
Mathematical Modeling Support for Lung Cancer Therapy—A Short Review


{kind=link}
Abstract
:1. Introduction
2. Lung Cancer Treatment and Associated Signaling Pathways
2.1. Treatment Options
2.2. Sample Molecular Players Involved in Tumor Response to Treatment
3. Modeling Cancer Growth and Response to Treatment at Different Levels
3.1. Cancer Growth
3.2. Therapy Modeling at the Cancer Cell Population Scale
3.2.1. Surgery
3.2.2. Chemotherapy
3.2.3. Radiotherapy
3.2.4. Antiangiogenic Treatment
3.2.5. Immunotherapy Models
3.2.6. Models of Combined Therapies
3.2.7. Therapy Resistance and Metastasis
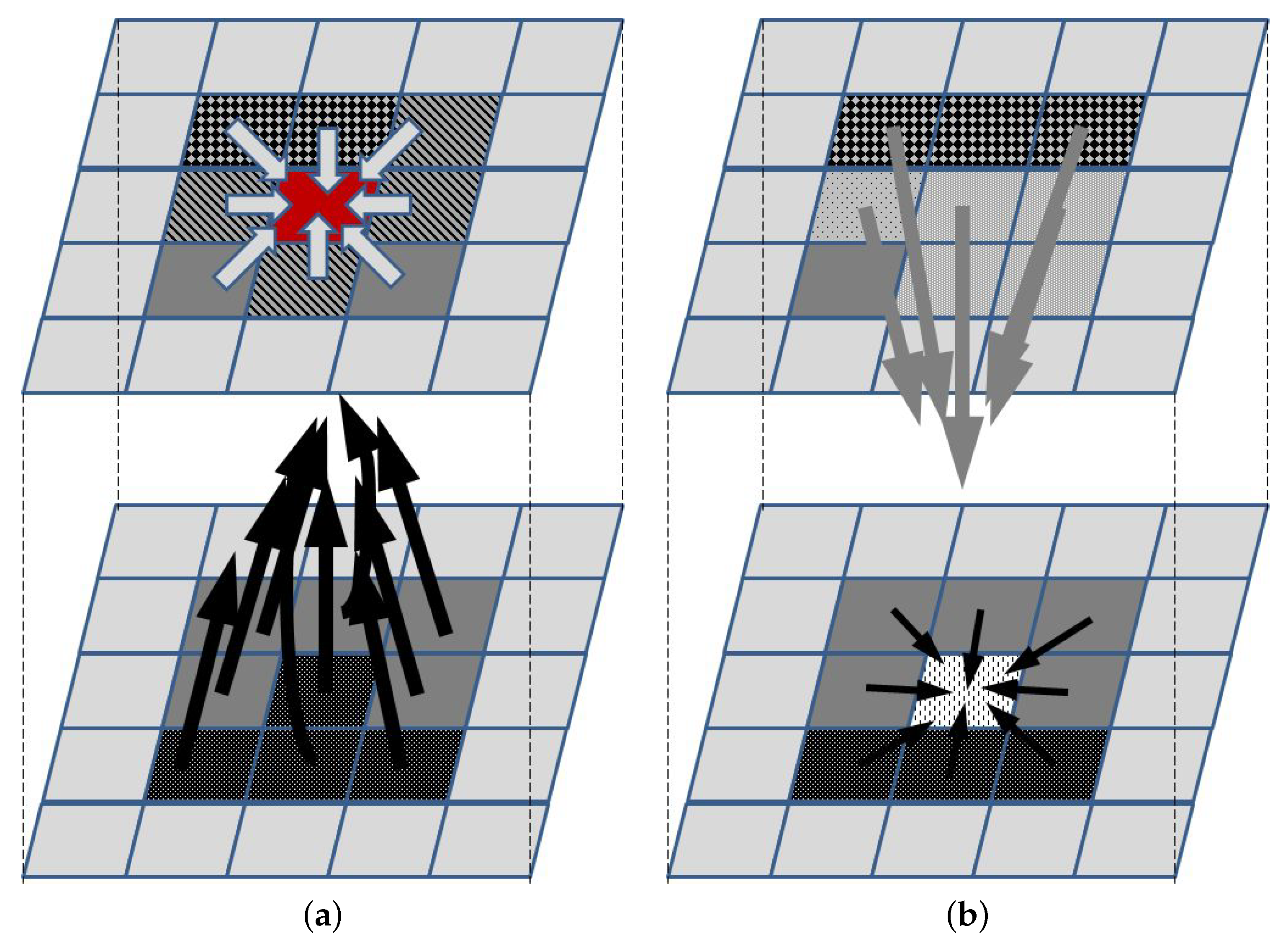
3.3. PDE Models
3.4. Agent-Based Models and Multiscale Modeling
3.5. Changing the Perspective from a Tumor in a Single Patient to a Patient Population
4. Modeling Intracellular Processes Associated with Cancer Growth and Its Responses to Treatment
4.1. General Modeling Remarks
4.2. Examples of Models of Intracellular Processes Related to Cancer Growth and Its Response to Therapy
5. Discussion
- Transforming structure, graph models of lung cancer metastasis, or regulatory networks involved in cellular responses to treatment into models describing the dynamics of these processes;
- Linking models describing synergistic effects of drugs in chemotherapy with models of cancer growth;
- Development of multiscale models, linking intracellular dynamics, specific for lung cancer, with models of metastasis or therapy resistance.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, J.; Deng, Y.; Tin, M.S.; Lok, V.; Ngai, C.H.; Zhang, L.; Lucero-Prisno, D.E., 3rd; Xu, W.; Zheng, Z.J.; Elcarte, E.; et al. Distribution, risk factors, and temporal trends for lung Cancer Incidence and mortality: A global analysis. Chest 2022, 161, 1101–1111. [Google Scholar] [CrossRef]
- Sharma, R. Mapping of global, regional and national incidence, mortality and mortality-to-incidence ratio of lung cancer in 2020 and 2050. Int. J. Clin. Oncol. 2022, 27, 665–675. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Eberhardt, W.; De Ruysscher, D.; Weder, W.; Le Péchoux, C.; De Leyn, P.; Hoffmann, H.; Westeel, V.; Stahel, R.; Felip, E.; Peters, S.; et al. 2nd ESMO Consensus Conference in Lung Cancer: Locally advanced stage III non-small-cell lung cancer. Ann. Oncol. 2015, 26, 1573–1588. [Google Scholar] [CrossRef]
- Alduais, Y.; Zhang, H.; Fan, F.; Chen, J.; Chen, B. Non-small cell lung cancer (NSCLC): A review of risk factors, diagnosis, and treatment. Medicine 2023, 102, e32899. [Google Scholar] [CrossRef]
- Claes, E.; Wener, R.; Neyrinck, A.P.; Coppens, A.; Van Schil, P.E.; Janssens, A.; Lapperre, T.S.; Snoeckx, A.; Wen, W.; Voet, H.; et al. Innovative invasive loco-regional techniques for the treatment of lung cancer. Cancers 2023, 15, 2244. [Google Scholar] [CrossRef]
- Ibodeng, G.O.; Uche, I.N.; Mokua, R.; Galo, M.; Odigwe, B.; Galeas, J.N.; Dasgupta, S. A snapshot of lung cancer: Where are we now?—A narrative review. Ann. Transl. Med. 2023, 11, 261. [Google Scholar] [CrossRef]
- Villaruz, L.C.; Socinski, M.A.; Weiss, J. Guidance for clinicians and patients with non-small cell lung cancer in the time of precision medicine. Front. Oncol. 2023, 13, 1124167. [Google Scholar] [CrossRef]
- NCI. Lung Cancers—Health Professional Version. 2023. Available online: https://www.cancer.gov/types/lung/hp (accessed on 15 May 2023).
- Cancer, B. Chemotherapy Protocols for Lung Cancer. 2023. Available online: http://www.bccancer.bc.ca/health-professionals/clinical-resources/chemotherapy-protocols/lung#Protocols (accessed on 15 May 2023).
- ESMO. ESMO Clinical Practice Guidelines: Lung and Chest Tumours. 2015. Available online: https://www.esmo.org/guidelines/guidelines-by-topic/lung-and-chest-tumours (accessed on 15 May 2023).
- Altorki, N.; Wang, X.; Kozono, D.; Watt, C.; Landrenau, R.; Wigle, D.; Port, J.; Jones, D.R.; Conti, M.; Ashrafi, A.S.; et al. Lobar or sublobar resection for peripheral stage IA non-small-cell lung cancer. N. Engl. J. Med. 2023, 388, 489–498. [Google Scholar] [CrossRef]
- Daly, M.E.; Beagen, P.; Madani, M.H. Nonsurgical therapy for early-stage lung cancer. Hematol. Oncol. Clin. N. Am. 2023, 37, 499–512. [Google Scholar] [CrossRef]
- Pirker, R. Chemotherapy remains a cornerstone in the treatment of nonsmall cell lung cancer. Curr. Opin. Oncol. 2020, 32, 63–67. [Google Scholar] [CrossRef]
- Moliner, L.; Zhang, B.; Lamberti, G.; Ardizzoni, A.; Byers, L.A.; Califano, R. Novel therapeutic strategies for recurrent SCLC. Crit. Rev. Oncol. Hematol. 2023, 186, 104017. [Google Scholar] [CrossRef]
- Grant, M.J.; Woodard, G.A.; Goldberg, S.B. The evolving role for systemic therapy in resectable non-small cell lung cancer. Hematol. Oncol. Clin. N. Am. 2023, 37, 513–531. [Google Scholar] [CrossRef]
- Monro, H.C.; Gaffney, E.A. Modelling chemotherapy resistance in palliation and failed cure. J. Theor. Biol. 2009, 257, 292–302. [Google Scholar] [CrossRef]
- Shu, Y.; Weng, S.; Zheng, S. Metronomic chemotherapy in non-small cell lung cancer. Oncol. Lett. 2020, 20, 307. [Google Scholar] [CrossRef]
- Min, H.Y.; Lee, H.Y. Mechanisms of resistance to chemotherapy in non-small cell lung cancer. Arch. Pharm. Res. 2021, 44, 146–164. [Google Scholar] [CrossRef]
- Wang, Z.; Xing, Y.; Li, B.; Li, X.; Liu, B. Molecular pathways, resistance mechanisms and targeted interventions in non-small-cell lung cancer. Mol. Biomed. 2022, 3, 42. [Google Scholar] [CrossRef]
- Merie, R.; Gee, H.; Hau, E.; Vinod, S. An overview of the role of radiotherapy in the treatment of small cell lung cancer—A mainstay of treatment or a modality in decline? Clin. Oncol. 2022, 34, 741–752. [Google Scholar] [CrossRef]
- Serrano, J.; Crespo, P.C.; Taboada, B.; Gonzalez, A.A.; García, R.G.; Caamaño, A.G.; Reyes, J.C.T.; Mielgo-Rubio, X.; Felipe, C. Postoperative radiotherapy in resected non-small cell lung cancer: The never-ending story. World J. Clin. Oncol. 2021, 12, 833–844. [Google Scholar] [CrossRef]
- Vinod, S.K.; Hau, E. Radiotherapy treatment for lung cancer: Current status and future directions. Respirology 2020, 25 (Suppl. 2), 61–71. [Google Scholar] [CrossRef]
- Yang, W.C.; Hsu, F.M.; Yang, P.C. Precision radiotherapy for non-small cell lung cancer. J. Biomed. Sci. 2020, 27, 82. [Google Scholar] [CrossRef] [PubMed]
- Laeseke, P.; Ng, C.; Ferko, N.; Naghi, A.; Wright, G.W.; Zhang, Y.; Laidlaw, A.; Kalsekar, I.; Laxmanan, B.; Ghosh, S.K.; et al. Stereotactic body radiation therapy and thermal ablation for treatment of NSCLC: A systematic literature review and meta-analysis. Lung Cancer 2023, 182, 107259. [Google Scholar] [CrossRef] [PubMed]
- Zayed, S.; Louie, A.V.; Breadner, D.A.; Palma, D.A.; Correa, R.J.M. Radiation and immune checkpoint inhibitors in the treatment of oligometastatic non-small-cell lung cancer: A practical review of rationale, recent data, and research questions. Ther. Adv. Med. Oncol. 2023, 15, 17588359231183668. [Google Scholar] [CrossRef] [PubMed]
- Allignet, B.; De Ruysscher, D.; Martel-Lafay, I.; Waissi, W. Stereotactic body radiation therapy in unresectable stage III non-small cell lung cancer: A systematic review. Cancer Treat. Rev. 2023, 118, 102573. [Google Scholar] [CrossRef] [PubMed]
- Huo, B.; Ji, Z.; He, C.; Yang, W.; Ma, Y.; Huo, X.; Wang, Z.; Zhao, X.; Dai, J.; Wang, H.; et al. Safety and efficacy of stereotactic ablative brachytherapy as a salvage therapy for recurrent chest wall cancer: A retrospective, multicenter study. Front. Oncol. 2023, 12, 957497. [Google Scholar] [CrossRef]
- Mutsaers, A.; Zhang, T.W.; Louie, A.; Rodrigues, G.; Palma, D.; Qu, M. Stereotactic or Conventional Radiation for Early-Stage Non-small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Cureus 2023, 15, e38198. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, L.Y.; He, J.Z.; Miao, Z.M.; Li, Y.Y.; Zhang, Y.M.; Liu, Z.W.; Zhang, S.Z.; Chen, Y.; Zhou, G.C.; et al. Review: Mechanisms and perspective treatment of radioresistance in non-small cell lung cancer. Front. Immunol. 2023, 14, 1133899. [Google Scholar] [CrossRef] [PubMed]
- Matschke, J.; Larafa, S.; Jendrossek, V. Metabolic reprograming of antioxidant defense: A precision medicine perspective for radiotherapy of lung cancer? Biochem. Soc. Trans. 2021, 49, 1265–1277. [Google Scholar] [CrossRef]
- Yu, X.Y.; Jin, X.; Shou, Z.X. Surface-engineered smart nanocarrier-based inhalation formulations for targeted lung cancer chemotherapy: A review of current practices. Drug Deliv. 2021, 28, 1995–2010. [Google Scholar] [CrossRef] [PubMed]
- McNamee, N.; da Silva, I.P.; Nagrial, A.; Gao, B. Small-cell lung cancer-an update on targeted and immunotherapies. Int. J. Mol. Sci. 2023, 24, 8129. [Google Scholar] [CrossRef]
- Salifu, I.; Singh, N.; Berraondo, M.; Remon, J.; Salifu, S.; Severson, E.; Quintana, A.; Peiró, S.; Ramkissoon, S.; Vidal, L.; et al. Antibody-drug conjugates, immune-checkpoint inhibitors, and their combination in advanced non-small cell lung cancer. Cancer Treat. Res. Commun. 2023, 36, 100713. [Google Scholar] [CrossRef]
- Shang, S.; Liu, J.; Verma, V.; Wu, M.; Welsh, J.; Yu, J.; Chen, D. Combined treatment of non-small cell lung cancer using radiotherapy and immunotherapy: Challenges and updates. Cancer Commun. 2021, 41, 1086–1099. [Google Scholar] [CrossRef]
- Reck, M.; Popat, S.; Grohé, C.; Corral, J.; Novello, S.; Gottfried, M.; Brueckl, W.; Radonjic, D.; Kaiser, R.; Heymach, J. Anti-angiogenic agents for NSCLC following first-line immunotherapy: Rationale, recent updates, and future perspectives. Lung Cancer 2023, 179, 107173. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhao, Z.; Tang, H. Current status and future of anti-angiogenic drugs in lung cancer. Clin. Exp. Med. 2023. [Google Scholar] [CrossRef]
- Yu, X.; Zhu, L.; Wang, T.; Li, L.; Liu, J.; Che, G.; Zhou, Q. Enhancing the anti-tumor response by combining DNA damage repair inhibitors in the treatment of solid tumors. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188910. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.; Mirzayans, R. Nonlinearities in the cellular response to ionizing radiation and the role of p53 therein. Int. J. Radiat. Biol. 2021, 97, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Ernst, M.; Poh, A.R. Multicellular effects of STAT3 in non-small cell lung cancer: Mechanistic insights and therapeutic opportunities. Cancers 2021, 13, 6228. [Google Scholar] [CrossRef]
- Toyokawa, G.; Bersani, F.; Bironzo, P.; Picca, F.; Tabbò, F.; Haratake, N.; Takenaka, T.; Seto, T.; Yoshizumi, T.; Novello, S.; et al. Tumor plasticity and therapeutic resistance in oncogene-addicted non-small cell lung cancer: From preclinical observations to clinical implications. Crit. Rev. Oncol. Hematol. 2023, 184, 103966. [Google Scholar] [CrossRef]
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef]
- Ashrafi, A.; Akter, Z.; Modareszadeh, P.; Modareszadeh, P.; Berisha, E.; Alemi, P.S.; Chacon Castro, M.D.C.; Deese, A.R.; Zhang, L. Current landscape of therapeutic resistance in lung cancer and promising strategies to overcome resistance. Cancers 2022, 14, 4562. [Google Scholar] [CrossRef]
- Nair, A.S.; Jayan, A.P.; Anandu, K.R.; Saiprabha, V.N.; Pappachen, L.K. Unraveling the prevalence of various signalling pathways in non-small-cell lung cancer: A review. Mol. Cell. Biochem. 2023. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.Y.; Li, Z.Y.; Gan, T.Q.; Fang, Y.Y.; Gan, B.L.; Chen, W.J.; Dang, Y.W.; Shi, K.; Feng, Z.B.; Chen, G. Downregulation of hsa-microRNA-204-5p and identification of its potential regulatory network in non-small cell lung cancer: RT-qPCR, bioinformatic- and meta-analyses. Respir. Res. 2020, 21, 60. [Google Scholar] [CrossRef] [PubMed]
- Samarth, N.; Gulhane, P.; Singh, S. Immunoregulatory framework and the role of miRNA in the pathogenesis of NSCLC—A systematic review. Front. Oncol. 2022, 12, 1089320. [Google Scholar] [CrossRef] [PubMed]
- Frydrychowicz, M.; Kuszel, Ł.; Dworacki, G.; Budna-Tukan, J. MicroRNA in lung cancer-a novel potential way for early diagnosis and therapy. J. Appl. Genet. 2023, 64, 459–477. [Google Scholar] [CrossRef]
- Kris, M.G.; Mitsudomi, T.; Peters, S. Adjuvant therapies in stages I-III epidermal growth factor receptor-mutated lung cancer: Current and future perspectives. Transl. Lung Cancer Res. 2023, 12, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Hoden, B.; DeRubeis, D.; Martinez-Moczygemba, M.; Ramos, K.S.; Zhang, D. Understanding the role of Toll-like receptors in lung cancer immunity and immunotherapy. Front. Immunol. 2022, 13, 1033483. [Google Scholar] [CrossRef]
- Jiang, R.; Huang, J.; Sun, X.; Chu, X.; Wang, F.; Zhou, J.; Fan, Q.; Pang, L. Construction of in vitro 3-D model for lung cancer-cell metastasis study. BMC Cancer 2022, 22, 438. [Google Scholar] [CrossRef]
- Swierniak, A.; Kimmel, M.; Smieja, J.; Puszynski, K.; Psiuk-Maksymowicz, K. System Engineering Approach to Planning Anticancer Therapies, 1st ed.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Skipper, H.; Schabel, F.J.; Wilcox, W. Experimental evaluation of potential anticancer agents. xiii. on the criteria and kinetics associated with curability of experimental leukemia. Cancer Chemother. Rep. 1964, 35, 1–111. [Google Scholar]
- Niu, J.; Wang, X.; Qu, J.; Mager, D.E.; Straubinger, R.M. Pharmacodynamic modeling of synergistic birinapant/paclitaxel interactions in pancreatic cancer cells. BMC Cancer 2020, 20, 1024. [Google Scholar] [CrossRef]
- Chen, D.; Liu, X.; Yang, Y.; Yang, H.; Lu, P. Systematic synergy modeling: Understanding drug synergy from a systems biology perspective. BMC Syst. Biol. 2015, 9, 56. [Google Scholar] [CrossRef]
- Lea, D.E.; Catcheside, D.G. The mechanism of the induction by radiation of chromosome aberrations inTradescantia. J. Genet. 1942, 44, 216–245. [Google Scholar] [CrossRef]
- Fowler, J.F. The linear-quadratic formula and progress in fractionated radiotherapy. Br. J. Radiol. 1989, 62, 679–694. [Google Scholar] [CrossRef]
- Fowler, J.F. 21 years of biologically effective dose. Br. J. Radiol. 2010, 83, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Victori, P.; Buffa, F.M. The many faces of mathematical modelling in oncology. Br. J. Radiol. 2019, 92, 20180856. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R. A microdosimetric-kinetic model of cell death from exposure to ionizing radiation of any LET, with experimental and clinical applications. Int. J. Radiat. Biol. 1996, 69, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Shiinoki, T.; Tanabe, S.; Utsunomiya, S.; Takizawa, T.; Kaidu, M.; Nishio, T.; Ishikawa, H. Mathematical model combined with microdosimetric kinetic model for tumor volume calculation in stereotactic body radiation therapy. Sci. Rep. 2023, 13, 10981. [Google Scholar] [CrossRef] [PubMed]
- Okawa, K. Comparison of dynamic tumor tracking error measurement methods for robotic radiosurgery. J. Appl. Clin. Med. Phys. 2023, 24, e14093. [Google Scholar] [CrossRef]
- Han, B.; Wu, B.; Hu, F.; Ma, Y.; Wang, H.; Han, X.; Liu, G.; Guo, Y. Simulation of dosimetric consequences of intrafraction variation of tumor drift in lung cancer stereotactic body radiotherapy. Front. Oncol. 2022, 12, 1010411. [Google Scholar] [CrossRef]
- Hahnfeldt, P.; Panigrahy, D.; Folkman, J.; Hlatky, L. Tumor development under angiogenic signaling: A dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999, 59, 4770–4775. [Google Scholar]
- Świerniak, A. Comparison of six models of antiangiogenic therapy. Appl. Math. 2009, 36, 333–348. [Google Scholar] [CrossRef]
- Depillis, L.; Gallegos, A.; Radunskaya, A. A model of dendritic cell therapy for melanoma. Front. Oncol. 2013, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Foryś, U.; Waniewski, J.; Zhivkov, P. Anti-tumor immunity and tumor anti-immunity in a mathematical model of tumor immunotherapy. J. Biol. Syst. 2006, 14, 13–30. [Google Scholar] [CrossRef]
- Woelke, A.L.; Murgueitio, M.S.; Preissner, R. Theoretical modeling techniques and their impact on tumor immunology. Clin. Dev. Immunol. 2010, 2010, 271794. [Google Scholar] [CrossRef]
- Bartha, L.; Eftimie, R. Mathematical investigation into the role of macrophage heterogeneity on the temporal and spatio-temporal dynamics of non-small cell lung cancers. J. Theor. Biol. 2022, 549, 111207. [Google Scholar] [CrossRef] [PubMed]
- Okuneye, K.; Bergman, D.; Bloodworth, J.C.; Pearson, A.T.; Sweis, R.F.; Jackson, T.L. A validated mathematical model of FGFR3-mediated tumor growth reveals pathways to harness the benefits of combination targeted therapy and immunotherapy in bladder cancer. Comput. Syst. Oncol. 2021, 1, e1019. [Google Scholar] [CrossRef] [PubMed]
- Butner, J.D.; Dogra, P.; Chung, C.; Pasqualini, R.; Arap, W.; Lowengrub, J.; Cristini, V.; Wang, Z. Mathematical modeling of cancer immunotherapy for personalized clinical translation. Nat. Comput. Sci. 2022, 2, 785–796. [Google Scholar] [CrossRef]
- Geng, C.; Paganetti, H.; Grassberger, C. Prediction of treatment response for combined chemo- and radiation therapy for Non-Small Cell Lung Cancer patients using a bio-mathematical model. Sci. Rep. 2017, 7, 13542. [Google Scholar] [CrossRef]
- El Haout, S.; Fatani, M.; Farha, N.A.; AlSawaftah, N.; Mortula, M.; Husseini, G.A. Modeling the effects of chemotherapy and immunotherapy on tumor growth. J. Biomed. Nanotechnol. 2021, 17, 2505–2518. [Google Scholar] [CrossRef]
- Khalili, P.; Vatankhah, R. Studying the importance of regulatory T cells in chemoimmunotherapy mathematical modeling and proposing new approaches for developing a mathematical dynamic of cancer. J. Theor. Biol. 2023, 563, 111437. [Google Scholar] [CrossRef]
- Friedrich, T.; Henthorn, N.; Durante, M. Modeling radioimmune response-current status and perspectives. Front. Oncol. 2021, 11, 647272. [Google Scholar] [CrossRef]
- Bekker, R.A.; Kim, S.; Pilon-Thomas, S.; Enderling, H. Mathematical modeling of radiotherapy and its impact on tumor interactions with the immune system. Neoplasia 2022, 28, 100796. [Google Scholar] [CrossRef] [PubMed]
- Powathil, G.G.; Adamson, D.J.A.; Chaplain, M.A.J. Towards predicting the response of a solid tumour to chemotherapy and radiotherapy treatments: Clinical insights from a computational model. PLoS Comput. Biol. 2013, 9, e1003120. [Google Scholar] [CrossRef] [PubMed]
- De Mendoza, A.M.; Michlíková, S.; Berger, J.; Karschau, J.; Kunz-Schughart, L.A.; McLeod, D.D. Mathematical model for the thermal enhancement of radiation response: Thermodynamic approach. Sci. Rep. 2021, 11, 5503. [Google Scholar] [CrossRef] [PubMed]
- Ciccolini, J.; Benzekry, S.; Barlesi, F. Deciphering the response and resistance to immune-checkpoint inhibitors in lung cancer with artificial intelligence-based analysis: When PIONeeR meets QUANTIC. Br. J. Cancer 2020, 123, 337–338. [Google Scholar] [CrossRef]
- Bodzioch, M.; Bajger, P.; Foryś, U. Angiogenesis and chemotherapy resistance: Optimizing chemotherapy scheduling using mathematical modeling. J. Cancer Res. Clin. Oncol. 2021, 147, 2281–2299. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Enta, A.; Maruya, Y.; Inomata, S.; Yamaguchi, H.; Mine, H.; Takagi, H.; Ozaki, Y.; Watanabe, M.; Inoue, T.; et al. Wnt/β-catenin signaling and resistance to immune checkpoint inhibitors: From non-small-cell lung cancer to other cancers. Biomedicines 2023, 11, 190. [Google Scholar] [CrossRef]
- Rhodes, A.; Hillen, T. A mathematical model for the immune-mediated theory of metastasis. J. Theor. Biol. 2019, 482, 109999. [Google Scholar] [CrossRef]
- Murray, J.D. Mathematical Biology. II: Spatial Models and Biomedical Applications, 3rd ed.; Springer: New York, NY, USA, 2003. [Google Scholar]
- Pouchol, C.; Clairambault, J.; Lorz, A.; Trélat, E. Asymptotic analysis and optimal control of an integro-differential system modelling healthy and cancer cells exposed to chemotherapy. J. Math. Pures Appl. 2018, 116, 268–308. [Google Scholar] [CrossRef]
- Iwata, K.; Kawasaki, K.; Shigesada, N. A dynamical model for the growth and size distribution of multiple metastatic tumors. J. Theor. Biol. 2000, 203, 177–186. [Google Scholar] [CrossRef]
- Ford Versypt, A.N. Multiscale modeling in disease. Curr. Opin. Syst. Biol. 2021, 27, 100340. [Google Scholar] [CrossRef]
- England, T.; Harper, P.; Crosby, T.; Gartner, D.; Arruda, E.F.; Foley, K.; Williamson, I. Modelling lung cancer diagnostic pathways using discrete event simulation. J. Simul. 2023, 17, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Jafari Nivlouei, S.; Soltani, M.; Shirani, E.; Salimpour, M.R.; Travasso, R.; Carvalho, J. A multiscale cell-based model of tumor growth for chemotherapy assessment and tumor-targeted therapy through a 3D computational approach. Cell Prolif. 2022, 55, e13187. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, J.C.L.; Grass, G.D.; Welsh, E.; Ahmed, K.A.; Teer, J.K.; Pilon-Thomas, S.; Harrison, L.B.; Cleveland, J.L.; Mulé, J.J.; Eschrich, S.A.; et al. Tumor-immune ecosystem dynamics define an individual Radiation Immune Score to predict pan-cancer radiocurability. Neoplasia 2021, 23, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of glioma formation: Iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Makaryan, S.Z.; Cess, C.G.; Finley, S.D. Modeling immune cell behavior across scales in cancer. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1484. [Google Scholar] [CrossRef]
- Giorgadze, T.; Fischel, H.; Tessier, A.; Norton, K.A. Investigating two modes of cancer-associated antigen heterogeneity in an agent-based model of chimeric antigen receptor T-cell therapy. Cells 2022, 11, 3165. [Google Scholar] [CrossRef]
- Agur, Z.; Elishmereni, M.; Foryś, U.; Kogan, Y. Accelerating the development of personalized cancer immunotherapy by integrating molecular patients’ profiles with dynamic mathematical models. Clin. Pharmacol. Ther. 2020, 108, 515–527. [Google Scholar] [CrossRef]
- Świerniak, A.; Śmieja, J.; Fujarewicz, K.; Suwiński, R. Towards Personalized Radio-Chemotherapy—Learning from Clinical Data vs. Model Optimization. In ACIIDS 2020: Intelligent Information and Database Systems, Proceedings of the 12th Asian Conference, Phuket, Thailand, 23–26 March 2020; Nguyen, N.T., Jearanaitanakij, K., Selamat, A., Trawiński, B., Chittayasothorn, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 371–379. [Google Scholar] [CrossRef]
- Kardynska, M.; Kogut, D.; Pacholczyk, M.; Smieja, J. Mathematical modeling of regulatory networks of intracellular processes—Aims and selected methods. Comput. Struct. Biotechnol. J. 2023, 21, 1523–1532. [Google Scholar] [CrossRef]
- Enciso, J.; Pelayo, R.; Villarreal, C. From discrete to continuous modeling of lymphocyte development and plasticity in chronic diseases. Front. Immunol. 2019, 10, 1927. [Google Scholar] [CrossRef]
- Gutowska, K.; Kogut, D.; Kardynska, M.; Formanowicz, P.; Smieja, J.; Puszynski, K. Petri nets and ODEs as complementary methods for comprehensive analysis on an example of the ATM-p53-NF-[Formula: See text]B signaling pathways. Sci. Rep. 2022, 12, 1135. [Google Scholar] [CrossRef]
- Handzlik, J.E.; Loh, Y.L.; Manu. Dynamic Modeling of Transcriptional Gene Regulatory Networks. In Modeling Transcriptional Regulation: Methods and Protocols; Mukhtar, S., Ed.; Springer: New York, NY, USA, 2021; pp. 67–97. [Google Scholar] [CrossRef]
- Paszek, A.; Kardyńska, M.; Bagnall, J.; Śmieja, J.; Spiller, D.G.; Widłak, P.; Kimmel, M.; Widlak, W.; Paszek, P. Heat shock response regulates stimulus-specificity and sensitivity of the pro-inflammatory NF-κB signalling. Cell Commun. Signal. 2020, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Kalliara, E.; Kardynska, M.; Bagnall, J.; Spiller, D.G.; Müller, W.; Ruckerl, D.; Śmieja, J.; Biswas, S.K.; Paszek, P. Post-transcriptional regulatory feedback encodes JAK-STAT signal memory of interferon stimulation. Front. Immunol. 2022, 13, 947213. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Beezhold, K.; Castranova, V. Tumor promoting or tumor suppressing of NF-κB, a matter of cell context dependency. Int. Rev. Immunol. 2008, 27, 183–204. [Google Scholar] [CrossRef] [PubMed]
- Kardynska, M.; Smieja, J.; Paszek, P.; Puszynski, K. Application of Sensitivity Analysis to Discover Potential Molecular Drug Targets. Int. J. Mol. Sci. 2022, 23, 6604. [Google Scholar] [CrossRef]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting cell death pathways for cancer therapy: Recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J. Hematol. Oncol. 2022, 15, 174. [Google Scholar] [CrossRef]
- Charan, K.; Giri, A.; Kar, S. Elucidating the implications of diverse dynamical responses in p53 protein. Chemphyschem 2023, 24, e202200537. [Google Scholar] [CrossRef]
- Konrath, F.; Loewer, A.; Wolf, J. Resolving Crosstalk Between Signaling Pathways Using Mathematical Modeling and Time-Resolved Single Cell Data. In Computational Modeling of Signaling Networks; Nguyen, L.K., Ed.; Springer: New York, NY, USA, 2023; pp. 267–284. [Google Scholar] [CrossRef]
- Lee, J.; Lee, D.; Kim, Y. Mathematical model of STAT signalling pathways in cancer development and optimal control approaches. R. Soc. Open Sci. 2021, 8, 210594. [Google Scholar] [CrossRef]
- Cheemanapalli, S.; Palaniappan, C.; Mahesh, Y.; Iyyappan, Y.; Yarrappagaari, S.; Kanagaraj, S. In vitro and in silico perspectives to explain anticancer activity of a novel syringic acid analog ((4-(1H-1,3-benzodiazol-2-yl)-2,6-dimethoxy phenol)) through apoptosis activation and NFkB inhibition in K562 leukemia cells. Comput. Biol. Med. 2023, 152, 106349. [Google Scholar] [CrossRef]
- Liu, S.; Pi, J.; Zhang, Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. 2022, 54, 102389. [Google Scholar] [CrossRef]
- Hochman, G.; Halevi-Tobias, K.; Kogan, Y.; Agur, Z. Extracellular inhibitors can attenuate tumorigenic Wnt pathway activity in adenomatous polyposis coli mutants: Predictions of a validated mathematical model. PLoS ONE 2017, 12, e0179888. [Google Scholar] [CrossRef]
- Tyson, J.J.; Novák, B. Time-keeping and decision-making in the cell cycle. Interface Focus 2022, 12, 20210075. [Google Scholar] [CrossRef]
- Salgia, R.; Mambetsariev, I.; Hewelt, B.; Achuthan, S.; Li, H.; Poroyko, V.; Wang, Y.; Sattler, M. Modeling small cell lung cancer (SCLC) biology through deterministic and stochastic mathematical models. Oncotarget 2018, 9, 26226–26242. [Google Scholar] [CrossRef]
- Curtis, L.T.; Frieboes, H.B. Modeling of combination chemotherapy and immunotherapy for lung cancer. In Proceedings of the 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Berlin, Germany, 23–27 July 2019. [Google Scholar]
- Benzekry, S.; Pasquier, E.; Barbolosi, D.; Lacarelle, B.; Barlési, F.; André, N.; Ciccolini, J. Metronomic reloaded: Theoretical models bringing chemotherapy into the era of precision medicine. Semin. Cancer Biol. 2015, 35, 53–61. [Google Scholar] [CrossRef]
- Bondarenko, M.; Le Grand, M.; Shaked, Y.; Raviv, Z.; Chapuisat, G.; Carrère, C.; Montero, M.P.; Rossi, M.; Pasquier, E.; Carré, M.; et al. Metronomic chemotherapy modulates clonal interactions to prevent drug resistance in non-small cell lung cancer. Cancers 2021, 13, 2239. [Google Scholar] [CrossRef] [PubMed]
- Siewe, N.; Friedman, A. Optimal timing of steroid initiation in response to CTLA-4 antibody in metastatic cancer: A mathematical model. PLoS ONE 2022, 17, e0277248. [Google Scholar] [CrossRef] [PubMed]
- Dionysiou, D.D.; Georgios, S. Stamatakos, J. Applying a 4D Multiscale In Vivo Tumor Growth Model to the Exploration of Radiotherapy Scheduling: The Effects of Weekend Treatment Gaps and P53 Gene Status on the Response of Fast Growing Solid Tumors. Cancer Inform. 2006, 2, 117693510600200001. [Google Scholar] [CrossRef]
- Skladowski, K.; Hutnik, M.; Wygoda, A.; Golen, M.; Pilecki, B.; Przeorek, W.; Rutkowski, T.; Lukaszczyk-Widel, B.; Heyda, A.; Suwinski, R.; et al. Radiation-free weekend rescued! Continuous accelerated irradiation of 7-days per week is equal to accelerated fractionation with concomitant boost of 7 fractions in 5-days per week: Report on phase 3 clinical trial in head-and-neck cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Barbolosi, D.; Ciccolini, J.; Meille, C.; Elharrar, X.; Faivre, C.; Lacarelle, B.; Andre, N.; Barlesi, F. Metronomics chemotherapy: Time for computational decision support. Cancer Chemother. Pharmacol. 2014, 74, 647–652. [Google Scholar] [CrossRef]
- Barlesi, F.; Deyme, L.; Imbs, D.C.; Cousin, E.; Barbolosi, M.; Bonnet, S.; Tomasini, P.; Greillier, L.; Galloux, M.; Testot-Ferry, A.; et al. Revisiting metronomic vinorelbine with mathematical modelling: A Phase I trial in lung cancer. Cancer Chemother. Pharmacol. 2022, 90, 149–160. [Google Scholar] [CrossRef]
- Creemers, J.H.A.; Ankan, A.; Roes, K.C.B.; Schröder, G.; Mehra, N.; Figdor, C.G.; de Vries, I.J.M.; Textor, J. In silico cancer immunotherapy trials uncover the consequences of therapy-specific response patterns for clinical trial design and outcome. Nat. Commun. 2023, 14, 2348. [Google Scholar] [CrossRef]
- Jafarnejad, M.; Gong, C.; Gabrielson, E.; Bartelink, I.H.; Vicini, P.; Wang, B.; Narwal, R.; Roskos, L.; Popel, A.S. A computational model of neoadjuvant PD-1 inhibition in non-small cell lung cancer. AAPS J. 2019, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.; Hillen, T. Implications of immune-mediated metastatic growth on metastatic dormancy, blow-up, early detection, and treatment. J. Math. Biol. 2020, 81, 799–843. [Google Scholar] [CrossRef]
- Pennisi, M.; Pappalardo, F.; Palladini, A.; Nicoletti, G.; Nanni, P.; Lollini, P.L.; Motta, S. Modeling the competition between lung metastases and the immune system using agents. BMC Bioinform. 2010, 11 (Suppl. 7), S13. [Google Scholar] [CrossRef]
- Schlicke, P.; Kuttler, C.; Schumann, C. How mathematical modeling could contribute to the quantification of metastatic tumor burden under therapy: Insights in immunotherapeutic treatment of non-small cell lung cancer. Theor. Biol. Med. Model. 2021, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.B.; Bowar, B.; Levine, E.; Esposito, A.; Garassino, M.C.; Bestvina, C.M. Targeted toxicities: Protocols for monitoring the adverse events of targeted therapies used in the treatment of non-small cell lung cancer. Int. J. Mol. Sci. 2023, 24, 9429. [Google Scholar] [CrossRef] [PubMed]
- Check, J.H.; Poretta, T.; Check, D.; Srivastava, M. Lung cancer-standard therapy and the use of a novel, highly effective, well tolerated, treatment with progesterone receptor modulators. Anticancer Res. 2023, 43, 951–965. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smieja, J. Mathematical Modeling Support for Lung Cancer Therapy—A Short Review. Int. J. Mol. Sci. 2023, 24, 14516. https://doi.org/10.3390/ijms241914516
Smieja J. Mathematical Modeling Support for Lung Cancer Therapy—A Short Review. International Journal of Molecular Sciences. 2023; 24(19):14516. https://doi.org/10.3390/ijms241914516
Chicago/Turabian StyleSmieja, Jaroslaw. 2023. "Mathematical Modeling Support for Lung Cancer Therapy—A Short Review" International Journal of Molecular Sciences 24, no. 19: 14516. https://doi.org/10.3390/ijms241914516
APA StyleSmieja, J. (2023). Mathematical Modeling Support for Lung Cancer Therapy—A Short Review. International Journal of Molecular Sciences, 24(19), 14516. https://doi.org/10.3390/ijms241914516