. 2021, 25, 100881. [Google Scholar] [CrossRef] [PubMed]
- Manju, V.; Balasubramanian, V.; Nalini, N. Oxidative stress and tumor markers in cervical cancer patients. J. Biochem. Mol. Biol. Biophys. 2002, 6, 387–390. [Google Scholar] [CrossRef]
- Manju, V.; Kalaivani Sailaja, J.; Nalini, N. Circulating lipid peroxidation and antioxidant status in cervical cancer patients: A case-control study. Clin. Biochem. 2002, 35, 621–625. [Google Scholar] [CrossRef]
- Bel’skaya, L.V.; Sarf, E.A.; Solomatin, D.V.; Kosenok, V.K. Metabolic Features of Saliva in Breast Cancer Patients. Metabolites 2022, 12, 166. [Google Scholar] [CrossRef]
- Cobanoglu, U.; Demir, H.; Duran, M.; Sehitogullari, A.; Mergan, D.; Demir, C. Erythrocyte catalase and carbonic anhydrase activities in lung cancer. Asian Pac. J. Cancer Prev. 2010, 11, 1377–1382. [Google Scholar]
- Skorska, K.B.; Placzkowska, S.; Prescha, A.; Porebska, I.; Kosacka, M.; Pawelczyk, K.; Zablocka-Slowinska, K. Serum Total SOD Activity and SOD1/2 Concentrations in Predicting All-Cause Mortality in Lung Cancer Patients. Pharmaceuticals 2021, 14, 1067. [Google Scholar] [CrossRef]
- Pirincci, N.; Gecit, I.; Gunes, M.; Yuksel, M.B.; Kaba, M.; Tanik, S.; Demir, H.; Aslan, M. Serum adenosine deaminase, catalase and carbonic anhydrase activities in patients with bladder cancer. Clinics 2012, 67, 1443–1446. [Google Scholar] [CrossRef]
- Cavallini, C.; Chignola, R.; Dando, I.; Perbellini, O.; Mimiola, E.; Lovato, O.; Laudanna, C.; Pizzolo, G.; Donadelli, M.; Scupoli, M.T. Low catalase expression confers redox hypersensitivity and identifies an indolent clinical behavior in CLL. Blood 2018, 131, 1942–1954. [Google Scholar] [CrossRef] [PubMed]
- Oltra, A.M.; Carbonell, F.; Tormos, C.; Iradi, A.; Saez, G.T. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic. Biol. Med. 2001, 30, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, Y.; Duan, W.; Song, J.; Wei, S.; Xia, S.; Wang, Y.; Du, X.; Li, E.; Ren, C.; et al. Glutathione peroxidase 4-dependent glutathione high-consumption drives acquired platinum chemoresistance in lung cancer-derived brain metastasis. Clin. Transl. Med. 2021, 11, e517. [Google Scholar] [CrossRef] [PubMed]
- Estrela, J.M.; Ortega, A.; Mena, S.; Sirerol, J.A.; Obrador, E. Glutathione in metastases: From mechanisms to clinical applications. Crit. Rev. Clin. Lab. Sci. 2016, 53, 253–267. [Google Scholar] [CrossRef]
- Hatem, E.; El Banna, N.; Huang, M.E. Multifaceted Roles of Glutathione and Glutathione-Based Systems in Carcinogenesis and Anticancer Drug Resistance. Antioxid. Redox Signal. 2017, 27, 1217–1234. [Google Scholar] [CrossRef]
- Nunes, S.C.; Serpa, J. Glutathione in Ovarian Cancer: A Double-Edged Sword. Int. J. Mol. Sci. 2018, 19, 1882. [Google Scholar] [CrossRef]
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar]
- Skrzydlewska, E.; Sulkowski, S.; Koda, M.; Zalewski, B.; Kanczuga-Koda, L.; Sulkowska, M. Lipid peroxidation and antioxidant status in colorectal cancer. World J. Gastroenterol. 2005, 11, 403–406. [Google Scholar] [CrossRef]
- Kontakiotis, T.; Katsoulis, K.; Hagizisi, O.; Kougioulis, M.; Gerou, S.; Papakosta, D. Bronchoalveolar lavage fluid alteration in antioxidant and inflammatory status in lung cancer patients. Eur. J. Intern. Med. 2011, 22, 522–526. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 2013, 288, 33542–33558. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M.; Herve, V.; Ben Khedher, M.R.; Rabanel, J.M.; Ramassamy, C. Glutathione: An Old and Small Molecule with Great Functions and New Applications in the Brain and in Alzheimer’s Disease. Antioxid. Redox Signal. 2021, 35, 270–292. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Gaestel, M. MAPK-Activated Protein Kinases: Servant or Partner? Annu. Rev. Biochem. 2022, 91, 505–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, M.; Gao, H.; Yu, G.; Zhao, Y.; Yao, F.; Yang, W. MAPK signalling-induced phosphorylation and subcellular translocation of PDHE1alpha promotes tumour immune evasion. Nat. Metab. 2022, 4, 374–388. [Google Scholar] [CrossRef]
- Behl, T.; Upadhyay, T.; Singh, S.; Chigurupati, S.; Alsubayiel, A.M.; Mani, V.; Vargas-De-La-Cruz, C.; Uivarosan, D.; Bustea, C.; Sava, C.; et al. Polyphenols Targeting MAPK Mediated Oxidative Stress and Inflammation in Rheumatoid Arthritis. Molecules 2021, 26, 6570. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert. Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117 Pt 20, 4619–4628. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef]
- Chakraborti, S.; Mandal, M.; Das, S.; Mandal, A.; Chakraborti, T. Regulation of matrix metalloproteinases: An overview. Mol. Cell. Biochem. 2003, 253, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu, S., II; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The relation between PI3K/AKT signalling pathway and cancer. Gene 2019, 698, 120–128. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front. Biosci. (Landmark Ed.) 2016, 21, 1084–1091. [Google Scholar]
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef]
- Shin, I.; Yakes, F.M.; Rojo, F.; Shin, N.Y.; Bakin, A.V.; Baselga, J.; Arteaga, C.L. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat. Med. 2002, 8, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Thakkar, H.; Tyan, F.; Gim, S.; Robinson, H.; Lee, C.; Pandey, S.K.; Nwokorie, C.; Onwudiwe, N.; Srivastava, R.K. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene 2001, 20, 6073–6083. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 11088. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, Y.; Tang, H.; Hu, X.; Hubert, S.M.; Li, Q.; Su, D.; Xu, H.; Fan, Y.; Yu, X.; et al. Activation of PI3K/AKT Pathway Is a Potential Mechanism of Treatment Resistance in Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Guo, C.; Zhao, Q.; Cao, J.; Huang, H.Y.; Yue, M.; Xue, Y.; Jin, Y.; Hu, L.; et al. PI3K/Akt/mTOR signaling orchestrates the phenotypic transition and chemo-resistance of small cell lung cancer. J. Genet. Genom. 2021, 48, 640–651. [Google Scholar] [CrossRef]
- Guerrero-Zotano, A.; Mayer, I.A.; Arteaga, C.L. PI3K/AKT/mTOR: Role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016, 35, 515–524. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- An, J.; Yang, L.; Pan, Y.; He, Y.; Xie, H.; Tao, Y.; Li, W.; Yan, Y.; Chen, S.; Liu, Y.; et al. SPAG5 Activates PI3K/AKT Pathway and Promotes the Tumor Progression and Chemo-Resistance in Gastric Cancer. DNA Cell Biol. 2022, 41, 893–902. [Google Scholar] [CrossRef]
- Sun, S.; Guo, C.; Gao, T.; Ma, D.; Su, X.; Pang, Q.; Zhang, R. Hypoxia Enhances Glioma Resistance to Sulfasalazine-Induced Ferroptosis by Upregulating SLC7A11 via PI3K/AKT/HIF-1alpha Axis. Oxid. Med. Cell. Longev. 2022, 2022, 7862430. [Google Scholar] [CrossRef]
- Xiang, M.; Liu, T.; Tian, C.; Ma, K.; Gou, J.; Huang, R.; Li, S.; Li, Q.; Xu, C.; Li, L.; et al. Kinsenoside attenuates liver fibro-inflammation by suppressing dendritic cells via the PI3K-AKT-FoxO1 pathway. Pharmacol. Res. 2022, 177, 106092. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.N.; Wu, C.B.; Chen, Z.M.; Zheng, P.P.; Liu, Y.Q.; Xiong, J.; Xu, J.Y.; Li, P.F.; Mamun, A.A.; Ye, L.B.; et al. DL-3-n-butylphthalide ameliorates diabetes-associated cognitive decline by enhancing PI3K/Akt signaling and suppressing oxidative stress. Acta Pharmacol. Sin. 2021, 42, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Fabbrin, S.B.; Girardi, B.A.; de Lorena Wendel, A.; Coelho Ilha Valin, C.; Pillat, M.M.; Viero, F.T.; Mello, C.F.; Rubin, M.A. Spermidine-induced improvement of memory consolidation involves PI3K/Akt signaling pathway. Brain Res. Bull. 2020, 164, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Knox, D.; Della Valle, R.; Mohammadmirzaei, N.; Shultz, B.; Biddle, M.; Farkash, A.; Chamness, M.; Moulton, E. PI3K-Akt Signaling in the Basolateral Amygdala Facilitates Traumatic Stress Enhancements in Fear Memory. Int. J. Neuropsychopharmacol. 2021, 24, 229–238. [Google Scholar] [CrossRef]
- Li, H.; Xue, X.; Li, L.; Li, Y.; Wang, Y.; Huang, T.; Wang, Y.; Meng, H.; Pan, B.; Niu, Q. Aluminum-Induced Synaptic Plasticity Impairment via PI3K-Akt-mTOR Signaling Pathway. Neurotox. Res. 2020, 37, 996–1008. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Wang, C.; Hao, J.; Liu, X.; Li, C.; Yuan, X.; Lee, R.J.; Bai, T.; Wang, D. Isoforsythiaside Attenuates Alzheimer’s Disease via Regulating Mitochondrial Function Through the PI3K/AKT Pathway. Int. J. Mol. Sci. 2020, 21, 5687. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Xu, Q.Q.; Xian, Y.F.; Lin, Z.X. Sulforaphene Ameliorates Neuroinflammation and Hyperphosphorylated Tau Protein via Regulating the PI3K/Akt/GSK-3beta Pathway in Experimental Models of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2020, 2020, 4754195. [Google Scholar] [CrossRef]
- Kumar, M.; Bansal, N. Implications of Phosphoinositide 3-Kinase-Akt (PI3K-Akt) Pathway in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 354–385. [Google Scholar] [CrossRef]
- Salem, M.A.; Budzynska, B.; Kowalczyk, J.; El Sayed, N.S.; Mansour, S.M. Tadalafil and bergapten mitigate streptozotocin-induced sporadic Alzheimer’s disease in mice via modulating neuroinflammation, PI3K/Akt, Wnt/beta-catenin, AMPK/mTOR signaling pathways. Toxicol. Appl. Pharmacol. 2021, 429, 115697. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Barnabei, L.; Laplantine, E.; Mbongo, W.; Rieux-Laucat, F.; Weil, R. NF-kappaB: At the Borders of Autoimmunity and Inflammation. Front. Immunol. 2021, 12, 716469. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.P.; Carmody, R.J. NF-kappaB and the Transcriptional Control of Inflammation. Int. Rev. Cell Mol. Biol. 2018, 335, 41–84. [Google Scholar] [PubMed]
- Mulero, M.C.; Huxford, T.; Ghosh, G. NF-kappaB, IkappaB, and IKK: Integral Components of Immune System Signaling. Adv. Exp. Med. Biol. 2019, 1172, 207–226. [Google Scholar] [PubMed]
- Su, S.Y.; Cheng, C.Y.; Tsai, T.H.; Hsiang, C.Y.; Ho, T.Y.; Hsieh, C.L. Paeonol attenuates H(2)O(2)-induced NF-kappaB-associated amyloid precursor protein expression. Am. J. Chin. Med. 2010, 38, 1171–1192. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.; Cang, H.; Gao, F.; Yamamoto, T.; Osaki, T.; Yi, J. The endogenous reactive oxygen species promote NF-kappaB activation by targeting on activation of NF-kappaB-inducing kinase in oral squamous carcinoma cells. Free Radic. Res. 2007, 41, 963–971. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-kappaB-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Du, Y.; Chen, X.; Wei, X.; Bales, K.R.; Berg, D.T.; Paul, S.M.; Farlow, M.R.; Maloney, B.; Ge, Y.W.; Lahiri, D.K. NF-(kappa)B mediates amyloid beta peptide-stimulated activity of the human apolipoprotein E gene promoter in human astroglial cells. Brain Res. Mol. Brain Res. 2005, 136, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Yadav, M.; Chauhan, N.; Huang, Y.; Luo, Y. Cancer Cell Metabolism Featuring Nrf2. Curr. Drug Discov. Technol. 2020, 17, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.S.; Jun, M.; Kong, A.N. Nrf2: A potential molecular target for cancer chemoprevention by natural compounds. Antioxid. Redox Signal. 2006, 8, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Sivinski, J.; Zhang, D.D.; Chapman, E. Targeting NRF2 to treat cancer. Semin. Cancer Biol. 2021, 76, 61–73. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef]
- Choi, B.H.; Kim, J.M.; Kwak, M.K. The multifaceted role of NRF2 in cancer progression and cancer stem cells maintenance. Arch. Pharm. Res. 2021, 44, 263–280. [Google Scholar] [CrossRef]
- George, M.; Tharakan, M.; Culberson, J.; Reddy, A.P.; Reddy, P.H. Role of Nrf2 in aging, Alzheimer’s and other neurodegenerative diseases. Ageing Res. Rev. 2022, 82, 101756. [Google Scholar] [CrossRef]
- Davies, D.A.; Adlimoghaddam, A.; Albensi, B.C. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells 2021, 10, 1884. [Google Scholar] [CrossRef] [PubMed]
- Riordan, R.; Rong, W.; Yu, Z.; Ross, G.; Valerio, J.; Dimas-Munoz, J.; Heredia, V.; Magnusson, K.; Galvan, V.; Perez, V.I. Effect of Nrf2 loss on senescence and cognition of tau-based P301S mice. Geroscience 2023, 45, 1451–1469. [Google Scholar] [CrossRef]
- Simoni, E.; Serafini, M.M.; Caporaso, R.; Marchetti, C.; Racchi, M.; Minarini, A.; Bartolini, M.; Lanni, C.; Rosini, M. Targeting the Nrf2/Amyloid-Beta Liaison in Alzheimer’s Disease: A Rational Approach. ACS Chem. Neurosci. 2017, 8, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Arias, E.; Diaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.J.; Qi, M.; Li, N.; Lei, Y.H.; Zhang, D.M.; Chen, J.X. Natural products and their derivatives: Promising modulators of tumor immunotherapy. J. Leukoc. Biol. 2020, 108, 493–508. [Google Scholar] [CrossRef]
- Lu, X.; Yang, F.; Chen, D.; Zhao, Q.; Chen, D.; Ping, H.; Xing, N. Quercetin reverses docetaxel resistance in prostate cancer via androgen receptor and PI3K/Akt signaling pathways. Int. J. Biol. Sci. 2020, 16, 1121–1134. [Google Scholar] [CrossRef]
- Jia, X.B.; Zhang, Q.; Xu, L.; Yao, W.J.; Wei, L. Lotus leaf flavonoids induce apoptosis of human lung cancer A549 cells through the ROS/p38 MAPK pathway. Biol. Res. 2021, 54, 7. [Google Scholar] [CrossRef]
- Reyes-Farias, M.; Carrasco-Pozo, C. The Anti-Cancer Effect of Quercetin: Molecular Implications in Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 3177. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shabestari, F.A.; Vaezi, S.; Abak, A.; Shoorei, H.; Karimi, A.; Taheri, M.; Basiri, A. Emerging impact of quercetin in the treatment of prostate cancer. Biomed. Pharmacother. 2021, 138, 111548. [Google Scholar] [CrossRef]
- Biswas, P.; Dey, D.; Biswas, P.K.; Rahaman, T.I.; Saha, S.; Parvez, A.; Khan, D.A.; Lily, N.J.; Saha, K.; Sohel, M.; et al. A Comprehensive Analysis and Anti-Cancer Activities of Quercetin in ROS-Mediated Cancer and Cancer Stem Cells. Int. J. Mol. Sci. 2022, 23, 11746. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.A.S.; Kalinina, E.V.; Tatarskiy, V.V.; Volodina, Y.L.; Petrova, S.; Novichkova, M.D.; Zhdanov, D.D.; Shtil, A.A. Suppression of the Antioxidant System and PI3K/Akt/mTOR Signaling Pathway in Cisplatin-Resistant Cancer Cells by Quercetin. Bull. Exp. Biol. Med. 2022, 173, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.; Mostafavi-Pour, Z.; Amiri, A.; Keshavarzi, F.; Nejabat, N.; Ramezani, F.; Sardarian, A.; Zal, F. Chemoprevention of Prostate Cancer Cells by Vitamin C plus Quercetin: Role of Nrf2 in Inducing Oxidative Stress. Nutr. Cancer 2021, 73, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi-Pour, Z.; Ramezani, F.; Keshavarzi, F.; Samadi, N. The role of quercetin and vitamin C in Nrf2-dependent oxidative stress production in breast cancer cells. Oncol. Lett. 2017, 13, 1965–1973. [Google Scholar] [CrossRef]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef]
- Lin, R.; Piao, M.; Song, Y.; Liu, C. Quercetin Suppresses AOM/DSS-Induced Colon Carcinogenesis through Its Anti-Inflammation Effects in Mice. J. Immunol. Res. 2020, 2020, 9242601. [Google Scholar] [CrossRef]
- Xu, D.; Hu, M.J.; Wang, Y.Q.; Cui, Y.L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef]
- Rishitha, N.; Muthuraman, A. Therapeutic evaluation of solid lipid nanoparticle of quercetin in pentylenetetrazole induced cognitive impairment of zebrafish. Life Sci. 2018, 199, 80–87. [Google Scholar] [CrossRef]
- Patil, C.S.; Singh, V.P.; Satyanarayan, P.S.; Jain, N.K.; Singh, A.; Kulkarni, S.K. Protective effect of flavonoids against aging- and lipopolysaccharide-induced cognitive impairment in mice. Pharmacology 2003, 69, 59–67. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Q.; Li, Z.; Dang, M.; Lin, Y.; Hou, X. Activation of Nrf2 signaling by sitagliptin and quercetin combination against beta-amyloid induced Alzheimer’s disease in rats. Drug Dev. Res. 2019, 80, 837–845. [Google Scholar] [CrossRef]
- Wang, L.; Sun, J.; Miao, Z.; Jiang, X.; Zheng, Y.; Yang, G. Quercitrin improved cognitive impairment through inhibiting inflammation induced by microglia in Alzheimer’s disease mice. Neuroreport 2022, 33, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, X.; Zhu, G.; Liu, H.; Chen, J.; Wang, Y.; He, X. Quercetin inhibits TNF-alpha induced HUVECs apoptosis and inflammation via downregulating NF-kB and AP-1 signaling pathway in vitro. Medicine 2020, 99, e22241. [Google Scholar] [CrossRef] [PubMed]
- Paco, A.; Bras, T.; Santos, J.O.; Sampaio, P.; Gomes, A.C.; Duarte, M.F. Anti-Inflammatory and Immunoregulatory Action of Sesquiterpene Lactones. Molecules 2022, 27, 1142. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhong, H.; Fang, S.; Zheng, Y.; Li, C.; Peng, G.; Shen, X. Potential Anti-inflammatory Sesquiterpene Lactones from Eupatorium lindleyanum. Planta Med. 2018, 84, 123–128. [Google Scholar] [CrossRef]
- Neganova, M.E.; Klochkov, S.G.; Pukhov, S.A.; Afanasieva, S.V.; Aleksandrova, Y.R.; Yandulova, E.Y.; Avila-Rodriguez, M.F.; Mikhaleva, L.M.; Nikolenko, V.N.; Somasundaram, S.G.; et al. Synthesis and Cytotoxic Activity of Azine Derivatives of 6-Hydroxyxanthanodiene. Curr. Cancer Drug Targets 2020, 20, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.; Semakov, A.; Aleksandrova, Y.; Yandulova, E.; Pukhov, S.; Anikina, L.; Klochkov, S. N-Alkylation of Anthracycline Antibiotics by Natural Sesquiterpene Lactones as a Way to Obtain Antitumor Agents with Reduced Side Effects. Biomedicines 2021, 9, 547. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.E.; Smirnova, E.V.; Sharova, E.V.; Artyushin, O.I.; Aleksandrova, Y.R.; Yandulova, E.Y.; Nikolaeva, N.S.; Brel, V.K. Design of Conjugates Based on Sesquiterpene Lactones with Polyalkoxybenzenes by “Click” Chemistry to Create Potential Anticancer Agents. Molecules 2022, 27, 8411. [Google Scholar] [CrossRef]
- Neganova, M.; Liu, J.; Aleksandrova, Y.; Vasilieva, N.; Semakov, A.; Yandulova, E.; Sukocheva, O.; Balakin, K.; Klochkov, S.; Fan, R. Development of Neuroprotective Agents for the Treatment of Alzheimer’s Disease Using Conjugates of Serotonin with Sesquiterpene Lactones. Curr. Med. Chem. 2022, 30, 529–551. [Google Scholar] [CrossRef]
- Talman, A.M.; Clain, J.; Duval, R.; Menard, R.; Ariey, F. Artemisinin Bioactivity and Resistance in Malaria Parasites. Trends Parasitol. 2019, 35, 953–963. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The birth of artemisinin. Pharmacol. Ther. 2020, 216, 107658. [Google Scholar] [CrossRef]
- Chen, G.Q.; Benthani, F.A.; Wu, J.; Liang, D.; Bian, Z.X.; Jiang, X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020, 27, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gan, S.; Zhuang, X.; Chen, Y.; Lu, L.; Wang, Y.; Qi, X.; Feng, Q.; Huang, Q.; Du, B.; et al. Artesunate Inhibits the Cell Growth in Colorectal Cancer by Promoting ROS-Dependent Cell Senescence and Autophagy. Cells 2022, 11, 2472. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yi, H.; Yao, H.; Lu, L.; He, G.; Wu, M.; Zheng, C.; Li, Y.; Chen, S.; Li, L.; et al. Artemisinin Derivatives Inhibit Non-small Cell Lung Cancer Cells Through Induction of ROS-dependent Apoptosis/Ferroptosis. J. Cancer 2021, 12, 4075–4085. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef]
- Nunes, J.J.; Pandey, S.K.; Yadav, A.; Goel, S.; Ateeq, B. Targeting NF-kappa B Signaling by Artesunate Restores Sensitivity of Castrate-Resistant Prostate Cancer Cells to Antiandrogens. Neoplasia 2017, 19, 333–345. [Google Scholar] [CrossRef]
- Yan, J.; Ma, H.; Lai, X.; Wu, J.; Liu, A.; Huang, J.; Sun, W.; Shen, M.; Zhang, Y. Artemisinin attenuated oxidative stress and apoptosis by inhibiting autophagy in MPP(+)-treated SH-SY5Y cells. J. Biol. Res. 2021, 28, 6. [Google Scholar] [CrossRef]
- Lv, J.; Zhu, J.; Wang, P.; Liu, T.; Yuan, J.; Yin, H.; Lan, Y.; Sun, Q.; Zhang, Z.; Ding, G.; et al. Artemisinin exerts a protective effect in the MPTP mouse model of Parkinson’s disease by inhibiting microglial activation via the TLR4/Myd88/NF-KB pathway. CNS Neurosci. Ther. 2023, 29, 1012–1023. [Google Scholar] [CrossRef]
- Lin, S.P.; Li, W.; Winters, A.; Liu, R.; Yang, S.H. Artemisinin Prevents Glutamate-Induced Neuronal Cell Death Via Akt Pathway Activation. Front. Cell Neurosci. 2018, 12, 108. [Google Scholar] [CrossRef]
- Okorji, U.P.; Velagapudi, R.; El-Bakoush, A.; Fiebich, B.L.; Olajide, O.A. Antimalarial Drug Artemether Inhibits Neuroinflammation in BV2 Microglia Through Nrf2-Dependent Mechanisms. Mol. Neurobiol. 2016, 53, 6426–6443. [Google Scholar] [CrossRef]
- Zhao, X.; Li, S.; Gaur, U.; Zheng, W. Artemisinin Improved Neuronal Functions in Alzheimer’s Disease Animal Model 3xtg Mice and Neuronal Cells via Stimulating the ERK/CREB Signaling Pathway. Aging Dis. 2020, 11, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Rasul, A.; Bao, R.; Malhi, M.; Zhao, B.; Tsuji, I.; Li, J.; Li, X. Induction of apoptosis by costunolide in bladder cancer cells is mediated through ROS generation and mitochondrial dysfunction. Molecules 2013, 18, 1418–1433. [Google Scholar] [CrossRef] [PubMed]
- Hua, P.; Sun, M.; Zhang, G.; Zhang, Y.; Song, G.; Liu, Z.; Li, X.; Zhang, X.; Li, B. Costunolide Induces Apoptosis through Generation of ROS and Activation of P53 in Human Esophageal Cancer Eca-109 Cells. J. Biochem. Mol. Toxicol. 2016, 30, 462–469. [Google Scholar] [CrossRef]
- Choi, Y.J.; Choi, Y.K.; Ko, S.G.; Cheon, C.; Kim, T.Y. Investigation of Molecular Mechanisms Involved in Sensitivity to the Anti-Cancer Activity of Costunolide in Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 4009. [Google Scholar] [CrossRef] [PubMed]
- Jeyamohan, S.; Moorthy, R.K.; Kannan, M.K.; Arockiam, A.J. Parthenolide induces apoptosis and autophagy through the suppression of PI3K/Akt signaling pathway in cervical cancer. Biotechnol. Lett. 2016, 38, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- D’Anneo, A.; Carlisi, D.; Lauricella, M.; Puleio, R.; Martinez, R.; Di Bella, S.; Di Marco, P.; Emanuele, S.; Di Fiore, R.; Guercio, A.; et al. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis. 2013, 4, e891. [Google Scholar] [CrossRef]
- Jorge, J.; Neves, J.; Alves, R.; Geraldes, C.; Goncalves, A.C.; Sarmento-Ribeiro, A.B. Parthenolide Induces ROS-Mediated Apoptosis in Lymphoid Malignancies. Int. J. Mol. Sci. 2023, 24, 9167. [Google Scholar] [CrossRef]
- Cheong, C.U.; Yeh, C.S.; Hsieh, Y.W.; Lee, Y.R.; Lin, M.Y.; Chen, C.Y.; Lee, C.H. Protective Effects of Costunolide against Hydrogen Peroxide-Induced Injury in PC12 Cells. Molecules 2016, 21, 898. [Google Scholar] [CrossRef]
- Fan, M.; Wang, C.; Zhao, X.; Jiang, Y.; Wang, C. Parthenolide alleviates microglia-mediated neuroinflammation via MAPK/TRIM31/NLRP3 signaling to ameliorate cognitive disorder. Int. Immunopharmacol. 2023, 120, 110287. [Google Scholar] [CrossRef]
- Arslan, M.E.; Turkez, H.; Sevim, Y.; Selvitopi, H.; Kadi, A.; Oner, S.; Mardinoglu, A. Costunolide and Parthenolide Ameliorate MPP+ Induced Apoptosis in the Cellular Parkinson’s Disease Model. Cells 2023, 12, 992. [Google Scholar] [CrossRef]
- Reyes, A.A.; Marcum, R.D.; He, Y. Structure and Function of Chromatin Remodelers. J. Mol. Biol. 2021, 433, 166929. [Google Scholar] [CrossRef]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. 2022, 83, 452–471. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wei, W.; Zhou, D.X. Histone Acetylation Enzymes Coordinate Metabolism and Gene Expression. Trends Plant Sci. 2015, 20, 614–621. [Google Scholar] [CrossRef]
- Peserico, A.; Simone, C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Y.; Yin, H. Recent advances in biosensor for histone acetyltransferase detection. Biosens. Bioelectron. 2021, 175, 112880. [Google Scholar] [CrossRef]
- Khangura, R.K.; Bali, A.; Jaggi, A.S.; Singh, N. Histone acetylation and histone deacetylation in neuropathic pain: An unresolved puzzle? Eur. J. Pharmacol. 2017, 795, 36–42. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Caron, C.; Boyault, C.; Khochbin, S. Regulatory cross-talk between lysine acetylation and ubiquitination: Role in the control of protein stability. Bioessays 2005, 27, 408–415. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.; Chatterji, B.P. HDAC Inhibitors as Novel Anti-Cancer Therapeutics. Recent. Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.J.; Shakespear, M.R.; Kamal, N.A.; Fairlie, D.P. HDAC inhibitors: Modulating leukocyte differentiation, survival, proliferation and inflammation. Immunol. Cell Biol. 2012, 90, 14–22. [Google Scholar] [CrossRef]
- Glauben, R.; Sonnenberg, E.; Zeitz, M.; Siegmund, B. HDAC inhibitors in models of inflammation-related tumorigenesis. Cancer Lett. 2009, 280, 154–159. [Google Scholar] [CrossRef]
- Leus, N.G.; Zwinderman, M.R.; Dekker, F.J. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-kappaB-mediated inflammation. Curr. Opin. Chem. Biol. 2016, 33, 160–168. [Google Scholar] [CrossRef]
- Robert, C.; Rassool, F.V. HDAC inhibitors: Roles of DNA damage and repair. Adv. Cancer Res. 2012, 116, 87–129. [Google Scholar]
- Zhao, C.; Dong, H.; Xu, Q.; Zhang, Y. Histone deacetylase (HDAC) inhibitors in cancer: A patent review (2017-present). Expert Opin. Ther. Pat. 2020, 30, 263–274. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Cao, L.L.; Song, X.; Pei, L.; Liu, L.; Wang, H.; Jia, M. Histone deacetylase HDAC1 expression correlates with the progression and prognosis of lung cancer: A meta-analysis. Medicine 2017, 96, e7663. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Hong, Y.S.; Choi, P.J.; Kim, N.Y.; Lee, K.E.; Roh, M.S. The Overexpression of Histone Deacetylase 1 and Its Relationship with p16INK4a Gene Hypermethylation in Pulmonary Squamous Cell Carcinoma and Adenocarcinoma. Korean J. Pathol. 2009, 43, 107–112. [Google Scholar] [CrossRef]
- Benard, A.; Goossens-Beumer, I.J.; van Hoesel, A.Q.; Horati, H.; de Graaf, W.; Putter, H.; Zeestraten, E.C.; Liefers, G.J.; van de Velde, C.J.; Kuppen, P.J. Nuclear expression of histone deacetylases and their histone modifications predicts clinical outcome in colorectal cancer. Histopathology 2015, 66, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Higashijima, J.; Kurita, N.; Miyatani, T.; Yoshikawa, K.; Morimoto, S.; Nishioka, M.; Iwata, T.; Shimada, M. Expression of histone deacetylase 1 and metastasis-associated protein 1 as prognostic factors in colon cancer. Oncol. Rep. 2011, 26, 343–348. [Google Scholar]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Mutze, K.; Langer, R.; Becker, K.; Ott, K.; Novotny, A.; Luber, B.; Hapfelmeier, A.; Gottlicher, M.; Hofler, H.; Keller, G. Histone deacetylase (HDAC) 1 and 2 expression and chemotherapy in gastric cancer. Ann. Surg. Oncol. 2010, 17, 3336–3343. [Google Scholar] [CrossRef]
- Wisnieski, F.; Calcagno, D.Q.; Leal, M.F.; Chen, E.S.; Gigek, C.O.; Santos, L.C.; Pontes, T.B.; Rasmussen, L.T.; Payao, S.L.; Assumpcao, P.P.; et al. Differential expression of histone deacetylase and acetyltransferase genes in gastric cancer and their modulation by trichostatin A. Tumour Biol. 2014, 35, 6373–6381. [Google Scholar] [CrossRef]
- Morine, Y.; Shimada, M.; Iwahashi, S.; Utsunomiya, T.; Imura, S.; Ikemoto, T.; Mori, H.; Hanaoka, J.; Miyake, H. Role of histone deacetylase expression in intrahepatic cholangiocarcinoma. Surgery 2012, 151, 412–419. [Google Scholar] [CrossRef]
- Rikimaru, T.; Taketomi, A.; Yamashita, Y.; Shirabe, K.; Hamatsu, T.; Shimada, M.; Maehara, Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 2007, 72, 69–74. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef]
- Sudo, T.; Mimori, K.; Nishida, N.; Kogo, R.; Iwaya, T.; Tanaka, F.; Shibata, K.; Fujita, H.; Shirouzu, K.; Mori, M. Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 2011, 26, 777–782. [Google Scholar] [PubMed]
- Jiang, Z.; Sun, X.; Zhang, Q.; Ji, X.; Yu, Q.; Huang, T.; Chen, D.; Chen, H.; Mei, X.; Wang, L.; et al. Identification of candidate biomarkers that involved in the epigenetic transcriptional regulation for detection gastric cancer by iTRAQ based quantitative proteomic analysis. Clin. Chim. Acta 2017, 471, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zeng, J.; Liu, H.; Wang, T.; Yu, Z.; Chen, J. Role of HDAC1 in the progression of gastric cancer and the correlation with lncRNAs. Oncol. Lett. 2019, 17, 3296–3304. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef]
- McCabe, E.M.; Rasmussen, T.P. lncRNA involvement in cancer stem cell function and epithelial-mesenchymal transitions. Semin. Cancer Biol. 2021, 75, 38–48. [Google Scholar] [CrossRef]
- Yan, H.; Bu, P. Non-coding RNA in cancer. Essays Biochem. 2021, 65, 625–639. [Google Scholar]
- Deng, R.; Zhang, P.; Liu, W.; Zeng, X.; Ma, X.; Shi, L.; Wang, T.; Yin, Y.; Chang, W.; Zhang, P.; et al. HDAC is indispensable for IFN-gamma-induced B7-H1 expression in gastric cancer. Clin. Epigenetics 2018, 10, 153. [Google Scholar] [CrossRef]
- Jiang, Z.; Yang, H.; Zhang, X.; Wang, Z.; Rong, R.; Wang, X. Histone deacetylase-1 as a prognostic factor and mediator of gastric cancer progression by enhancing glycolysis. Hum. Pathol. 2019, 85, 194–201. [Google Scholar] [CrossRef]
- Guo, Q.; Cheng, K.; Wang, X.; Li, X.; Yu, Y.; Hua, Y.; Yang, Z. Expression of HDAC1 and RBBP4 correlate with clinicopathologic characteristics and prognosis in breast cancer. Int. J. Clin. Exp. Pathol. 2020, 13, 563–572. [Google Scholar]
- Tang, Z.; Ding, S.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. HDAC1 triggers the proliferation and migration of breast cancer cells via upregulation of interleukin-8. Biol. Chem. 2017, 398, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, C.; Sanmamed, M.F.; Rodriguez-Ruiz, M.E.; Teijeira, A.; Onate, C.; Gonzalez, A.; Ponz, M.; Schalper, K.A.; Perez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Lukina, E.; Friedemann, M.; Menschikowski, M.; Hagelgans, A.; Aliev, G. The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: Current findings and future perspectives. Semin. Cancer Biol. 2022, 82, 35–59. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Ashby, C.R., Jr.; Assaraf, Y.G.; Chen, Z.S.; Liu, H.M. Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jiang, Z.; Yin, P.; Li, Q.; Liu, J. Role for Class I histone deacetylases in multidrug resistance. Exp. Cell Res. 2012, 318, 177–186. [Google Scholar] [CrossRef]
- Duan, H.; Zhou, K.; Zhang, Y.; Yue, P.; Wang, T.; Li, Y.; Qiu, D.; Hua, Y.; Wang, C. HDAC1 was involved in placental breast cancer resistance protein regulation in vitro: A preliminary study. J. Cell. Mol. Med. 2019, 23, 5818–5821. [Google Scholar] [CrossRef]
- Wang, C.; Ma, D.; Hua, Y.; Duan, H. Modulation of Placental Breast Cancer Resistance Protein by HDAC1 in Mice: Implications for Optimization of Pharmacotherapy During Pregnancy. Reprod. Sci. 2021, 28, 3540–3546. [Google Scholar] [CrossRef]
- Burdelski, C.; Ruge, O.M.; Melling, N.; Koop, C.; Simon, R.; Steurer, S.; Sauter, G.; Kluth, M.; Hube-Magg, C.; Minner, S.; et al. HDAC1 overexpression independently predicts biochemical recurrence and is associated with rapid tumor cell proliferation and genomic instability in prostate cancer. Exp. Mol. Pathol. 2015, 98, 419–426. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, S.N.; Kim, Y.K. Involvement of HDAC1 in E-cadherin expression in prostate cancer cells; its implication for cell motility and invasion. Biochem. Biophys. Res. Commun. 2011, 404, 915–921. [Google Scholar] [CrossRef]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Torigoe, T.; Asanuma, H.; Hisasue, S.I.; Suzuki, K.; Tsukamoto, T.; Satoh, M.; Sato, N. Cytosolic overexpression of p62 sequestosome 1 in neoplastic prostate tissue. Histopathology 2006, 48, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Shankar, E.; Pandey, M.; Verma, S.; Abbas, A.; Candamo, M.; Kanwal, R.; Shukla, S.; MacLennan, G.T.; Gupta, S. Role of class I histone deacetylases in the regulation of maspin expression in prostate cancer. Mol. Carcinog. 2020, 59, 955–966. [Google Scholar] [CrossRef]
- Bodenstine, T.M.; Seftor, R.E.; Khalkhali-Ellis, Z.; Seftor, E.A.; Pemberton, P.A.; Hendrix, M.J. Maspin: Molecular mechanisms and therapeutic implications. Cancer Metastasis Rev. 2012, 31, 529–551. [Google Scholar] [CrossRef]
- Dzinic, S.H.; Bernardo, M.M.; Oliveira, D.S.; Wahba, M.; Sakr, W.; Sheng, S. Tumor suppressor maspin as a modulator of host immune response to cancer. Bosn. J. Basic Med. Sci. 2015, 15, 1–6. [Google Scholar] [CrossRef]
- Jin, K.L.; Pak, J.H.; Park, J.Y.; Choi, W.H.; Lee, J.Y.; Kim, J.H.; Nam, J.H. Expression profile of histone deacetylases 1, 2 and 3 in ovarian cancer tissues. J. Gynecol. Oncol. 2008, 19, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhou, P.; Zhang, L.; Gong, W.; Huang, G.; Zheng, Y.; He, F. HDAC1/DNMT3A-containing complex is associated with suppression of Oct4 in cervical cancer cells. Biochemistry 2012, 77, 934–940. [Google Scholar] [CrossRef]
- Yano, M.; Yasuda, M.; Sakaki, M.; Nagata, K.; Fujino, T.; Arai, E.; Hasebe, T.; Miyazawa, M.; Miyazawa, M.; Ogane, N.; et al. Association of histone deacetylase expression with histology and prognosis of ovarian cancer. Oncol. Lett. 2018, 15, 3524–3531. [Google Scholar] [CrossRef]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef]
- Hooks, S.B.; Murph, M.M. Cellular deficiency in the RGS10 protein facilitates chemoresistant ovarian cancer. Future Med. Chem. 2015, 7, 1483–1489. [Google Scholar] [CrossRef]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E. Histone Deacetylase-1-mediated Suppression of FAS in Chemoresistant Ovarian Cancer Cells. Anticancer Res. 2016, 36, 2819–2826. [Google Scholar] [PubMed]
- Liu, X.; Yu, Y.; Zhang, J.; Lu, C.; Wang, L.; Liu, P.; Song, H. HDAC1 Silencing in Ovarian Cancer Enhances the Chemotherapy Response. Cell Physiol. Biochem. 2018, 48, 1505–1518. [Google Scholar] [CrossRef]
- Zhu, Y.; Piao, C.; Zhang, Z.; Jiang, Y.; Kong, C. The potential role of c-MYC and polyamine metabolism in multiple drug resistance in bladder cancer investigated by metabonomics. Genomics 2022, 114, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Krysan, K.; Kusko, R.; Grogan, T.; O’Hearn, J.; Reckamp, K.L.; Walser, T.C.; Garon, E.B.; Lenburg, M.E.; Sharma, S.; Spira, A.E.; et al. PGE2-driven expression of c-Myc and oncomiR-17-92 contributes to apoptosis resistance in NSCLC. Mol. Cancer Res. 2014, 12, 765–774. [Google Scholar] [CrossRef]
- Yang, Y.; Yuan, H.; Zhao, L.; Guo, S.; Hu, S.; Tian, M.; Nie, Y.; Yu, J.; Zhou, C.; Niu, J.; et al. Targeting the miR-34a/LRPPRC/MDR1 axis collapse the chemoresistance in P53 inactive colorectal cancer. Cell Death Differ. 2022, 29, 2177–2189. [Google Scholar] [CrossRef]
- Li, W.J.; Wang, Y.; Liu, R.; Kasinski, A.L.; Shen, H.; Slack, F.J.; Tang, D.G. MicroRNA-34a: Potent Tumor Suppressor, Cancer Stem Cell Inhibitor, and Potential Anticancer Therapeutic. Front. Cell Dev. Biol. 2021, 9, 640587. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, H.; Wang, C.; Zhang, W.; Wen, X.; Long, H.; Xu, Z.; Guo, H.; Liu, Y.; Wei, D.; et al. Clinicopathological and prognostic significance of octamer-binding transcription factor 4 in patients with gastric cancer: A systematic review and meta-analysis. Biomark. Med. 2019, 13, 219–234. [Google Scholar] [CrossRef]
- Gao, Z.Y.; Liu, X.B.; Yang, F.M.; Liu, L.; Zhao, J.Z.; Gao, B.; Li, S.B. Octamer binding transcription factor-4 expression is associated with cervical cancer malignancy and histological differentiation: A systematic review and meta-analysis. Biosci. Rep. 2019, 39, BSR20182328. [Google Scholar] [CrossRef]
- Zhao, X.; Lu, H.; Sun, Y.; Liu, L.; Wang, H. Prognostic value of octamer binding transcription factor 4 for patients with solid tumors: A meta-analysis. Medicine 2020, 99, e22804. [Google Scholar] [CrossRef]
- Upadhyay, V.A.; Shah, K.A.; Makwana, D.P.; Raval, A.P.; Shah, F.D.; Rawal, R.M. Putative stemness markers octamer-binding transcription factor 4, sex-determining region Y-box 2, and NANOG in non-small cell lung carcinoma: A clinicopathological association. J. Cancer Res. Ther. 2020, 16, 804–810. [Google Scholar] [PubMed]
- Yokoi, E.; Mabuchi, S.; Shimura, K.; Komura, N.; Kozasa, K.; Kuroda, H.; Takahashi, R.; Sasano, T.; Kawano, M.; Matsumoto, Y.; et al. Lurbinectedin (PM01183), a selective inhibitor of active transcription, effectively eliminates both cancer cells and cancer stem cells in preclinical models of uterine cervical cancer. Investig. New Drugs 2019, 37, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.X.; Chen, W.S.; Zeng, W.; Cheng, X.F.; Lin, M.B.; Wang, J.S. Roles of HDAC2, eIF5, and eIF6 in Lung Cancer Tumorigenesis. Curr. Med. Sci. 2021, 41, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.W.; Guan, X.Y.; Xie, D. Roles of eukaryotic initiation factor 5A2 in human cancer. Int. J. Biol. Sci. 2013, 9, 1013–1020. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, K.; Ning, L.; Chen, D.; Hao, F.; Li, P. Clinical significance of eukaryotic translation initiation factor 5A2 in papillary thyroid cancer. Bioengineered 2020, 11, 1325–1333. [Google Scholar] [CrossRef]
- Gantenbein, N.; Bernhart, E.; Anders, I.; Golob-Schwarzl, N.; Krassnig, S.; Wodlej, C.; Brcic, L.; Lindenmann, J.; Fink-Neuboeck, N.; Gollowitsch, F.; et al. Influence of eukaryotic translation initiation factor 6 on non-small cell lung cancer development and progression. Eur. J. Cancer 2018, 101, 165–180. [Google Scholar] [CrossRef]
- Golob-Schwarzl, N.; Wodlej, C.; Kleinegger, F.; Gogg-Kamerer, M.; Birkl-Toeglhofer, A.M.; Petzold, J.; Aigelsreiter, A.; Thalhammer, M.; Park, Y.N.; Haybaeck, J. Eukaryotic translation initiation factor 6 overexpression plays a major role in the translational control of gallbladder cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2699–2711. [Google Scholar] [CrossRef]
- Li, L.; Mei, D.T.; Zeng, Y. HDAC2 promotes the migration and invasion of non-small cell lung cancer cells via upregulation of fibronectin. Biomed. Pharmacother. 2016, 84, 284–290. [Google Scholar] [CrossRef]
- Kumra, H.; Reinhardt, D.P. Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef]
- Rick, J.W.; Chandra, A.; Dalle Ore, C.; Nguyen, A.T.; Yagnik, G.; Aghi, M.K. Fibronectin in malignancy: Cancer-specific alterations, protumoral effects, and therapeutic implications. Semin. Oncol. 2019, 46, 284–290. [Google Scholar] [CrossRef]
- Lin, T.C.; Yang, C.H.; Cheng, L.H.; Chang, W.T.; Lin, Y.R.; Cheng, H.C. Fibronectin in Cancer: Friend or Foe. Cells 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Shen, X.Y.; Pan, L.Y.; Li, Z.; Chen, C.J.; Yao, Y.S.; Tang, D.F.; Gao, W. The HDAC2-MTA3 interaction induces nonsmall cell lung cancer cell migration and invasion by targeting c-Myc and cyclin D1. In Molecular Carcinogenesis; Wiley: Hoboken, NJ, USA, 2023. [Google Scholar]
- Conte, M.; Di Mauro, A.; Capasso, L.; Montella, L.; De Simone, M.; Nebbioso, A.; Altucci, L. Targeting HDAC2-Mediated Immune Regulation to Overcome Therapeutic Resistance in Mutant Colorectal Cancer. Cancers 2023, 15, 1960. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.P.; Yalikong, A.; Zhang, J.W.; Cai, S.L.; Li, B.; Di, S.; Lv, Z.T.; Xu, E.P.; Zhong, Y.S.; Zhou, P.H. HDAC2 promotes the EMT of colorectal cancer cells and via the modular scaffold function of ENSG00000274093.1. J. Cell. Mol. Med. 2021, 25, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Krishna, A.; Singh, V.; Singh, S.; Kumar, S.; Kumar, V.; Mehrotra, D.; Singh, U.S.; Mahdi, A.A. Upregulated histone deacetylase 2 gene correlates with the progression of oral squamous cell carcinoma. Cancer Biomark. 2020, 29, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, F.; Qu, Y.; Chen, X.; Gao, M.; Yang, J.; Zhang, D.; Zhang, N.; Li, W.; Liu, H. HDAC2 regulates cell proliferation, cell cycle progression and cell apoptosis in esophageal squamous cell carcinoma EC9706 cells. Oncol. Lett. 2017, 13, 403–409. [Google Scholar] [CrossRef][Green Version]
- Du, X.; Zhao, H.; Zang, L.; Song, N.; Yang, T.; Dong, R.; Yin, J.; Wang, C.; Lu, J. Overexpression of histone deacetylase 2 predicts unfavorable prognosis in human gallbladder carcinoma. Pathol. Oncol. Res. 2013, 19, 397–403. [Google Scholar] [CrossRef]
- Shan, W.; Jiang, Y.; Yu, H.; Huang, Q.; Liu, L.; Guo, X.; Li, L.; Mi, Q.; Zhang, K.; Yang, Z. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am. J. Cancer Res. 2017, 7, 1213–1226. [Google Scholar]
- Ma, L.; Qi, L.; Li, S.; Yin, Q.; Liu, J.; Wang, J.; She, C.; Li, P.; Liu, Q.; Wang, X.; et al. Aberrant HDAC3 expression correlates with brain metastasis in breast cancer patients. Thorac. Cancer 2020, 11, 2493–2505. [Google Scholar] [CrossRef]
- Hu, G.; He, N.; Cai, C.; Cai, F.; Fan, P.; Zheng, Z.; Jin, X. HDAC3 modulates cancer immunity via increasing PD-L1 expression in pancreatic cancer. Pancreatology 2019, 19, 383–389. [Google Scholar] [CrossRef]
- Edderkaoui, M.; Xu, S.; Chheda, C.; Morvaridi, S.; Hu, R.W.; Grippo, P.J.; Mascarinas, E.; Principe, D.R.; Knudsen, B.; Xue, J.; et al. HDAC3 mediates smoking-induced pancreatic cancer. Oncotarget 2016, 7, 7747–7760. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Curtis, S.D.; Brun, S.N.; McGuire, C.K.; Gushterova, I.; Baumgart, J.T.; Trefts, E.; Ross, D.S.; Rymoff, T.J.; Shaw, R.J. HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non-small cell lung cancer. Sci. Adv. 2023, 9, eadd3243. [Google Scholar] [CrossRef] [PubMed]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Motoyama, S.; Ogawa, J. Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumour Biol. 2010, 31, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.J.; Huang, F.X.; Sun, Z.T.; Zhang, R.X.; Huang, S.F.; Wang, J. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in nonsmall cell lung cancer cells. Tumour Biol. 2014, 35, 7097–7103. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Q.; Chen, C.S.; Chen, J.J.; Zhou, L.P.; Xu, H.L.; Jin, W.W.; Wu, J.B.; Gao, S.M. Histone deacetylases inhibitor trichostatin A increases the expression of Dleu2/miR-15a/16-1 via HDAC3 in non-small cell lung cancer. Mol. Cell. Biochem. 2013, 383, 137–148. [Google Scholar] [CrossRef]
- McLeod, A.B.; Stice, J.P.; Wardell, S.E.; Alley, H.M.; Chang, C.Y.; McDonnell, D.P. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate 2018, 78, 266–277. [Google Scholar] [CrossRef]
- Zheng, Y.; Wu, C.; Yang, J.; Zhao, Y.; Jia, H.; Xue, M.; Xu, D.; Yang, F.; Fu, D.; Wang, C.; et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 53. [Google Scholar] [CrossRef]
- Wu, L.M.; Yang, Z.; Zhou, L.; Zhang, F.; Xie, H.Y.; Feng, X.W.; Wu, J.; Zheng, S.S. Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS ONE 2010, 5, e14460. [Google Scholar] [CrossRef]
- Liu, C.; Liu, L.; Shan, J.; Shen, J.; Xu, Y.; Zhang, Q.; Yang, Z.; Wu, L.; Xia, F.; Bie, P.; et al. Histone deacetylase 3 participates in self-renewal of liver cancer stem cells through histone modification. Cancer Lett. 2013, 339, 60–69. [Google Scholar] [CrossRef]
- Xu, G.; Zhu, H.; Zhang, M.; Xu, J. Histone deacetylase 3 is associated with gastric cancer cell growth via the miR-454-mediated targeting of CHD5. Int. J. Mol. Med. 2018, 41, 155–163. [Google Scholar] [CrossRef]
- Nemati, M.; Ajami, N.; Estiar, M.A.; Rezapour, S.; Gavgani, R.R.; Hashemzadeh, S.; Kafil, H.S.; Sakhinia, E. Deregulated expression of HDAC3 in colorectal cancer and its clinical significance. Adv. Clin. Exp. Med. 2018, 27, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Hu, H.; Han, T.; Zhuo, M.; Yuan, C.; Yang, H.; Wang, L.; Wang, L. Aberrant expression of nuclear HDAC3 and cytoplasmic CDH1 predict a poor prognosis for patients with pancreatic cancer. Oncotarget 2016, 7, 16505–16516. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fan, Y.; Wu, B.; Wang, Y.; Jiang, S.; Ge, J.; Hua, C.; Zhao, G.; Chen, Y.; Xu, H. HDAC3 Expression Correlates with the Prognosis and Grade of Patients with Glioma: A Diversification Analysis Based on Transcriptome and Clinical Evidence. World Neurosurg. 2018, 119, e145–e158. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Romanski, A.; Mustafa, A.M.; Pons, M.; Buchler, I.; Vogel, A.; Pautz, A.; Sellmer, A.; Schneider, G.; Bug, G.; et al. HDAC3 Activity is Essential for Human Leukemic Cell Growth and the Expression of beta-catenin, MYC, and WT1. Cancers 2019, 11, 1436. [Google Scholar] [CrossRef]
- Godman, C.A.; Joshi, R.; Tierney, B.R.; Greenspan, E.; Rasmussen, T.P.; Wang, H.W.; Shin, D.G.; Rosenberg, D.W.; Giardina, C. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol. Ther. 2008, 7, 1570–1580. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, J.; Luo, H.; Meng, X.; Chen, M.; Zhu, D. Wnt signaling pathway in cancer immunotherapy. Cancer Lett. 2022, 525, 84–96. [Google Scholar] [CrossRef]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef]
- Jiao, F.; Hu, H.; Yuan, C.; Jin, Z.; Guo, Z.; Wang, L.; Wang, L. Histone deacetylase 3 promotes pancreatic cancer cell proliferation, invasion and increases drug-resistance through histone modification of P27, P53 and Bax. Int. J. Oncol. 2014, 45, 1523–1530. [Google Scholar] [CrossRef]
- Ren, H.; Tang, L. HDAC3-mediated lncRNA-LOC101928316 contributes to cisplatin resistance in gastric cancer via activating the PI3K-Akt-mTOR pathway. Neoplasma 2021, 68, 1043–1051. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. Pathological Role of HDAC8: Cancer and Beyond. Cells 2022, 11, 3161. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3beta/Snail signals. Oncogene 2020, 39, 4956–4969. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, L.; Ma, J.; Yue, C.; Zhu, D.; An, R.; Wang, X.; Guo, Y.; Gu, B. HDAC8 Promotes Liver Metastasis of Colorectal Cancer via Inhibition of IRF1 and Upregulation of SUCNR1. Oxid. Med. Cell. Longev. 2022, 2022, 2815187. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Du, C.; Lv, Z.; Ding, C.; Cheng, J.; Xie, H.; Zhou, L.; Zheng, S. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig. Dis. Sci. 2013, 58, 3545–3553. [Google Scholar] [CrossRef]
- Song, S.; Wang, Y.; Xu, P.; Yang, R.; Ma, Z.; Liang, S.; Zhang, G. The inhibition of histone deacetylase 8 suppresses proliferation and inhibits apoptosis in gastric adenocarcinoma. Int. J. Oncol. 2015, 47, 1819–1828. [Google Scholar] [CrossRef]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.; de Paula Queiroz, R.; Valera, E.T.; da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Yoon, J.H. Histone deacetylase 8 as a novel therapeutic target in oral squamous cell carcinoma. Oncol. Rep. 2017, 37, 540–546. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Colebatch, A.J.; Kakavand, H.; Shang, P.; Carlino, M.S.; Thompson, J.F.; Long, G.V.; Scolyer, R.A.; Hersey, P. Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod. Pathol. 2015, 28, 884–894. [Google Scholar] [CrossRef]
- Mormino, A.; Cocozza, G.; Fontemaggi, G.; Valente, S.; Esposito, V.; Santoro, A.; Bernardini, G.; Santoni, A.; Fazi, F.; Mai, A.; et al. Histone-deacetylase 8 drives the immune response and the growth of glioma. Glia 2021, 69, 2682–2698. [Google Scholar] [CrossRef]
- Lopez, G.; Bill, K.L.; Bid, H.K.; Braggio, D.; Constantino, D.; Prudner, B.; Zewdu, A.; Batte, K.; Lev, D.; Pollock, R.E. HDAC8, A Potential Therapeutic Target for the Treatment of Malignant Peripheral Nerve Sheath Tumors (MPNST). PLoS ONE 2015, 10, e0133302. [Google Scholar] [CrossRef]
- Gao, S.M.; Chen, C.Q.; Wang, L.Y.; Hong, L.L.; Wu, J.B.; Dong, P.H.; Yu, F.J. Histone deacetylases inhibitor sodium butyrate inhibits JAK2/STAT signaling through upregulation of SOCS1 and SOCS3 mediated by HDAC8 inhibition in myeloproliferative neoplasms. Exp. Hematol. 2013, 41, 261–270.e4. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Wu, W.Z.; Zhang, Y.P.; Huang, W.J. Gene polymorphisms of SOCS1 and SOCS2 and acute lymphoblastic leukemia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5564–5572. [Google Scholar] [PubMed]
- Zhang, R.; Shen, M.; Wu, C.; Chen, Y.; Lu, J.; Li, J.; Zhao, L.; Meng, H.; Zhou, X.; Huang, G.; et al. HDAC8-dependent deacetylation of PKM2 directs nuclear localization and glycolysis to promote proliferation in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 1036. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.; Wang, Y.; Yu, Z.; Li, X.; Deng, Y.; Wang, Y.; Yang, D.; Li, Q.; Zeng, X.; Ju, J.; et al. AURKB promotes gastric cancer progression via activation of CCND1 expression. Aging 2020, 12, 1304–1321. [Google Scholar] [CrossRef]
- Zhu, D.; Huang, J.; Liu, N.; Li, W.; Yan, L. PSMC2/CCND1 axis promotes development of ovarian cancer through regulating cell growth, apoptosis and migration. Cell Death Dis. 2021, 12, 730. [Google Scholar] [CrossRef]
- Valla, M.; Klaestad, E.; Ytterhus, B.; Bofin, A.M. CCND1 Amplification in Breast Cancer -associations With Proliferation, Histopathological Grade, Molecular Subtype and Prognosis. J. Mammary Gland Biol. Neoplasia 2022, 27, 67–77. [Google Scholar] [CrossRef]
- Tang, X.; Li, G.; Su, F.; Cai, Y.; Shi, L.; Meng, Y.; Liu, Z.; Sun, J.; Wang, M.; Qian, M.; et al. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 2020, 48, 2912–2923. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. HDAC8 Deacetylates HIF-1alpha and Enhances Its Protein Stability to Promote Tumor Growth and Migration in Melanoma. Cancers 2023, 15, 1123. [Google Scholar] [CrossRef]
- Vanaja, G.R.; Ramulu, H.G.; Kalle, A.M. Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun. Signal 2018, 16, 20. [Google Scholar] [CrossRef]
- Wu, S.; Luo, Z.; Yu, P.J.; Xie, H.; He, Y.W. Suberoylanilide hydroxamic acid (SAHA) promotes the epithelial mesenchymal transition of triple negative breast cancer cells via HDAC8/FOXA1 signals. Biol. Chem. 2016, 397, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sheng, S.; Qin, C. The role of HDAC6 in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Mahady, L.; Nadeem, M.; Malek-Ahmadi, M.; Chen, K.; Perez, S.E.; Mufson, E.J. Frontal Cortex Epigenetic Dysregulation During the Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 115–131. [Google Scholar] [CrossRef]
- Bai, P.; Mondal, P.; Bagdasarian, F.A.; Rani, N.; Liu, Y.; Gomm, A.; Tocci, D.R.; Choi, S.H.; Wey, H.Y.; Tanzi, R.E.; et al. Development of a potential PET probe for HDAC6 imaging in Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 3891–3904. [Google Scholar] [CrossRef]
- Li, L.; Yang, X.J. Tubulin acetylation: Responsible enzymes, biological functions and human diseases. Cell. Mol. Life Sci. 2015, 72, 4237–4255. [Google Scholar] [CrossRef]
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2021, 163, 105274. [Google Scholar] [CrossRef]
- Dent, E.W. Of microtubules and memory: Implications for microtubule dynamics in dendrites and spines. Mol. Biol. Cell 2017, 28, 1–8. [Google Scholar] [CrossRef]
- Pena-Ortega, F.; Robles-Gomez, A.A.; Xolalpa-Cueva, L. Microtubules as Regulators of Neural Network Shape and Function: Focus on Excitability, Plasticity and Memory. Cells 2022, 11, 923. [Google Scholar] [CrossRef]
- Boiarska, Z.; Passarella, D. Microtubule-targeting agents and neurodegeneration. Drug Discov. Today 2021, 26, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kwon, Y.; Kim, S.; Jo, M.; Jeon, Y.M.; Cheon, M.; Lee, S.; Kim, S.R.; Kim, K.; Kim, H.J. The Role of HDAC6 in TDP-43-Induced Neurotoxicity and UPS Impairment. Front. Cell Dev. Biol. 2020, 8, 581942. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; Yen, S.H.; Nakamoto, Y. Autophagy and Tau Protein. Int. J. Mol. Sci. 2021, 22, 7475. [Google Scholar] [CrossRef]
- Xu, Y.; Propson, N.E.; Du, S.; Xiong, W.; Zheng, H. Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc. Natl. Acad. Sci. USA 2021, 118, e2023418118. [Google Scholar] [CrossRef]
- Choi, H.; Kim, H.J.; Yang, J.; Chae, S.; Lee, W.; Chung, S.; Kim, J.; Choi, H.; Song, H.; Lee, C.K.; et al. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 2020, 19, e13081. [Google Scholar] [CrossRef]
- Balmik, A.A.; Chidambaram, H.; Dangi, A.; Marelli, U.K.; Chinnathambi, S. HDAC6 ZnF UBP as the Modifier of Tau Structure and Function. Biochemistry 2020, 59, 4546–4562. [Google Scholar] [CrossRef]
- Qureshi, T.; Chinnathambi, S. Histone deacetylase-6 modulates Tau function in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119275. [Google Scholar] [CrossRef]
- Tseng, J.H.; Xie, L.; Song, S.; Xie, Y.; Allen, L.; Ajit, D.; Hong, J.S.; Chen, X.; Meeker, R.B.; Cohen, T.J. The Deacetylase HDAC6 Mediates Endogenous Neuritic Tau Pathology. Cell Rep. 2017, 20, 2169–2183. [Google Scholar] [CrossRef]
- Cook, C.; Gendron, T.F.; Scheffel, K.; Carlomagno, Y.; Dunmore, J.; DeTure, M.; Petrucelli, L. Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation. Hum. Mol. Genet. 2012, 21, 2936–2945. [Google Scholar] [CrossRef]
- Sreenivasmurthy, S.G.; Iyaswamy, A.; Krishnamoorthi, S.; Reddi, R.N.; Kammala, A.K.; Vasudevan, K.; Senapati, S.; Zhu, Z.; Su, C.F.; Liu, J.; et al. Bromo-protopine, a novel protopine derivative, alleviates tau pathology by activating chaperone-mediated autophagy for Alzheimer’s disease therapy. Front. Mol. Biosci. 2022, 9, 1030534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.; Li, C.; Zhu, T.; Gao, J.; Zhou, H.; Zheng, Y.; Chang, Q.; Wang, M.; Wu, J.; et al. S100A11 Promotes Liver Steatosis via FOXO1-Mediated Autophagy and Lipogenesis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 697–724. [Google Scholar] [CrossRef]
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 Inhibition Promotes Transcription Factor EB Activation and Is Protective in Experimental Kidney Disease. Front. Pharmacol. 2018, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Li, H.; Hu, H.; Li, Y.; Wang, T. The Role of HDAC6 in Autophagy and NLRP3 Inflammasome. Front. Immunol. 2021, 12, 763831. [Google Scholar] [CrossRef] [PubMed]
- Mazzetti, S.; De Leonardis, M.; Gagliardi, G.; Calogero, A.M.; Basellini, M.J.; Madaschi, L.; Costa, I.; Cacciatore, F.; Spinello, S.; Bramerio, M.; et al. Phospho-HDAC6 Gathers Into Protein Aggregates in Parkinson’s Disease and Atypical Parkinsonisms. Front. Neurosci. 2020, 14, 624. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Choi, J.; Sim, H.R.; Kim, J.; Jun, J.H.; Kyung, J.; Ha, N.; Kim, S.; Ryu, K.H.; Chung, S.S.; et al. A novel HDAC6 inhibitor, CKD-504, is effective in treating preclinical models of huntington’s disease. BMB Rep. 2023, 56, 178–183. [Google Scholar] [CrossRef]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Graff, J.; Rei, D.; Guan, J.S.; Wang, W.Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef]
- Jawerka, M.; Colak, D.; Dimou, L.; Spiller, C.; Lagger, S.; Montgomery, R.L.; Olson, E.N.; Wurst, W.; Gottlicher, M.; Gotz, M. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 2010, 6, 93–107. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, S.; Yu, L.; Jin, J.; Ye, X.; Liu, Y.; Xu, Y. HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer’s disease. Aging Cell 2017, 16, 1073–1082. [Google Scholar] [CrossRef]
- Bardai, F.H.; D’Mello, S.R. Selective toxicity by HDAC3 in neurons: Regulation by Akt and GSK3beta. J. Neurosci. 2011, 31, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, H.L.; Schwartz, S.; Given, F.M.; Scott, M.J. The histone deacetylase HDAC4 regulates long-term memory in Drosophila. PLoS ONE 2013, 8, e83903. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, Y.; Munkuev, A.; Mozhaitsev, E.; Suslov, E.; Tsypyshev, D.; Chaprov, K.; Begunov, R.; Volcho, K.; Salakhutdinov, N.; Neganova, M. Elaboration of the Effective Multi-Target Therapeutic Platform for the Treatment of Alzheimer’s Disease Based on Novel Monoterpene-Derived Hydroxamic Acids. Int. J. Mol. Sci. 2023, 24, 9743. [Google Scholar] [CrossRef]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Osipov, V.N.; Avdeev, D.V.; Pukhov, S.A.; Gromyko, A.V.; Aliev, G. New Spirocyclic Hydroxamic Acids as Effective Antiproliferative Agents. Anticancer Agents Med. Chem. 2021, 21, 597–610. [Google Scholar] [CrossRef]
- Neganova, M.; Aleksandrova, Y.; Suslov, E.; Mozhaitsev, E.; Munkuev, A.; Tsypyshev, D.; Chicheva, M.; Rogachev, A.; Sukocheva, O.; Volcho, K.; et al. Novel Multitarget Hydroxamic Acids with a Natural Origin CAP Group against Alzheimer’s Disease: Synthesis, Docking and Biological Evaluation. Pharmaceutics 2021, 13, 1893. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. The Hydroxamic Acids as Potential Anticancer and Neuroprotective Agents. Curr. Med. Chem. 2021, 28, 8139–8162. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar] [CrossRef]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): Trial results and interleukin-6 analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 2009, 115, 5541–5549. [Google Scholar]
- Quinn, D.I.; Tsao-Wei, D.D.; Twardowski, P.; Aparicio, A.M.; Frankel, P.; Chatta, G.; Wright, J.J.; Groshen, S.G.; Khoo, S.; Lenz, H.J.; et al. Phase II study of the histone deacetylase inhibitor vorinostat (Suberoylanilide Hydroxamic Acid; SAHA) in recurrent or metastatic transitional cell carcinoma of the urothelium—an NCI-CTEP sponsored: California Cancer Consortium trial, NCI 6879. Investig. New Drugs 2021, 39, 812–820. [Google Scholar] [CrossRef]
- DuBois, S.G.; Granger, M.M.; Groshen, S.; Tsao-Wei, D.; Ji, L.; Shamirian, A.; Czarnecki, S.; Goodarzian, F.; Berkovich, R.; Shimada, H.; et al. Randomized Phase II Trial of MIBG Versus MIBG, Vincristine, and Irinotecan Versus MIBG and Vorinostat for Patients With Relapsed or Refractory Neuroblastoma: A Report From NANT Consortium. J. Clin. Oncol. 2021, 39, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.E.S.; Kilburn, L.; Geyer, J.R.; Kocak, M.; Huang, J.; Smith, K.S.; Hadley, J.; Ermoian, R.; MacDonald, T.J.; Goldman, S.; et al. Vorinostat and isotretinoin with chemotherapy in young children with embryonal brain tumors: A report from the Pediatric Brain Tumor Consortium (PBTC-026). Neuro Oncol. 2022, 24, 1178–1190. [Google Scholar] [CrossRef]
- Arora, S.P.; Tenner, L.; Sarantopoulos, J.; Morris, J.; Liu, Q.; Mendez, J.A.; Curiel, T.; Michalek, J.; Mahalingam, D. Modulation of autophagy: A Phase II study of vorinostat plus hydroxychloroquine versus regorafenib in chemotherapy-refractory metastatic colorectal cancer (mCRC). Br. J. Cancer 2022, 127, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Hurez, V.; Nawrocki, S.T.; Goros, M.; Michalek, J.; Sarantopoulos, J.; Curiel, T.; Mahalingam, D. Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 2016, 7, 59087–59097. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Janku, F.; Piha-Paul, S.; Hess, K.; Broaddus, R.; Liu, L.; Shi, N.; Overman, M.; Kopetz, S.; Subbiah, V.; et al. Phase I studies of vorinostat with ixazomib or pazopanib imply a role of antiangiogenesis-based therapy for TP53 mutant malignancies. Sci. Rep. 2020, 10, 3080. [Google Scholar] [CrossRef] [PubMed]
- Teknos, T.N.; Grecula, J.; Agrawal, A.; Old, M.O.; Ozer, E.; Carrau, R.; Kang, S.; Rocco, J.; Blakaj, D.; Diavolitsis, V.; et al. A phase 1 trial of Vorinostat in combination with concurrent chemoradiation therapy in the treatment of advanced staged head and neck squamous cell carcinoma. Investig. New Drugs 2019, 37, 702–710. [Google Scholar] [CrossRef]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef]
- Chen, S.H.; Wu, H.M.; Ossola, B.; Schendzielorz, N.; Wilson, B.C.; Chu, C.H.; Chen, S.L.; Wang, Q.; Zhang, D.; Qian, L.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Br. J. Pharmacol. 2012, 165, 494–505. [Google Scholar] [CrossRef]
- Nuutinen, T.; Suuronen, T.; Kauppinen, A.; Salminen, A. Valproic acid stimulates clusterin expression in human astrocytes: Implications for Alzheimer’s disease. Neurosci. Lett. 2010, 475, 64–68. [Google Scholar] [CrossRef]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.E.; La, H.; Plise, E.; Chen, Y.H.; Ding, X.; Hanania, T.; Sabath, E.V.; Alexandrov, V.; Brunner, D.; Leahy, E.; et al. SAHA enhances synaptic function and plasticity in vitro but has limited brain availability in vivo and does not impact cognition. PLoS ONE 2013, 8, e69964. [Google Scholar] [CrossRef] [PubMed]
- Sarathlal, K.C.; Kakoty, V.; Krishna, K.V.; Dubey, S.K.; Chitkara, D.; Taliyan, R. Neuroprotective Efficacy of Co-Encapsulated Rosiglitazone and Vorinostat Nanoparticle on Streptozotocin Induced Mice Model of Alzheimer Disease. ACS Chem. Neurosci. 2021, 12, 1528–1541. [Google Scholar]
- Kakoty, V.; Sarathlal, K.C.; Dubey, S.K.; Yang, C.H.; Marathe, S.A.; Taliyan, R. Epigenetic regulation and autophagy modulation debilitates insulin resistance associated Alzheimer’s disease condition in rats. Metab. Brain Dis. 2022, 37, 927–944. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sanzhez-Arias, J.; Mederos, S.; Rabal, O.; Ugarte, A.; Franco, R.; Pascual-Lucas, M.; Segura, V.; Perea, G.; et al. Concomitant histone deacetylase and phosphodiesterase 5 inhibition synergistically prevents the disruption in synaptic plasticity and it reverses cognitive impairment in a mouse model of Alzheimer’s disease. Clin. Epigenetics 2015, 7, 108. [Google Scholar] [CrossRef]
- More, S.S.; Itsara, M.; Yang, X.; Geier, E.G.; Tadano, M.K.; Seo, Y.; Vanbrocklin, H.F.; Weiss, W.A.; Mueller, S.; Haas-Kogan, D.A.; et al. Vorinostat increases expression of functional norepinephrine transporter in neuroblastoma in vitro and in vivo model systems. Clin. Cancer Res. 2011, 17, 2339–2349. [Google Scholar] [CrossRef]
- DuBois, S.G.; Groshen, S.; Park, J.R.; Haas-Kogan, D.A.; Yang, X.; Geier, E.; Chen, E.; Giacomini, K.; Weiss, B.; Cohn, S.L.; et al. Phase I Study of Vorinostat as a Radiation Sensitizer with 131I-Metaiodobenzylguanidine (131I-MIBG) for Patients with Relapsed or Refractory Neuroblastoma. Clin. Cancer Res. 2015, 21, 2715–2721. [Google Scholar] [CrossRef]
- Sinha, S.; Cheng, K.; Schaffer, A.A.; Aldape, K.; Schiff, E.; Ruppin, E. In vitro and in vivo identification of clinically approved drugs that modify ACE2 expression. Mol. Syst. Biol. 2020, 16, e9628. [Google Scholar] [CrossRef]
- Pinto, N.; DuBois, S.G.; Marachelian, A.; Diede, S.J.; Taraseviciute, A.; Glade Bender, J.L.; Tsao-Wei, D.; Groshen, S.G.; Reid, J.M.; Haas-Kogan, D.A.; et al. Phase I study of vorinostat in combination with isotretinoin in patients with refractory/recurrent neuroblastoma: A new approaches to Neuroblastoma Therapy (NANT) trial. Pediatr. Blood Cancer 2018, 65, e27023. [Google Scholar] [CrossRef]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef]
- Fu, S.; Hou, M.M.; Naing, A.; Janku, F.; Hess, K.; Zinner, R.; Subbiah, V.; Hong, D.; Wheler, J.; Piha-Paul, S.; et al. Phase I study of pazopanib and vorinostat: A therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Ann. Oncol. 2015, 26, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, R.; Ma, Z. Autophagy and Energy Metabolism. Adv. Exp. Med. Biol. 2019, 1206, 329–357. [Google Scholar] [PubMed]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Sun, X.; Peng, Y.; Zhao, J.; Xie, Z.; Lei, X.; Tang, G. Discovery and development of tumor glycolysis rate-limiting enzyme inhibitors. Bioorg Chem. 2021, 112, 104891. [Google Scholar] [CrossRef]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Altered metabolism in cancer. BMC Biol. 2010, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Jiang, H.; Sun, X.; Xu, H.; Wei, X.; Wei, Y.; Xiao, G.; Song, Z.; Zhou, F. Mitochondrial dysfunction induces radioresistance in colorectal cancer by activating [Ca(2+)](m)-PDP1-PDH-histone acetylation retrograde signaling. Cell Death Dis. 2021, 12, 837. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, Q.; Chen, S.; Fang, B.; Yang, Q.; Chen, C.; Miele, L.; Sarkar, F.H.; Xia, J.; Wang, Z. Mitochondrial dysfunction promotes breast cancer cell migration and invasion through HIF1alpha accumulation via increased production of reactive oxygen species. PLoS ONE 2013, 8, e69485. [Google Scholar]
- Lee, H.C.; Huang, K.H.; Yeh, T.S.; Chi, C.W. Somatic alterations in mitochondrial DNA and mitochondrial dysfunction in gastric cancer progression. World J. Gastroenterol. 2014, 20, 3950–3959. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef]
- Almaguel, F.A.; Sanchez, T.W.; Ortiz-Hernandez, G.L.; Casiano, C.A. Alpha-Enolase: Emerging Tumor-Associated Antigen, Cancer Biomarker, and Oncotherapeutic Target. Front. Genet. 2020, 11, 614726. [Google Scholar] [CrossRef]
- Kohnken, R.; Kodigepalli, K.M.; Wu, L. Regulation of deoxynucleotide metabolism in cancer: Novel mechanisms and therapeutic implications. Mol. Cancer 2015, 14, 176. [Google Scholar] [CrossRef]
- Le, T.M.; Poddar, S.; Capri, J.R.; Abt, E.R.; Kim, W.; Wei, L.; Uong, N.T.; Cheng, C.M.; Braas, D.; Nikanjam, M.; et al. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat. Commun. 2017, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Efimova, E.V.; Takahashi, S.; Shamsi, N.A.; Wu, D.; Labay, E.; Ulanovskaya, O.A.; Weichselbaum, R.R.; Kozmin, S.A.; Kron, S.J. Linking Cancer Metabolism to DNA Repair and Accelerated Senescence. Mol. Cancer Res. 2016, 14, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Backos, D.S.; Franklin, C.C.; Reigan, P. The role of glutathione in brain tumor drug resistance. Biochem. Pharmacol. 2012, 83, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef]
- Deng, D.; Yan, N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Wu, C.H.; Hong, C.H.; Chang, K.L.; Lee, C.H. GLUT-1 Enhances Glycolysis, Oxidative Stress, and Fibroblast Proliferation in Keloid. Life 2021, 11, 505. [Google Scholar] [CrossRef]
- Reckzeh, E.S.; Waldmann, H. Small-Molecule Inhibition of Glucose Transporters GLUT-1-4. Chembiochem 2020, 21, 45–52. [Google Scholar] [CrossRef]
- Carvalho, K.C.; Cunha, I.W.; Rocha, R.M.; Ayala, F.R.; Cajaiba, M.M.; Begnami, M.D.; Vilela, R.S.; Paiva, G.R.; Andrade, R.G.; Soares, F.A. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics 2011, 66, 965–972. [Google Scholar] [CrossRef]
- Yin, C.; Gao, B.; Yang, J.; Wu, J. Glucose Transporter-1 (GLUT-1) Expression is Associated with Tumor Size and Poor Prognosis in Locally Advanced Gastric Cancer. Med. Sci. Monit. Basic. Res. 2020, 26, e920778. [Google Scholar] [CrossRef]
- Higashi, T.; Tamaki, N.; Torizuka, T.; Nakamoto, Y.; Sakahara, H.; Kimura, T.; Honda, T.; Inokuma, T.; Katsushima, S.; Ohshio, G.; et al. FDG uptake, GLUT-1 glucose transporter and cellularity in human pancreatic tumors. J. Nucl. Med. 1998, 39, 1727–1735. [Google Scholar] [PubMed]
- Gasinska, A.; Jaszczynski, J.; Rychlik, U.; Luczynska, E.; Pogodzinski, M.; Palaczynski, M. Prognostic Significance of Serum PSA Level and Telomerase, VEGF and GLUT-1 Protein Expression for the Biochemical Recurrence in Prostate Cancer Patients after Radical Prostatectomy. Pathol. Oncol. Res. 2020, 26, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Carreno, D.; Corro, N.; Torres-Estay, V.; Veliz, L.P.; Jaimovich, R.; Cisternas, P.; San Francisco, I.F.; Sotomayor, P.C.; Tanasova, M.; Inestrosa, N.C.; et al. Fructose and prostate cancer: Toward an integrated view of cancer cell metabolism. Prostate Cancer Prostatic Dis. 2019, 22, 49–58. [Google Scholar] [CrossRef]
- Mogi, A.; Koga, K.; Aoki, M.; Hamasaki, M.; Uesugi, N.; Iwasaki, A.; Shirakusa, T.; Tamura, K.; Nabeshima, K. Expression and role of GLUT-1, MCT-1, and MCT-4 in malignant pleural mesothelioma. Virchows Arch. 2013, 462, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Khabaz, M.N.; Qureshi, I.A.; Al-Maghrabi, J.A. GLUT 1 expression is a supportive mean in predicting prognosis and survival estimates of endometrial carcinoma. Ginekol. Pol. 2019, 90, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Canpolat, T.; Ersoz, C.; Uguz, A.; Vardar, M.A.; Altintas, A. GLUT-1 Expression in Proliferative Endometrium, Endometrial Hyperplasia, Endometrial Adenocarcinoma and the Relationship Between GLUT-1 Expression and Prognostic Parameters in Endometrial Adenocarcinoma. Turk. Patoloji Derg. 2016, 32, 141–147. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kubo, T.; Shimose, S.; Fujimori, J.; Furuta, T.; Arihiro, K.; Ochi, M. Does expression of glucose transporter protein-1 relate to prognosis and angiogenesis in osteosarcoma? Clin. Orthop. Relat. Res. 2015, 473, 305–310. [Google Scholar] [CrossRef] [PubMed]
- van de Nes, J.A.; Griewank, K.G.; Schmid, K.W.; Grabellus, F. Immunocytochemical analysis of glucose transporter protein-1 (GLUT-1) in typical, brain invasive, atypical and anaplastic meningioma. Neuropathology 2015, 35, 24–36. [Google Scholar] [CrossRef]
- Luo, X.M.; Zhou, S.H.; Fan, J. Glucose transporter-1 as a new therapeutic target in laryngeal carcinoma. J. Int. Med. Res. 2010, 38, 1885–1892. [Google Scholar] [CrossRef]
- Xi, J.; Wang, Y.; Liu, H. GLUT-1 participates in the promotion of LncRNA CASC9 in proliferation and metastasis of laryngeal carcinoma cells. Gene 2020, 726, 144194. [Google Scholar] [CrossRef]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zou, J.; Deng, T.; Liu, J. Clinicopathological and prognostic significance of GLUT1 in breast cancer: A meta-analysis. Medicine 2018, 97, e12961. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Chun, Y.K.; Hur, M.H.; Lee, H.K.; Kim, Y.J.; Hong, S.R.; Lee, J.H.; Lee, S.G.; Park, Y.K. Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Jpn. J. Cancer Res. 2002, 93, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Hussein, Y.R.; Bandyopadhyay, S.; Semaan, A.; Ahmed, Q.; Albashiti, B.; Jazaerly, T.; Nahleh, Z.; Ali-Fehmi, R. Glut-1 Expression Correlates with Basal-like Breast Cancer. Transl. Oncol. 2011, 4, 321–327. [Google Scholar] [CrossRef]
- Fukushi, A.; Kim, H.D.; Chang, Y.C.; Kim, C.H. Revisited Metabolic Control and Reprogramming Cancers by Means of the Warburg Effect in Tumor Cells. Int. J. Mol. Sci. 2022, 23, 10037. [Google Scholar] [CrossRef]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef]
- Garcia, S.N.; Guedes, R.C.; Marques, M.M. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr. Med. Chem. 2019, 26, 7285–7322. [Google Scholar] [CrossRef]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhang, M.; Cong, Q.; Zhang, M.X.; Zhang, M.Y.; Lu, Y.Y.; Xu, C.J. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef]
- Tian, X.; Liu, D.; Zuo, X.; Sun, X.; Wu, M.; Li, X.; Teng, Y. Hexokinase 2 promoted cell motility and proliferation by activating Akt1/p-Akt1 in human ovarian cancer cells. J. Ovarian Res. 2022, 15, 92. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yan, X.; Chen, J.; Zhan, Q.; Hua, Y.; Xu, S.; Li, Z.; Wang, Z.; Dong, Y.; Zuo, D.; et al. Hexokinase 2 discerns a novel circulating tumor cell population associated with poor prognosis in lung cancer patients. Proc. Natl. Acad. Sci. USA 2021, 118, e2012228118. [Google Scholar] [CrossRef]
- Deng, Y.; Lu, J. Targeting hexokinase 2 in castration-resistant prostate cancer. Mol. Cell. Oncol. 2015, 2, e974465. [Google Scholar] [CrossRef] [PubMed]
- Rho, M.; Kim, J.; Jee, C.D.; Lee, Y.M.; Lee, H.E.; Kim, M.A.; Lee, H.S.; Kim, W.H. Expression of type 2 hexokinase and mitochondria-related genes in gastric carcinoma tissues and cell lines. Anticancer Res. 2007, 27, 251–258. [Google Scholar]
- Sato-Tadano, A.; Suzuki, T.; Amari, M.; Takagi, K.; Miki, Y.; Tamaki, K.; Watanabe, M.; Ishida, T.; Sasano, H.; Ohuchi, N. Hexokinase II in breast carcinoma: A potent prognostic factor associated with hypoxia-inducible factor-1alpha and Ki-67. Cancer Sci. 2013, 104, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, H.; Yu, W.; Qiao, F.; Su, X.; Xu, H. Downregulation of hexokinase 2 improves radiosensitivity of breast cancer. Transl. Cancer Res. 2019, 8, 290–297. [Google Scholar] [CrossRef]
- Shangguan, X.; He, J.; Ma, Z.; Zhang, W.; Ji, Y.; Shen, K.; Yue, Z.; Li, W.; Xin, Z.; Zheng, Q.; et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat. Commun. 2021, 12, 1812. [Google Scholar] [CrossRef]
- Wang, J.; Shao, F.; Yang, Y.; Wang, W.; Yang, X.; Li, R.; Cheng, H.; Sun, S.; Feng, X.; Gao, Y.; et al. A non-metabolic function of hexokinase 2 in small cell lung cancer: Promotes cancer cell stemness by increasing USP11-mediated CD133 stability. Cancer Commun. 2022, 42, 1008–1027. [Google Scholar] [CrossRef]
- Thomas, G.E.; Egan, G.; Garcia-Prat, L.; Botham, A.; Voisin, V.; Patel, P.S.; Hoff, F.W.; Chin, J.; Nachmias, B.; Kaufmann, K.B.; et al. The metabolic enzyme hexokinase 2 localizes to the nucleus in AML and normal haematopoietic stem and progenitor cells to maintain stemness. Nat. Cell Biol. 2022, 24, 872–884. [Google Scholar]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar]
- Yalcin, A.; Solakoglu, T.H.; Ozcan, S.C.; Guzel, S.; Peker, S.; Celikler, S.; Balaban, B.D.; Sevinc, E.; Gurpinar, Y.; Chesney, J.A. 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor beta1-enhanced invasion of Panc1 cells in vitro. Biochem. Biophys. Res. Commun. 2017, 484, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Hong, L.L.; Ling, Z.N.; Zhong, Y.; Hu, X.Y.; Li, P.; Ling, Z.Q. A Potential Oncogenic Role for PFKFB3 Overexpression in Gastric Cancer Progression. Clin. Transl. Gastroenterol. 2021, 12, e00377. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Jin, W.J.; Kwak, J.H.; Kim, H.J.; Yun, M.J.; Kim, J.W.; Park, S.W.; Kim, K.S. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem. J. 2011, 433, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar]
- Alvarez, R.; Mandal, D.; Chittiboina, P. Canonical and Non-Canonical Roles of PFKFB3 in Brain Tumors. Cells 2021, 10, 2913. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar]
- Xia, Y.; Wang, X.; Liu, Y.; Shapiro, E.; Lepor, H.; Tang, M.S.; Sun, T.T.; Wu, X.R. PKM2 Is Essential for Bladder Cancer Growth and Maintenance. Cancer Res. 2022, 82, 571–585. [Google Scholar] [CrossRef]
- Yu, S.; Zang, W.; Qiu, Y.; Liao, L.; Zheng, X. Deubiquitinase OTUB2 exacerbates the progression of colorectal cancer by promoting PKM2 activity and glycolysis. Oncogene 2022, 41, 46–56. [Google Scholar]
- Zhou, S.; Li, D.; Xiao, D.; Wu, T.; Hu, X.; Zhang, Y.; Deng, J.; Long, J.; Xu, S.; Wu, J.; et al. Inhibition of PKM2 Enhances Sensitivity of Olaparib to Ovarian Cancer Cells and Induces DNA Damage. Int. J. Biol. Sci. 2022, 18, 1555–1568. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, L.M.; Xie, J.Y.; Han, H.; Zhu, B.F.; Wang, L.J.; Wang, W.J. High Expression of PKM2 Was Associated with the Poor Prognosis of Acute Leukemia. Cancer Manag. Res. 2021, 13, 7851–7858. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, L.; Yang, Z.; Tang, Y.; Tao, Y.; Zhan, Q.; Lei, L.; Jing, Y.; Jiang, X.; Jin, H.; et al. Glycolytic Enzyme PKM2 Mediates Autophagic Activation to Promote Cell Survival in NPM1-Mutated Leukemia. Int. J. Biol. Sci. 2019, 15, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Ardanaz, C.G.; Ramirez, M.J.; Solas, M. Brain Metabolic Alterations in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 3785. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Lan, M.J.; Brown, P.J.; Sublette, E.M.; Burke, A.; Mann, J.J. Brain cytochrome-c-oxidase as a marker of mitochondrial function: A pilot study in major depression using NIRS. Depress. Anxiety 2019, 36, 766–779. [Google Scholar] [CrossRef]
- Bergman, O.; Karry, R.; Milhem, J.; Ben-Shachar, D. NDUFV2 pseudogene (NDUFV2P1) contributes to mitochondrial complex I deficits in schizophrenia. Mol. Psychiatry 2020, 25, 805–820. [Google Scholar] [CrossRef]
- Rice, M.W.; Smith, K.L.; Roberts, R.C.; Perez-Costas, E.; Melendez-Ferro, M. Assessment of cytochrome C oxidase dysfunction in the substantia nigra/ventral tegmental area in schizophrenia. PLoS ONE 2014, 9, e100054. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Dong, Y.; Brewer, G.J. Global Metabolic Shifts in Age and Alzheimer’s Disease Mouse Brains Pivot at NAD+/NADH Redox Sites. J. Alzheimers Dis. 2019, 71, 119–140. [Google Scholar] [CrossRef]
- Ohta, S.; Ohsawa, I. Dysfunction of mitochondria and oxidative stress in the pathogenesis of Alzheimer’s disease: On defects in the cytochrome c oxidase complex and aldehyde detoxification. J. Alzheimers Dis. 2006, 9, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Ibrahim, Z.; Proitsi, P.; Lourdusamy, A.; Newhouse, S.; Sattlecker, M.; Furney, S.; Saleem, M.; Soininen, H.; Kloszewska, I.; et al. Mitochondrial dysfunction and immune activation are detectable in early Alzheimer’s disease blood. J. Alzheimers Dis. 2012, 30, 685–710. [Google Scholar] [CrossRef] [PubMed]
- Jaroudi, W.; Garami, J.; Garrido, S.; Hornberger, M.; Keri, S.; Moustafa, A.A. Factors underlying cognitive decline in old age and Alzheimer’s disease: The role of the hippocampus. Rev. Neurosci. 2017, 28, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, L.M.; Yokomizo, J.E.; Bottino, C.M.; Fuentes, D. Frontal Lobe Degeneration in Adults with Down Syndrome and Alzheimer’s Disease: A Review. Dement. Geriatr. Cogn. Disord. 2016, 41, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Berron, D.; van Westen, D.; Ossenkoppele, R.; Strandberg, O.; Hansson, O. Medial temporal lobe connectivity and its associations with cognition in early Alzheimer’s disease. Brain 2020, 143, 1233–1248. [Google Scholar] [CrossRef]
- Ahullo-Fuster, M.A.; Ortiz, T.; Varela-Donoso, E.; Nacher, J.; Sanchez-Sanchez, M.L. The Parietal Lobe in Alzheimer’s Disease and Blindness. J. Alzheimers Dis. 2022, 89, 1193–1202. [Google Scholar] [CrossRef]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta 2016, 1862, 1871–1882. [Google Scholar] [CrossRef]
- Francis, B.M.; Yang, J.; Song, B.J.; Gupta, S.; Maj, M.; Bazinet, R.P.; Robinson, B.; Mount, H.T. Reduced levels of mitochondrial complex I subunit NDUFB8 and linked complex I + III oxidoreductase activity in the TgCRND8 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 347–355. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef]
- Emmerzaal, T.L.; Rodenburg, R.J.; Tanila, H.; Verweij, V.; Kiliaan, A.J.; Kozicz, T. Age-Dependent Decrease of Mitochondrial Complex II Activity in a Familial Mouse Model for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 66, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Van Eldik, L.J. Comprehensive behavioral characterization of an APP/PS-1 double knock-in mouse model of Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP alternations in an Alzheimer’s disease transgenic mouse model. J. Alzheimers Dis. 2015, 44, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Deak, F.; Freeman, W.M.; Ungvari, Z.; Csiszar, A.; Sonntag, W.E. Recent Developments in Understanding Brain Aging: Implications for Alzheimer’s Disease and Vascular Cognitive Impairment. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 13–20. [Google Scholar] [CrossRef]
- Kim, D.I.; Lee, K.H.; Oh, J.Y.; Kim, J.S.; Han, H.J. Relationship Between beta-Amyloid and Mitochondrial Dynamics. Cell. Mol. Neurobiol. 2017, 37, 955–968. [Google Scholar] [CrossRef]
- Shi, C.; Zhu, X.; Wang, J.; Long, D. Intromitochondrial IkappaB/NF-kappaB signaling pathway is involved in amyloid beta peptide-induced mitochondrial dysfunction. J. Bioenerg. Biomembr. 2014, 46, 371–376. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Fujii, S. Extracellular ATP modulates synaptic plasticity induced by activation of metabotropic glutamate receptors in the hippocampus. Biomed. Res. 2015, 36, 1–9. [Google Scholar] [CrossRef]
- Recuero, M.; Munoz, T.; Aldudo, J.; Subias, M.; Bullido, M.J.; Valdivieso, F. A free radical-generating system regulates APP metabolism/processing. FEBS Lett. 2010, 584, 4611–4618. [Google Scholar] [CrossRef]
- Leuner, K.; Schutt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, J.; Roy Chowdhury, S.; Snow, W.M.; Perez, C.; Cadonic, C.; Fernyhough, P.; Albensi, B.C. Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice. Cells 2020, 9, 1541. [Google Scholar] [CrossRef] [PubMed]
- Joh, Y.; Choi, W.S. Mitochondrial Complex I Inhibition Accelerates Amyloid Toxicity. Dev. Reprod. 2017, 21, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Drose, S.; Brandt, U.; Muller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef]
- Gotz, J.; Chen, F.; Barmettler, R.; Nitsch, R.M. Tau filament formation in transgenic mice expressing P301L tau. J. Biol. Chem. 2001, 276, 529–534. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Yamada, E.S.; Respondek, G.; Mussner, S.; de Andrade, A.; Hollerhage, M.; Depienne, C.; Rastetter, A.; Tarze, A.; Friguet, B.; Salama, M.; et al. Annonacin, a natural lipophilic mitochondrial complex I inhibitor, increases phosphorylation of tau in the brain of FTDP-17 transgenic mice. Exp. Neurol. 2014, 253, 113–125. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Peng, Y.; Xing, S.N.; Tang, H.Y.; Wang, C.D.; Yi, F.P.; Liu, G.L.; Wu, X.M. Influence of glucose transporter 1 activity inhibition on neuroblastoma in vitro. Gene 2019, 689, 11–17. [Google Scholar] [CrossRef]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.S.; Li, Z.K.; Liu, J.; Deng, Y.T.; Jiang, Y. WZB117 enhanced the anti-tumor effect of apatinib against melanoma via blocking STAT3/PKM2 axis. Front. Pharmacol. 2022, 13, 976117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Ming, J.; Zhou, Y.; Fan, L. Inhibition of Glut1 by WZB117 sensitizes radioresistant breast cancer cells to irradiation. Cancer Chemother. Pharmacol. 2016, 77, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, A.; Zhang, S.; Kim, J.; Xia, J.; Zhang, F.; Wang, D.; Wang, Q.; Wang, J. Paclitaxel-loaded ginsenoside Rg3 liposomes for drug-resistant cancer therapy by dual targeting of the tumor microenvironment and cancer cells. J. Adv. Res. 2023, 49, 159–173. [Google Scholar] [CrossRef]
- Klippel, S.; Jakubikova, J.; Delmore, J.; Ooi, M.; McMillin, D.; Kastritis, E.; Laubach, J.; Richardson, P.G.; Anderson, K.C.; Mitsiades, C.S. Methyljasmonate displays in vitro and in vivo activity against multiple myeloma cells. Br. J. Haematol. 2012, 159, 340–351. [Google Scholar] [CrossRef]
- Li, J.; Chen, K.; Wang, F.; Dai, W.; Li, S.; Feng, J.; Wu, L.; Liu, T.; Xu, S.; Xia, Y.; et al. Methyl jasmonate leads to necrosis and apoptosis in hepatocellular carcinoma cells via inhibition of glycolysis and represses tumor growth in mice. Oncotarget 2017, 8, 45965–45980. [Google Scholar] [CrossRef]
- Jiang, Y.X.; Siu, M.K.Y.; Wang, J.J.; Leung, T.H.Y.; Chan, D.W.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. PFKFB3 Regulates Chemoresistance, Metastasis and Stemness via IAP Proteins and the NF-kappaB Signaling Pathway in Ovarian Cancer. Front. Oncol. 2022, 12, 748403. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, F.; Nan, Y.; Qu, L.; Na, W.; Jia, C.; Chen, X. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int. J. Biol. Sci. 2018, 14, 1883–1891. [Google Scholar] [CrossRef]
- Dai, Y.; Liu, Y.; Li, J.; Jin, M.; Yang, H.; Huang, G. Shikonin inhibited glycolysis and sensitized cisplatin treatment in non-small cell lung cancer cells via the exosomal pyruvate kinase M2 pathway. Bioengineered 2022, 13, 13906–13918. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, Q.; Zheng, S.; Liu, T.; Yang, L.; Han, X.; Lu, X. Shikonin Inhibits Tumor Growth of ESCC by suppressing PKM2 mediated Aerobic Glycolysis and STAT3 Phosphorylation. J. Cancer 2021, 12, 4830–4840. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Yakisich, J.S.; Azad, N.; Kaushik, V.; Iyer, A.K.V. The Biguanides Metformin and Buformin in Combination with 2-Deoxy-glucose or WZB-117 Inhibit the Viability of Highly Resistant Human Lung Cancer Cells. Stem Cells Int. 2019, 2019, 6254269. [Google Scholar] [CrossRef]
- Li, Y.L.; Weng, H.C.; Hsu, J.L.; Lin, S.W.; Guh, J.H.; Hsu, L.C. The Combination of MK-2206 and WZB117 Exerts a Synergistic Cytotoxic Effect Against Breast Cancer Cells. Front. Pharmacol. 2019, 10, 1311. [Google Scholar] [CrossRef]
- Liu, W.; Fang, Y.; Wang, X.T.; Liu, J.; Dan, X.; Sun, L.L. Overcoming 5-Fu resistance of colon cells through inhibition of Glut1 by the specific inhibitor WZB117. Asian Pac. J. Cancer Prev. 2014, 15, 7037–7041. [Google Scholar] [CrossRef]
- Chen, Q.; Meng, Y.Q.; Xu, X.F.; Gu, J. Blockade of GLUT1 by WZB117 resensitizes breast cancer cells to adriamycin. Anticancer Drugs 2017, 28, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Ancey, P.B.; Contat, C.; Boivin, G.; Sabatino, S.; Pascual, J.; Zangger, N.; Perentes, J.Y.; Peters, S.; Abel, E.D.; Kirsch, D.G.; et al. GLUT1 Expression in Tumor-Associated Neutrophils Promotes Lung Cancer Growth and Resistance to Radiotherapy. Cancer Res. 2021, 81, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhao, S.; Duan, Y.; Ma, Y.; Wang, Y.; Ji, H.; Zhang, Q. GLUT1 participates in tamoxifen resistance in breast cancer cells through autophagy regulation. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Shima, T.; Taniguchi, K.; Tokumaru, Y.; Inomata, Y.; Arima, J.; Lee, S.W.; Takabe, K.; Yoshida, K.; Uchiyama, K. Glucose transporter-1 inhibition overcomes imatinib resistance in gastrointestinal stromal tumor cells. Oncol. Rep. 2022, 47, 7. [Google Scholar] [CrossRef]
- Zeng, Z.; Nian, Q.; Chen, N.; Zhao, M.; Zheng, Q.; Zhang, G.; Zhao, Z.; Chen, Y.; Wang, J.; Zeng, J.; et al. Ginsenoside Rg3 inhibits angiogenesis in gastric precancerous lesions through downregulation of Glut1 and Glut4. Biomed. Pharmacother. 2022, 145, 112086. [Google Scholar] [CrossRef]
- Chen, C.; Xia, J.; Ren, H.; Wang, A.; Zhu, Y.; Zhang, R.; Gan, Z.; Wang, J. Effect of the structure of ginsenosides on the in vivo fate of their liposomes. Asian J. Pharm. Sci. 2022, 17, 219–229. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Y.; He, C.; Gao, G.; Li, J.; Qiu, L.; Wang, X.; Gao, Y.; Qi, Y.; Sun, K.; et al. Identification of a novel GLUT1 inhibitor with in vitro and in vivo anti-tumor activity. Int. J. Biol. Macromol. 2022, 216, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta 2016, 1866, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Shutkov, I.A.; Okulova, Y.N.; Mazur, D.M.; Melnichuk, N.A.; Babkov, D.A.; Sokolova, E.V.; Spasov, A.A.; Milaeva, E.R.; Nazarov, A.A. New Organometallic Ru(II) Compounds with Lonidamine Motif as Antitumor Agents. Pharmaceutics 2023, 15, 1366. [Google Scholar] [CrossRef]
- Muhammad, N.; Tan, C.P.; Nawaz, U.; Wang, J.; Wang, F.X.; Nasreen, S.; Ji, L.N.; Mao, Z.W. Multiaction Platinum(IV) Prodrug Containing Thymidylate Synthase Inhibitor and Metabolic Modifier against Triple-Negative Breast Cancer. Inorg. Chem. 2020, 59, 12632–12642. [Google Scholar] [CrossRef]
- Goldin, N.; Arzoine, L.; Heyfets, A.; Israelson, A.; Zaslavsky, Z.; Bravman, T.; Bronner, V.; Notcovich, A.; Shoshan-Barmatz, V.; Flescher, E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 2008, 27, 4636–4643. [Google Scholar] [CrossRef] [PubMed]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef]
- Boyd, S.; Brookfield, J.L.; Critchlow, S.E.; Cumming, I.A.; Curtis, N.J.; Debreczeni, J.; Degorce, S.L.; Donald, C.; Evans, N.J.; Groombridge, S.; et al. Structure-Based Design of Potent and Selective Inhibitors of the Metabolic Kinase PFKFB3. J. Med. Chem. 2015, 58, 3611–3625. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P.; Van Wielendaele, P.; Lambeir, A.M.; Abdali, A.; Bellosta, S.; Monaco, G.; Bultynck, G.; Martinet, W.; De Meyer, G.R.Y. Small molecule 3PO inhibits glycolysis but does not bind to 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3). FEBS Lett. 2020, 594, 3067–3075. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Van Wielendaele, P.; Delibegovic, M.; Martinet, W.; De Meyer, G.R.Y. The PFKFB3 Inhibitor AZ67 Inhibits Angiogenesis Independently of Glycolysis Inhibition. Int. J. Mol. Sci. 2021, 22, 5970. [Google Scholar] [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Mahnken, J.D.; Welch, P.; Bothwell, R.; Koppel, S.; Jackson, R.L.; Burns, J.M.; Swerdlow, R.H. A Mitochondrial Biomarker-Based Study of S-Equol in Alzheimer’s Disease Subjects: Results of a Single-Arm, Pilot Trial. J. Alzheimers Dis. 2017, 59, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Lin, S.H.; Hidayah, K.; Lin, C.I. Equol Pretreatment Protection of SH-SY5Y Cells against Abeta (25-35)-Induced Cytotoxicity and Cell-Cycle Reentry via Sustaining Estrogen Receptor Alpha Expression. Nutrients 2019, 11, 2356. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Q.; Li, W.S.; Liu, Z.; Zhang, H.L.; Ba, Y.G.; Zhang, R.X. Metformin therapy and cognitive dysfunction in patients with type 2 diabetes: A meta-analysis and systematic review. Medicine 2020, 99, e19378. [Google Scholar] [CrossRef] [PubMed]






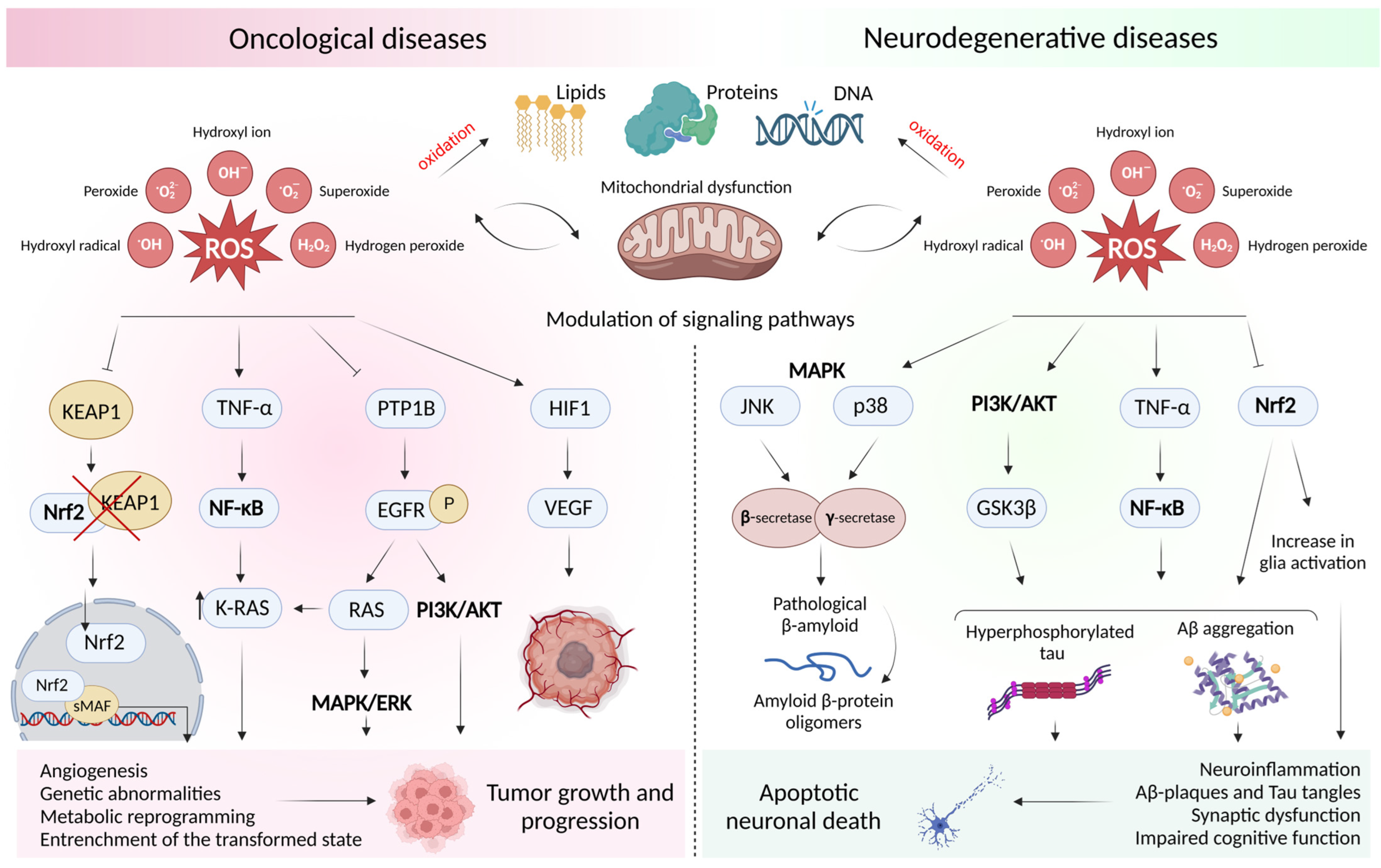{kind=link}
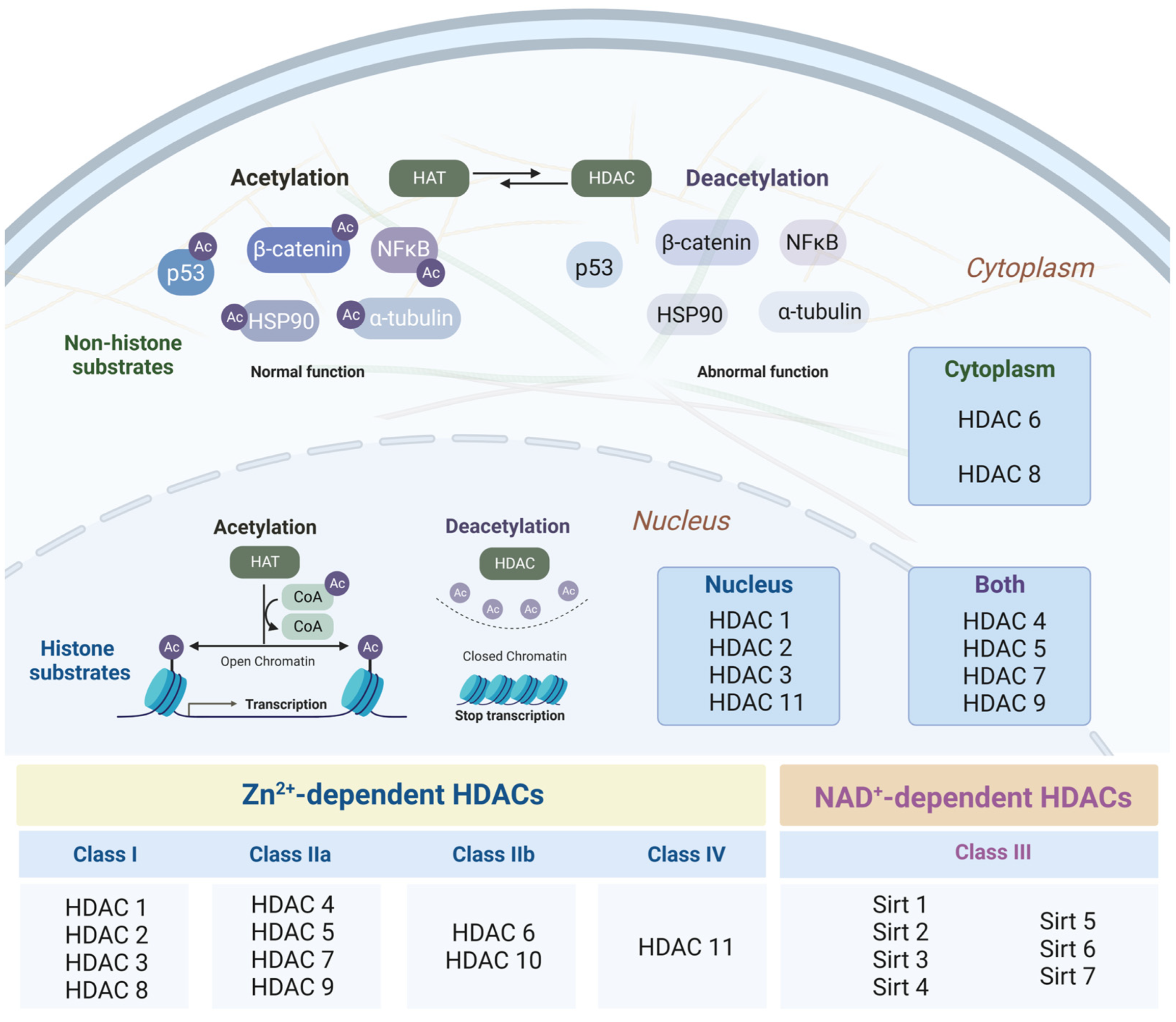{kind=link}
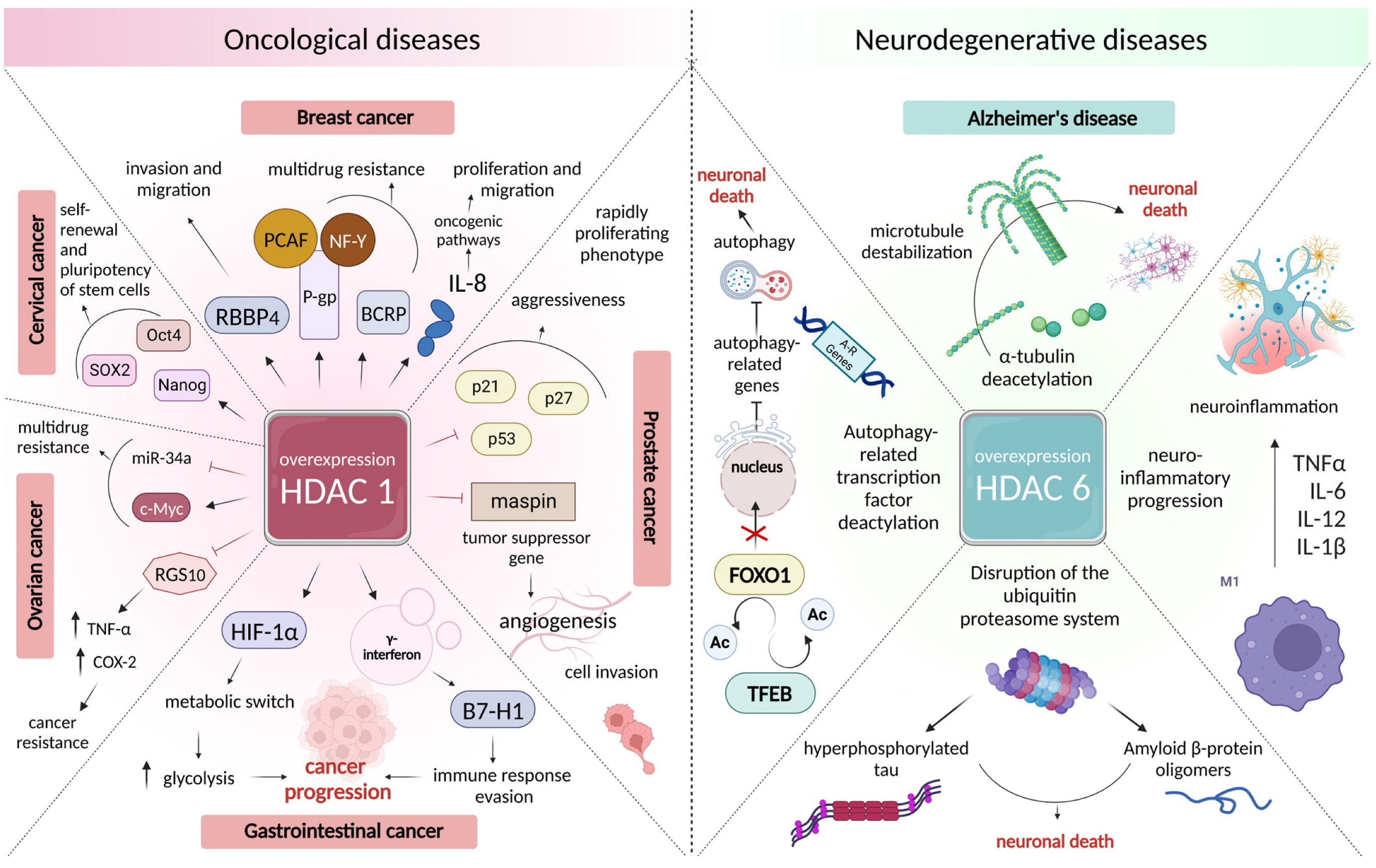{kind=link}
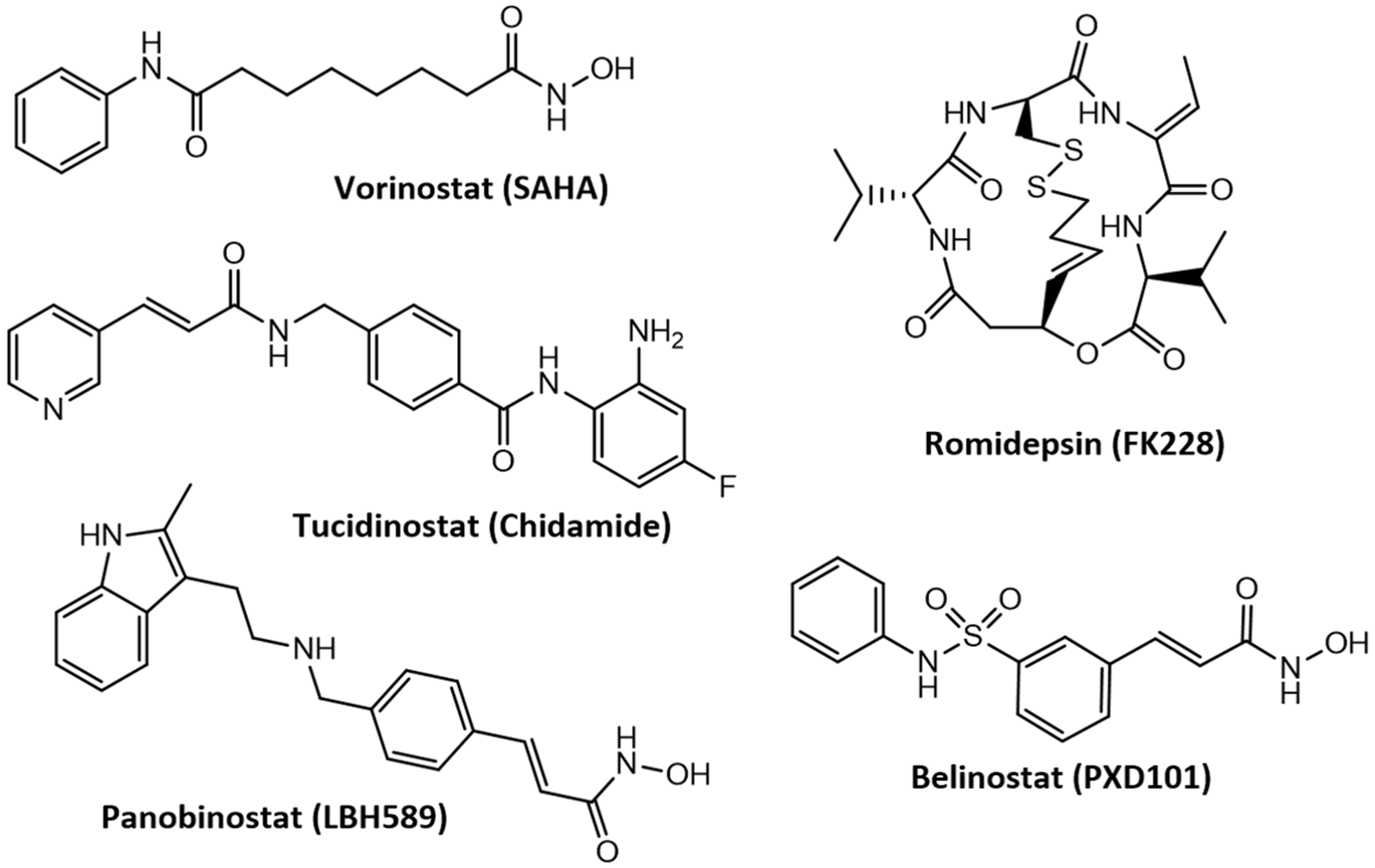{kind=link}
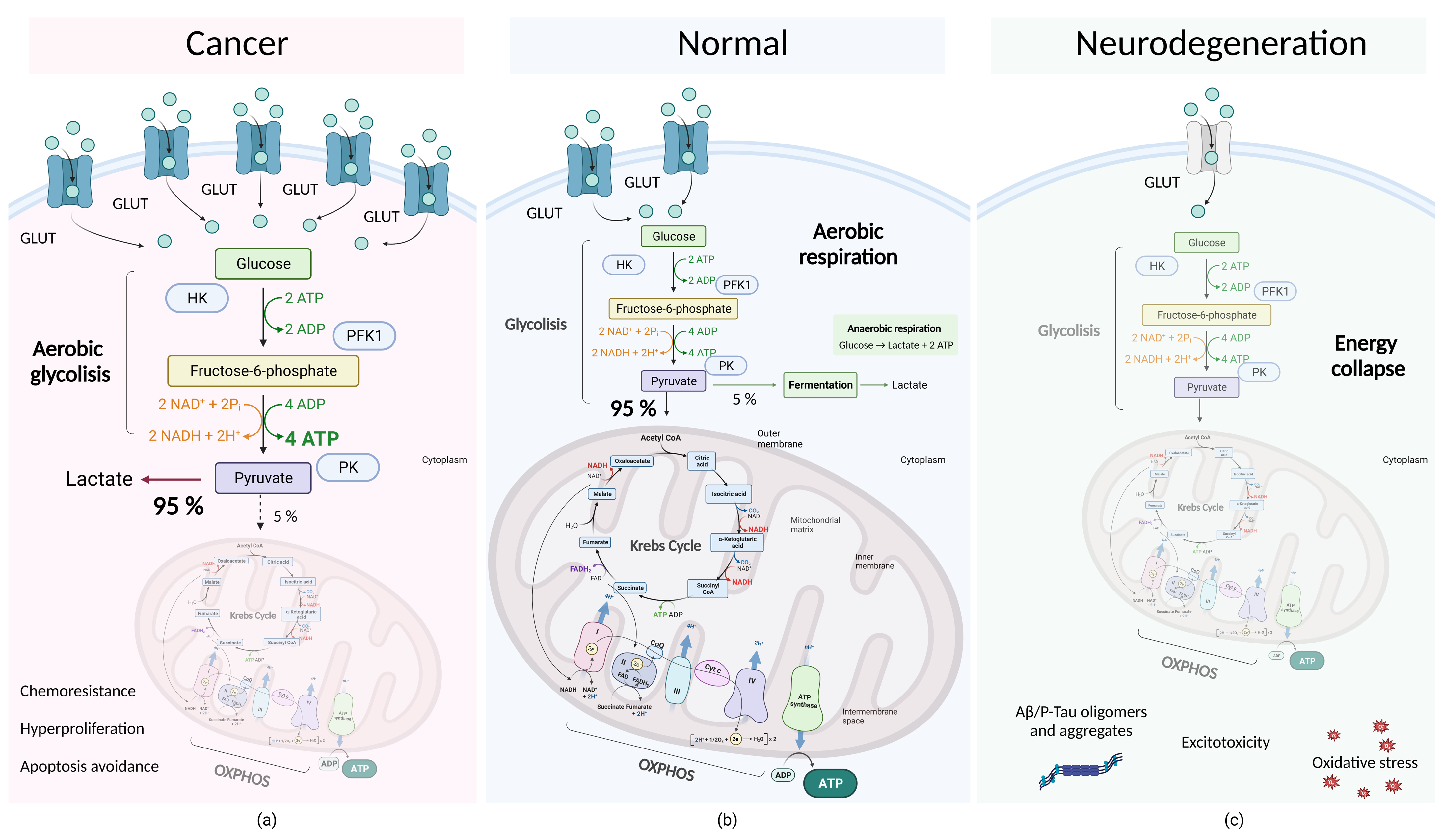{kind=link}
| Therapeutic Agent | Molecular Mechanisms of Action | Prognostic Significance | Disease | |
|---|---|---|---|---|
| Quercetin | Inhibition of the PI3K/Akt signaling pathway | Restoration of sensitivity of transformed cells to docetaxel action [197] | PC | Oncological diseases |
| Reduction in the expression of oxidative stress markers and restoration of the leukocyte count | Reduction in inflammation, reduction in tumor size [206] | CC | ||
| Regulation of the PI3K/Akt/mTOR signaling pathway and inhibition of Nrf2 expression | Reversing drug resistance to cisplatin [202] | EOC | ||
| Quercetin + Vitamin C | Decreased Nrf2 expression and activity of glutathione enzymes | Stimulation of tumor cell death [203,204] | BC | |
| Artesunate | ROS-dependent cell cycle arrest due to changes in cyclin D3, E2F-1, and p21 expression | Antiproliferative effect [223] | EOC | |
| Induction of ROS-dependent apoptosis by reducing the VDAC and increasing the cleavage of caspase 3 | NSCLC | |||
| Cell cycle arrest due to an increase in p16, p21, p-IRE1a, and LC3B and a decrease in Ki67 and cyclin D1 | CC | |||
| Inhibition of NF-kB, ubiquitin-mediated degradation of castration-resistant prostate cancer cells | Reversal of cell resistance to androgen receptor antagonists [226] | PC | ||
| Costunolid | Triggering of apoptosis as a result of ROS hyperproduction and φm violation | Transformed cell death [232,233,234] | BCa | |
| ESCC | ||||
| BC | ||||
| Parthenolide | PI3K/Akt pathway blocking, ROS hyperproduction | Antiproliferative action [235,236,237] | CCa | |
| GSH depletion, NF-kB shutdown | BC | |||
| Increase in ROS production, decreased GSH activity | LN | |||
| Quercetin | Inhibition of LPO, increase in GSH expression | Danio rerio cognitive dysfunction restoration in PTZ-induced neurodegeneration [208] | AD | Neurodegenerative diseases |
| Reducing the COX-2 level | Improvement of memory parameters in mice with LPS-induced neurodegeneration [209] | |||
| Activation of Nrf2, decrease in the MDA level, increased expression of SOD, CAT, and GSH | Preventing neuronal damage, leveling cognitive impairment in rats with Alzheimer’s disease model stimulated by toxic Aβ1-42 forms [210] | |||
| Artemisinin | Decrease in the MDA level, increased SOD and GSH expression | SH-SY5Y cell death inhibition in MPP+-induced neurotoxicity [227], reduction in damage to dopaminergic neurons with MPTP-induced toxicity [228] | PD | |
| Blocking of ROS production as a result of Akt signaling pathway activation | Increased HT-22 survival with glutamate-induced neurotoxicity [229] | AD | ||
| Activating the ERK/CREB pathway | Inhibition of SH-SY5Y cell death in Aβ1-42 toxicity, 3xTg transgenic mice cognitive function improvement [231] | |||
| Artemeter | Activation of the Nrf2 signaling pathway, decrease in the level of inflammatory mediators, Aβ levels, and activity of β-secretase 1 | Inhibition of neuroinflammation in LPS-stimulated BV2 microglia [230] | ||
| Costunolid | Decrease in the intracellular ROS level and caspase 3 expression | Preventing damage to the PC12 cell line by H2O2 [238] | ||
| Parthenolide | Blocking of the AKT/MAPK/NF-kB signaling pathway, neuroinflammation reduction | Improvement of memory indicators in the APP/PS1 transgenic mice line [239] | ||
| Inhibition of MAO B activity | Cell death decrease in MPP+-induced toxicity [240] | PD | ||
| Therapeutic Agent in Combination | Molecular Mechanisms of Action | Prognostic Significance | Disease | |
|---|---|---|---|---|
| 131I-methaiodbenzylguanidine | Increase in human NET protein expression | Increased radioligand absorption and frequency of true response [412,427,428] | NB | Oncological diseases |
| Isotretinoin | Modulation of APF2 levels | Five-year progression-free and overall survival improvement [413,429,430] | MB | |
| Hydroxychloroquine | Autophagy inhibition due to increased cathepsin D and p62 levels | Strengthening of antitumor immunity [414,415,431] | CC | |
| Pazopanib | Degradation of mutant p53, increased VEGF expression, decreased HIF-1α levels | Increase in the average duration of overall survival and life without progression of the disease [416,432] | EOC | |
| BC | ||||
| CC | ||||
| GC | ||||
| HNSCC | ||||
| NSCLC | ||||
| Chemoradiotherapy | Increased apoptosis rate due to increased Bax and p21 expression | Improved overall survival [417,418,419] | HNSCC | |
| PC | ||||
| GB | ||||
| Rosiglitazone | Increased expression of neurotrophic factor genes | Reduced biochemical, cellular, and behavioral disorders in the STZ mouse model of Alzheimer’s disease [424] | AD | Neurodegenerative diseases |
| Rapamycin | Decreased APP due to increased expression of Beclin 1, neurotrophic factors GDNF, BDNF, NGF, and neuronal markers MAP2 and LAMP2 | Relief of cognitive dysfunction in rats with an insulin resistance and intracerebroventricular injection Aβ1-42 [425] | ||
| Tadalafil | Restoration of long-term potentiation, Aβ, and tau pathology relief through the Akt/GSK3β pathway | Restoration of cognitive functions in APP/PS1 transgenic mice [426] | ||
| Therapeutic Agent | Key Target | Molecular Mechanisms of Action | Prognostic Significance | Disease | |
|---|---|---|---|---|---|
| WZB117 | GLUT1 | Cell cycle arrest, necrotic cell death | Tumor size reduction in a xenograft mouse model [540] | NSCLC | Oncological diseases |
| Decreased cell viability [541] | NB | ||||
| Self-renewal of stem cell obstruction | Tumor initiation inhibition in a xenograft mouse model [542] | PC | |||
| Blocking the STAT3/PKM2 pathway | Overcoming resistance to apatinib [543] | M | |||
| AMPK activation, blocking the mTOR pathway, increased Bax and Bcl-2 translocation in mitochondria | Increased sensitivity to Adriamycin and radiation [544] | BC | |||
| Decreased AKT and Bcl-2 expression | Overcoming resistance to imatinib [543] | GIST | |||
| GRg3 | GLUT1, GLUT4 | IL-6/STAT3/p-STAT3 pathway inhibition, MDSC suppression, CAF and collagen fibers decrease, cell death | Overcoming resistance to paclitaxel in an in vivo xenograft model [545] | BC | |
| Methyl jasmonate | HK2 | Decrease in the level of AKR1C1 | Induction of cell death, overcoming resistance to bortezomib [546] | MM | |
| Opening of the mPTP due to dissociation of the HK2/VDAC1 complex, triggering apoptotic cell death | Increased cell sensitivity to 5-fluorouracil, Adriamycin, and sorafenib in a xenograft mouse model [547] | HCC | |||
| 3PO | PFKFB3 | Decreased survivin expression, c-IAP1 and c-IAP2, NF-κB activation | Cell death induction [548] | EOC | |
| Shikonin | PKM2 | Decrease in Bcl-2 expression, apoptotic cell death | Increasing the therapeutic effect of cisplatin [549,550] | BC | |
| Exosome secretion inhibition | NSCLC | ||||
| DNA damage, decreased in BRCA1 | Overcoming resistance to Olaparib [501] | EOC | |||
| PKM2/STAT3 pathway inhibition | Reduced tumor growth in an in vivo xenograft model [551] | ESCC | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleksandrova, Y.; Neganova, M. Deciphering the Mysterious Relationship between the Cross-Pathogenetic Mechanisms of Neurodegenerative and Oncological Diseases. Int. J. Mol. Sci. 2023, 24, 14766. https://doi.org/10.3390/ijms241914766
Aleksandrova Y, Neganova M. Deciphering the Mysterious Relationship between the Cross-Pathogenetic Mechanisms of Neurodegenerative and Oncological Diseases. International Journal of Molecular Sciences. 2023; 24(19):14766. https://doi.org/10.3390/ijms241914766
Chicago/Turabian StyleAleksandrova, Yulia, and Margarita Neganova. 2023. "Deciphering the Mysterious Relationship between the Cross-Pathogenetic Mechanisms of Neurodegenerative and Oncological Diseases" International Journal of Molecular Sciences 24, no. 19: 14766. https://doi.org/10.3390/ijms241914766
APA StyleAleksandrova, Y., & Neganova, M. (2023). Deciphering the Mysterious Relationship between the Cross-Pathogenetic Mechanisms of Neurodegenerative and Oncological Diseases. International Journal of Molecular Sciences, 24(19), 14766. https://doi.org/10.3390/ijms241914766