
NTHL1 Gene Mutations in Polish Polyposis Patients—Weighty Player or Vague Background?

 , , , and
, , , and
Abstract
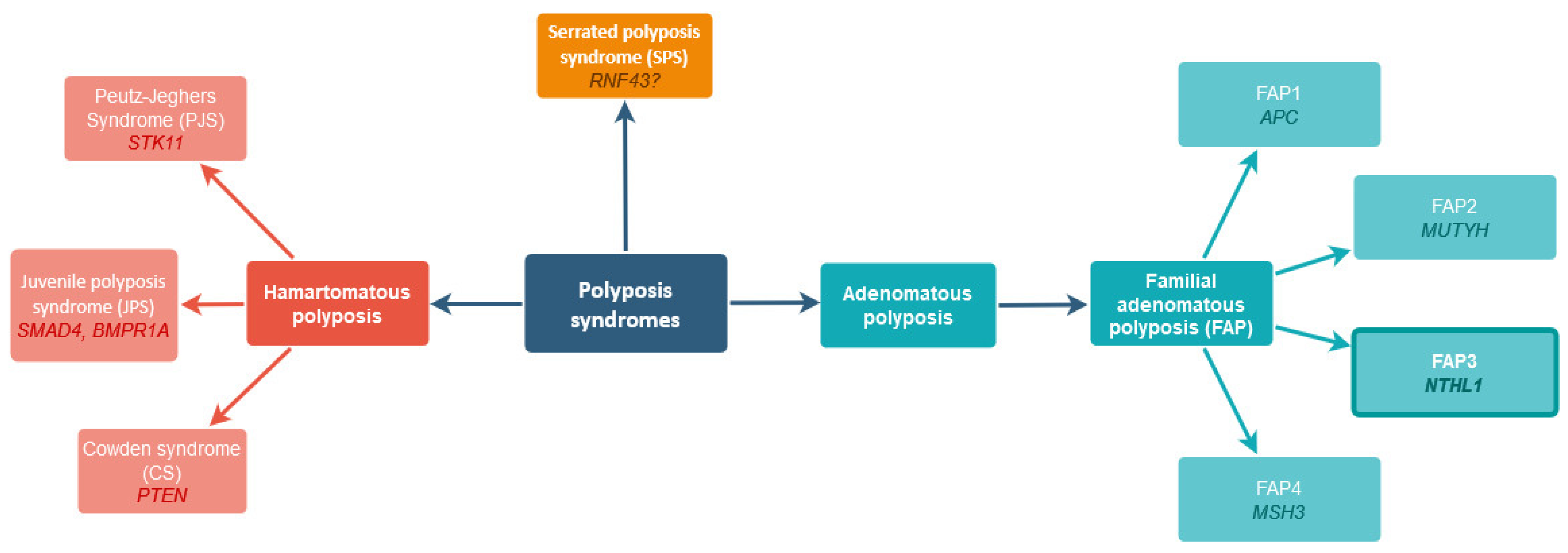
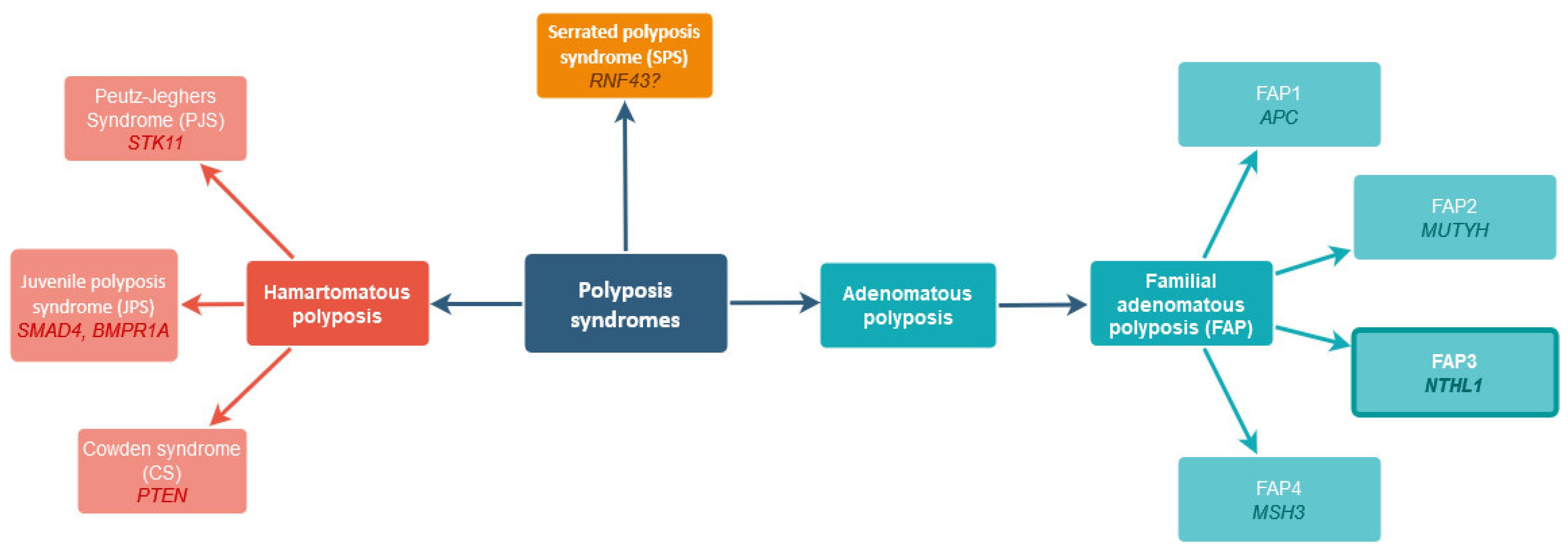
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Genetic Analyses
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.Y.; Siegel, D.A.; Demb, J.; Richardson, L.C.; Gupta, S. Increasing Colorectal Cancer Incidence Before and After Age 50: Implications for Screening Initiation and Promotion of “On-Time” Screening. Dig. Dis. Sci. 2021, 67, 4086–4091. [Google Scholar] [CrossRef] [PubMed]
- Didkowska, J.; Wojciechowska, U.; Michalek, I.M.; dos Santos, F.L.C. Cancer incidence and mortality in Poland in 2019. Sci. Rep. 2022, 12, 10875. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Duong, H. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy (Review). Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; de Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Asp. Med. 2019, 69, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef]
- Yamagishi, H.; Kuroda, H.; Imai, Y.; Hiraishi, H. Molecular pathogenesis of sporadic colorectal cancers. Chin. J. Cancer 2016, 35, 4. [Google Scholar] [CrossRef]
- Czene, K.; Lichtenstein, P.; Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish family-cancer database. Int. J. Cancer 2002, 99, 260–266. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and Heritable Factors in the Causation of Cancer—Analyses of Cohorts of Twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Stoffel, E.M.; Boland, C.R. Genetics and Genetic Testing in Hereditary Colorectal Cancer. Gastroenterology 2015, 149, 1191–1203.e2. [Google Scholar] [CrossRef]
- Hryhorowicz, S.; Kaczmarek-Ryś, M.; Lis-Tanaś, E.; Porowski, J.; Szuman, M.; Grot, N.; Kryszczyńska, A.; Paszkowski, J.; Banasiewicz, T.; Pławski, A. Strong Hereditary Predispositions to Colorectal Cancer. Genes 2022, 13, 2326. [Google Scholar] [CrossRef] [PubMed]
- Lorans, M.; Dow, E.; Macrae, F.A.; Winship, I.M.; Buchanan, D.D. Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clin. Color. Cancer 2018, 17, e293–e305. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.; Wise, P.E. Hereditary Colorectal Cancer Syndromes. Surg. Clin. North Am. 2017, 97, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, N.S.; Wilding, J.L.; Bodmer, W.F. Genetics of colorectal cancer: Hereditary aspects and overview of colorectal tumorigenesis. Br. Med. Bull. 2002, 64, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Available online: Https://Omim.Org/Entry/175100 (accessed on 15 May 2023).
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; De Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef]
- Belhadj, S.; Mur, P.; Navarro, M.; González, S.; Moreno, V.; Capellá, G.; Valle, L. Delineating the Phenotypic Spectrum of the NTHL1-Associated Polyposis. Clin. Gastroenterol. Hepatol. 2016, 15, 461–462. [Google Scholar] [CrossRef]
- Imai, K.; Sarker, A.H.; Akiyama, K.; Ikeda, S.; Yao, M.; Tsutsui, K.; Shohmori, T.; Seki, S. Genomic structure and sequence of a human homologue (NTHL1/NTH1) of Escherichia coli endonuclease III with those of the adjacent parts of TSC2 and SLC9A3R2 genes. Gene 1998, 222, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Lorca, V.; Garre, P. Current status of the genetic susceptibility in attenuated adenomatous polyposis. World J. Gastrointest. Oncol. 2019, 11, 1101–1114. [Google Scholar] [CrossRef]
- Elsayed, F.A.; Grolleman, J.E.; Ragunathan, A.; Buchanan, D.D.; van Wezel, T.; de Voer, R.M.; Boot, A.; Stojovska, M.S.; Mahmood, K.; Clendenning, M.; et al. Monoallelic NTHL1 Loss-of-Function Variants and Risk of Polyposis and Colorectal Cancer. Gastroenterology 2020, 159, 2241–2243.e6. [Google Scholar] [CrossRef]
- Carroll, B.L.; Zahn, K.E.; Hanley, J.P.; Wallace, S.S.; Dragon, J.A.; Doublié, S. Caught in motion: Human NTHL1 undergoes interdomain rearrangement necessary for catalysis. Nucleic Acids Res. 2021, 49, 13165–13178. [Google Scholar] [CrossRef]
- Weren, R.D.; Ligtenberg, M.J.; van Kessel, A.G.; De Voer, R.M.; Hoogerbrugge, N.; Kuiper, R.P. NTHL1 and MUTYH polyposis syndromes: Two sides of the same coin? J. Pathol. 2017, 244, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Quintana, V.G.; Sweasy, J.B. NTHL1 in genomic integrity, aging and cancer. DNA Repair 2020, 93, 102920. [Google Scholar] [CrossRef] [PubMed]
- Rivera, B.; Castellsagué, E.; Bah, I.; van Kempen, L.C.; Foulkes, W.D. Biallelic NTHL1 Mutations in a Woman with Multiple Primary Tumors. N. Engl. J. Med. 2015, 373, 1985–1986. [Google Scholar] [CrossRef] [PubMed]
- Grolleman, J.E.; de Voer, R.M.; Elsayed, F.A.; Nielsen, M.; Weren, R.D.; Palles, C.; Ligtenberg, M.J.; Vos, J.R.; Broeke, S.W.T.; de Miranda, N.F.; et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-tumor Phenotype. Cancer Cell 2019, 35, 256–266.e5. [Google Scholar] [CrossRef] [PubMed]
- Fostira, F.; Kontopodis, E.; Apostolou, P.; Fragkaki, M.; Androulakis, N.; Yannoukakos, D.; Konstantopoulou, I.; Saloustros, E. Extending the clinical phenotype associated with biallelic NTHL1 germline mutations. Clin. Genet. 2018, 94, 588–589. [Google Scholar] [CrossRef]
- Groves, A.; Gleeson, M.; Spigelman, A.D. NTHL1-associate polyposis: First Australian case report. Fam. Cancer 2019, 18, 179–182. [Google Scholar] [CrossRef]
- Altaraihi, M.; Gerdes, A.M.; Wadt, K. A new family with a homozygous nonsense variant in NTHL1 further delineated the clinical phenotype of NTHL1-associated polyposis. Hum. Genome Var. 2019, 6, 1–3. [Google Scholar] [CrossRef]
- Available online: https://Gnomad.Broadinstitute.Org/Variant/16-2096239-G-A?Dataset=gnomad_r2_1_controls2023 (accessed on 22 May 2023).
- Słomski, R. Analiza DNA: Teoria i praktyka; Wydawnictwo Uniwersytetu Przyrodniczego: Poznan, Poland, 2008. [Google Scholar]
- MedCalc Software Ltd. Odds Ratio Calculator. Available online: https://www.Medcalc.Org/Calc/Odds_ratio.Php2023 (accessed on 22 May 2023).

{kind=link}
{kind=link}
| NTHL1 Mutation | Genotypes n (%) | Alleles n (%) | |||
|---|---|---|---|---|---|
| Group | xx | Xx | XX | x | X |
| Polyposis patients whole group (n = 644) | 639 (99.22%) | 2 (0.31%) | 3 (0.47%) | 1280 (99.38%) | 8 (0.62%) |
| Polyposis patients (no identified genetic background) * (n = 316) | 313 (99.05%) | 1 (0.32%) | 2 (0.63%) | 627 (99.21%) | 5 (0.79%) |
| Polyposis patients with an identified mutation (n = 328) | 326 (99.39%) | 1 (0.305%) | 1 (0.305%) | 653 (99.54%) | 3 (0.46%) |
| Controls (n = 634) | 630 (99.37%) | 4 (0.63%) | n.o. | 1264 (99.68%) | 4 (0.32%) |
| [XX] vs. [xx + Xx] | [XX + Xx] vs. [xx] | [XX] vs. [xx] | [x] vs. [X] | [X] vs. [x] | |
| Polyposis patients (whole group) vs. controls p value OR (95% CI) | OR = 6.92 CI = [0.36–134.32] p value = 0.201 | OR = 1.23 CI = [0.33–4.61] p value = 0.756 | OR = 6.90 CI = [0.36–133.89] p value = 0.202 | OR = 0.45 CI = [0.14–1.46] | OR = 1.97 CI = [ 0.59–6.58] |
| p value = 0.267 | |||||
| Polyposis patients (no identified genetic background) vs. controls p value OR (95% CI) | OR = 10.09 CI = [0.48–210.76] p value = 0.136 | OR = 1.51 CI = [0.34–6.79] p value = 0.591 | OR = 10.06 CI = [0.48–210.10] p value = 0.137 | OR = 0.40 CI = [0.11–1.48] | OR = 2.52 CI = [ 0.67–9.42] |
| p value = 0.169 | |||||
| Polyposis patients with an identified mutation vs. controls p value OR (95% CI) | OR = 5.81 CI = [0.24–143.08] p value = 0.282 | OR = 0.97 CI = [0.18–5.30] p value = 0.969 | OR = 5.79 CI = [0.24–142.62] p value = 0.283 | OR = 0.69 CI = [0.15–3.09] | OR = 1.45 CI = [0.32–6.51] |
| p value = 0.626 | |||||
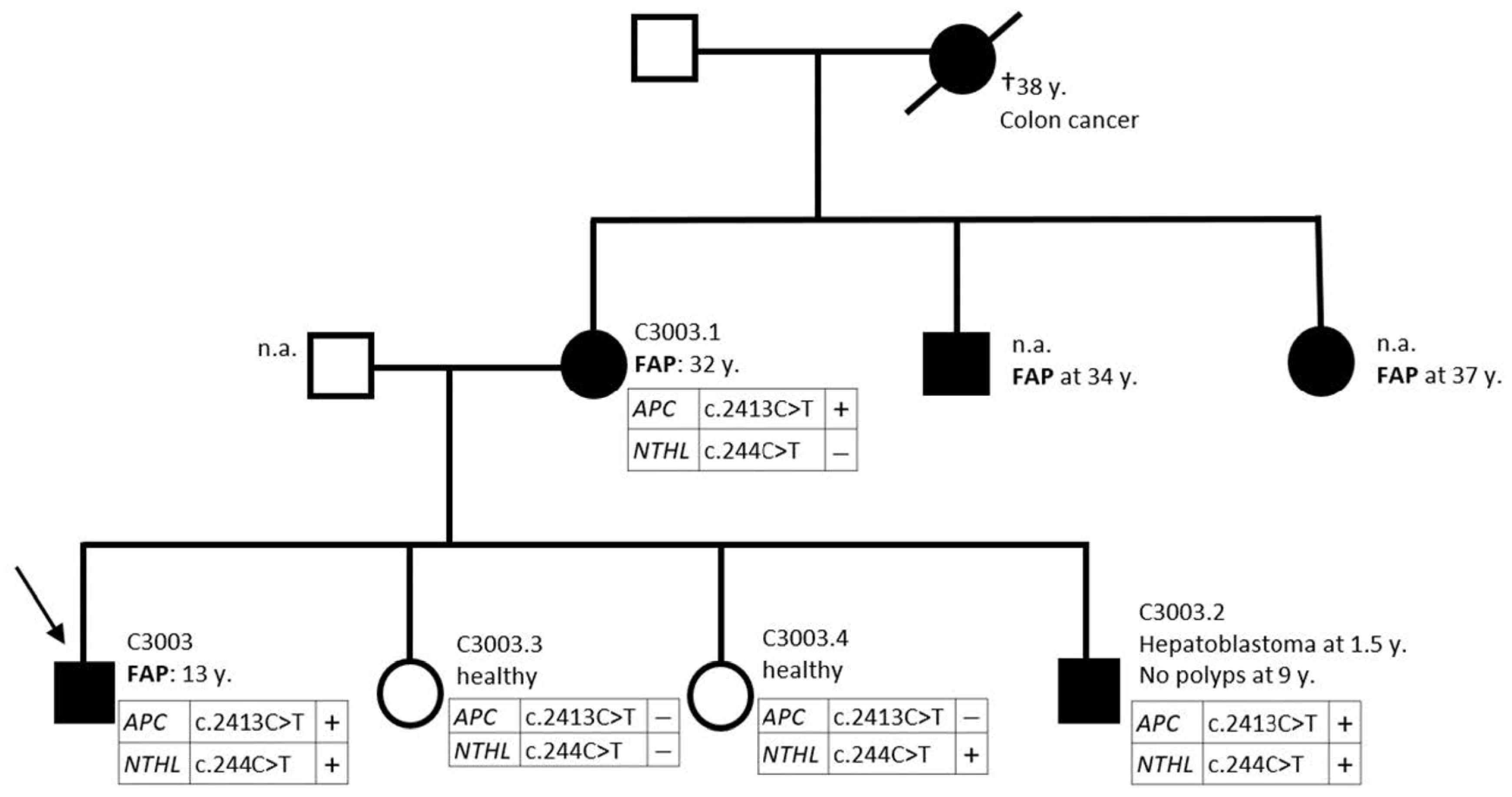
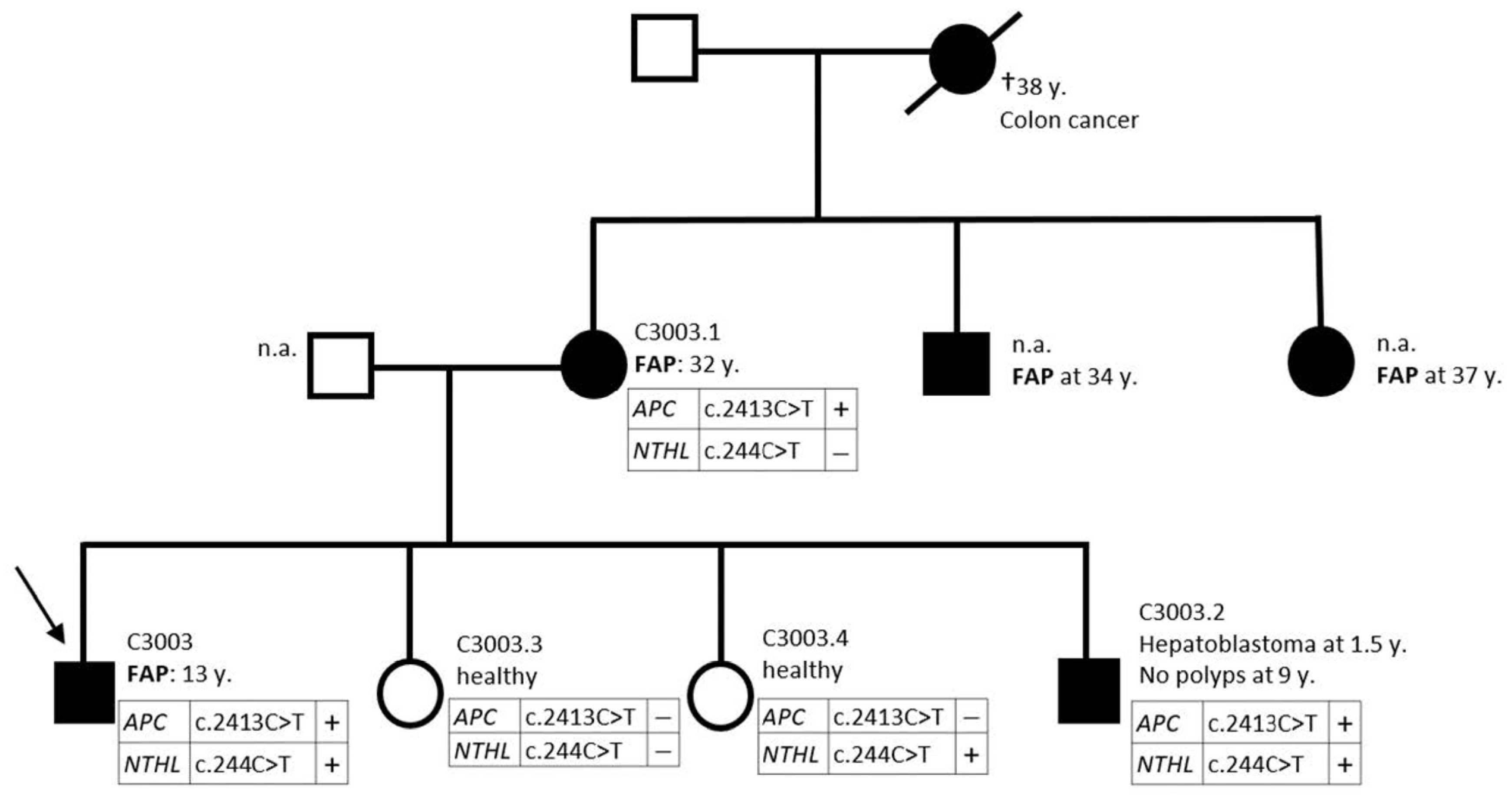
| Family | Patient ID | NTHL1 Variant | Allelic State | Amino Acid Change | Malignancies | Polyps | Co-Occurrent Mutation |
|---|---|---|---|---|---|---|---|
| 1 | 9119 | c.244C>T | homozygous | p.Q82* | no accessible data | adenomatous | APC c.4129_4130delGT |
| 2 | 9434 | c.244C>T | homozygous | p.Q82* | papillary thyroid cancer at 60 | tubular, hyperplastic, serrated, above 100 | n.d. |
| 3 | 9671 | c.244C>T | homozygous | p.Q82* | occipital meningioma, two meningiomas of the left frontal lobe diagnosed at the age of 69; at 43, hysterectomy due to fibroids and heavy bleeding | histopathologically villous and tubulovillous adenomas at the age of 61 | n.d. |
| 4 | C1167 | c.274C>T | heterozygous | p.R92C | no family history | dozen adenomatous polyps at 60 | n.d. |
| 5 | C3003 | c.244C>T | heterozygous | p.Q82* | adenomatous at 13 | APC c.2413C>T | |
| C3003.1 | c.244C>T | - | - | adenomatous at 32 | APC c.2413C>T | ||
| C3003.2 | c.244C>T | heterozygous | p.Q82* | hepatoblastoma at 1.5 | no polyps at 9 | APC c.2413C>T | |
| C3003.3 | n.d. | - | - | healthy | APC c.2413C>T excluded | ||
| C3003.4 | c.244C>T | heterozygous | p.Q82* | healthy | APC c.2413C>T excluded |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grot, N.; Kaczmarek-Ryś, M.; Lis-Tanaś, E.; Kryszczyńska, A.; Nowakowska, D.; Jakubiuk-Tomaszuk, A.; Paszkowski, J.; Banasiewicz, T.; Hryhorowicz, S.; Pławski, A. NTHL1 Gene Mutations in Polish Polyposis Patients—Weighty Player or Vague Background? Int. J. Mol. Sci. 2023, 24, 14548. https://doi.org/10.3390/ijms241914548
Grot N, Kaczmarek-Ryś M, Lis-Tanaś E, Kryszczyńska A, Nowakowska D, Jakubiuk-Tomaszuk A, Paszkowski J, Banasiewicz T, Hryhorowicz S, Pławski A. NTHL1 Gene Mutations in Polish Polyposis Patients—Weighty Player or Vague Background? International Journal of Molecular Sciences. 2023; 24(19):14548. https://doi.org/10.3390/ijms241914548
Chicago/Turabian StyleGrot, Natalia, Marta Kaczmarek-Ryś, Emilia Lis-Tanaś, Alicja Kryszczyńska, Dorota Nowakowska, Anna Jakubiuk-Tomaszuk, Jacek Paszkowski, Tomasz Banasiewicz, Szymon Hryhorowicz, and Andrzej Pławski. 2023. "NTHL1 Gene Mutations in Polish Polyposis Patients—Weighty Player or Vague Background?" International Journal of Molecular Sciences 24, no. 19: 14548. https://doi.org/10.3390/ijms241914548
APA StyleGrot, N., Kaczmarek-Ryś, M., Lis-Tanaś, E., Kryszczyńska, A., Nowakowska, D., Jakubiuk-Tomaszuk, A., Paszkowski, J., Banasiewicz, T., Hryhorowicz, S., & Pławski, A. (2023). NTHL1 Gene Mutations in Polish Polyposis Patients—Weighty Player or Vague Background? International Journal of Molecular Sciences, 24(19), 14548. https://doi.org/10.3390/ijms241914548