
EpiPro, a Novel, Synthetic, Activity-Regulated Promoter That Targets Hyperactive Neurons in Epilepsy for Gene Therapy Applications
 , ,
, ,
Abstract
:1. Introduction
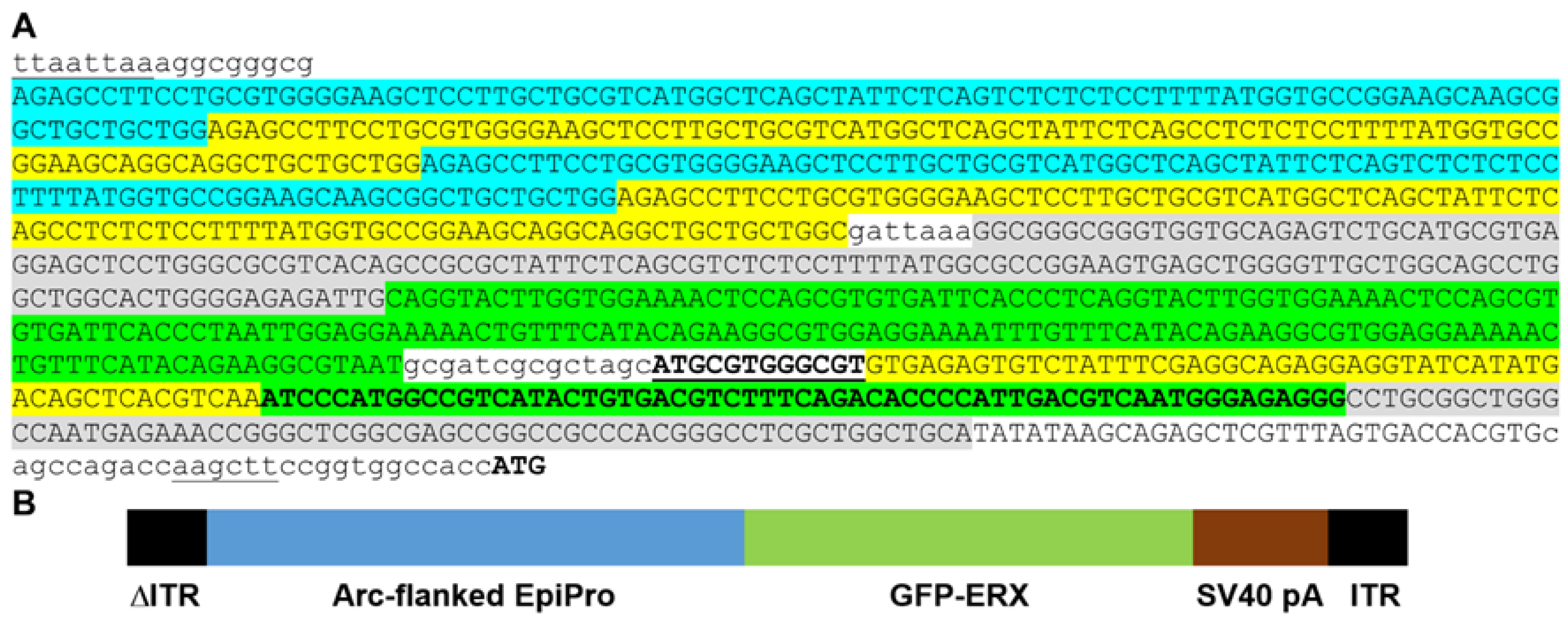
2. Results
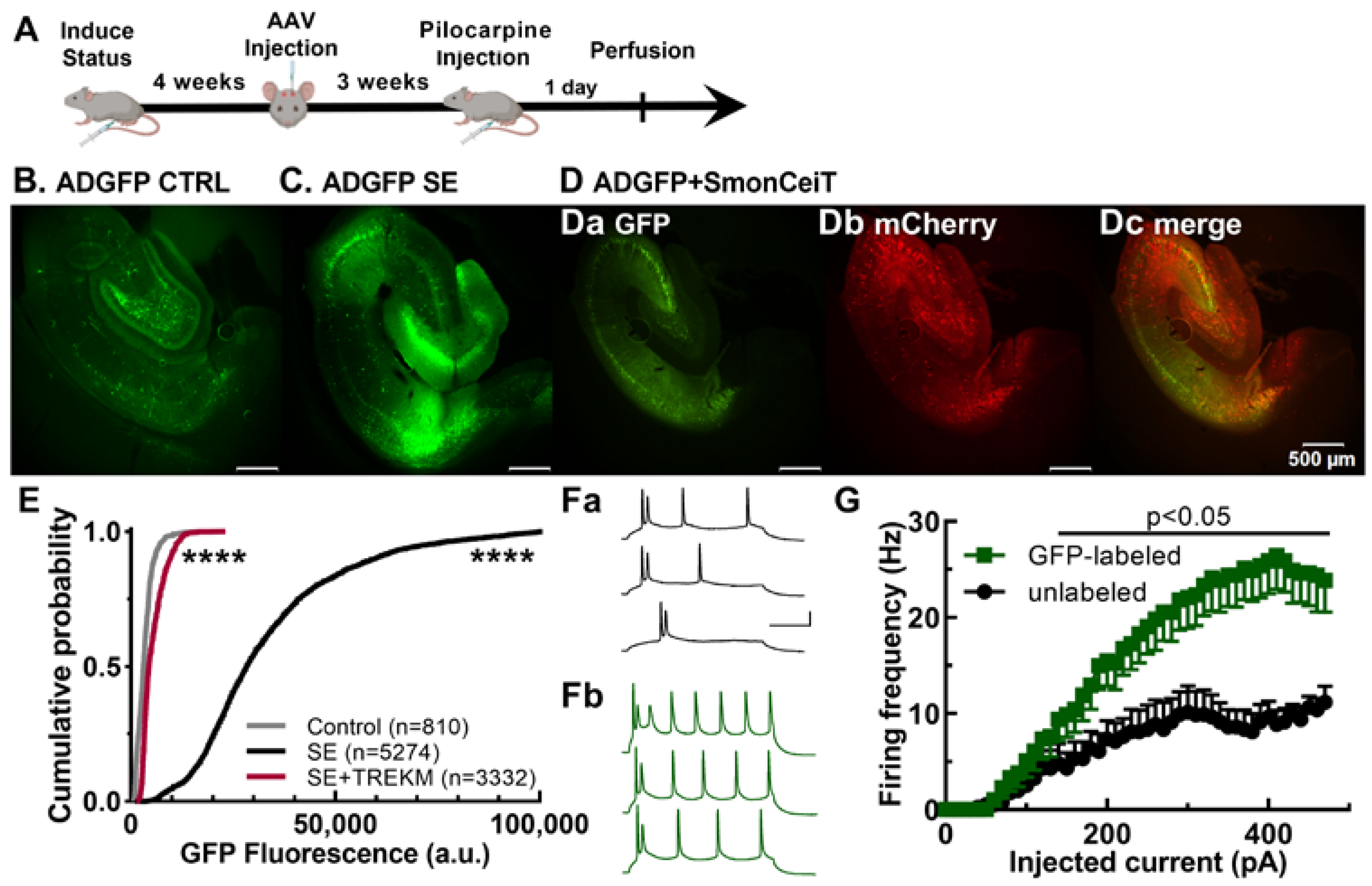
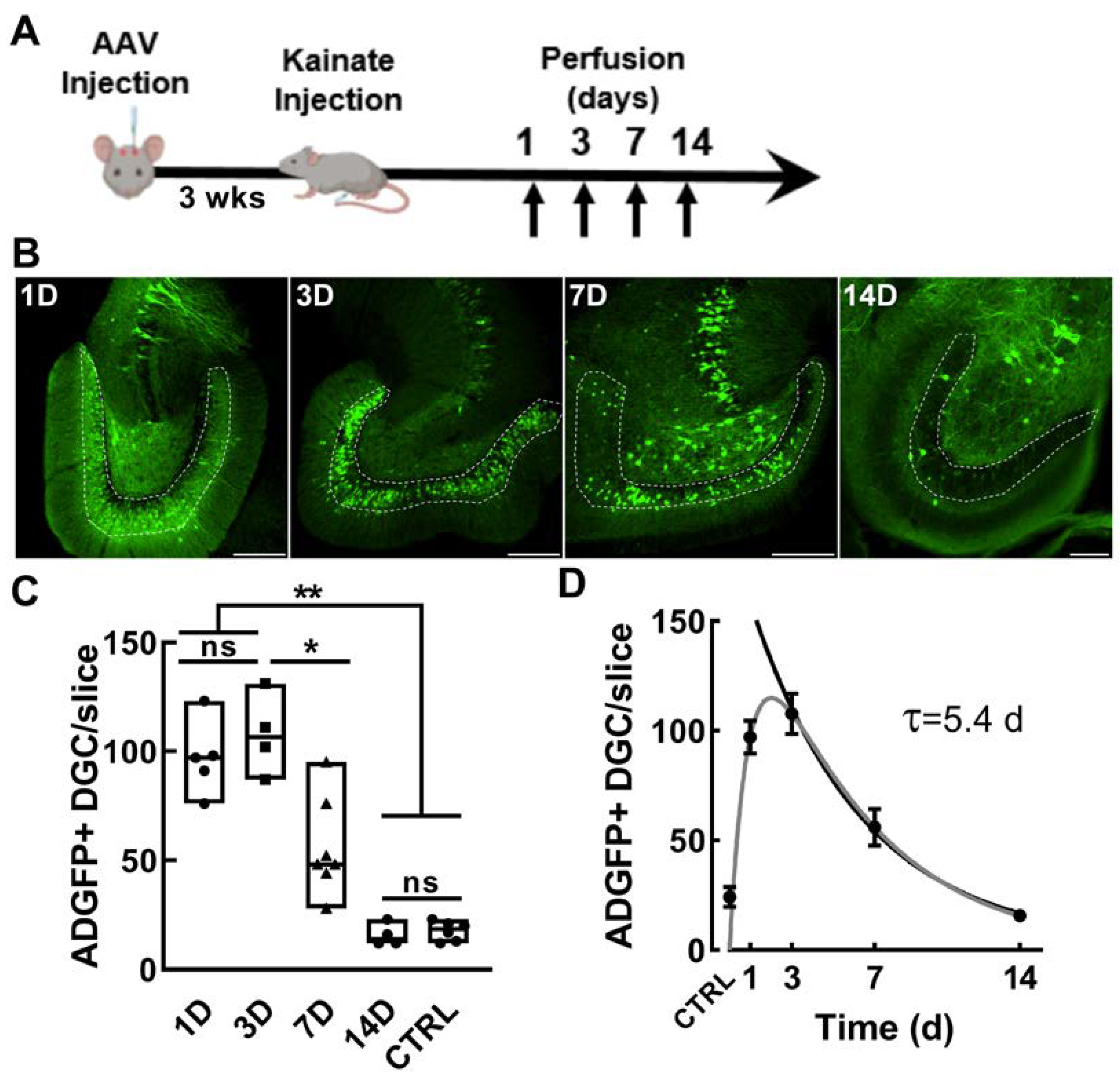
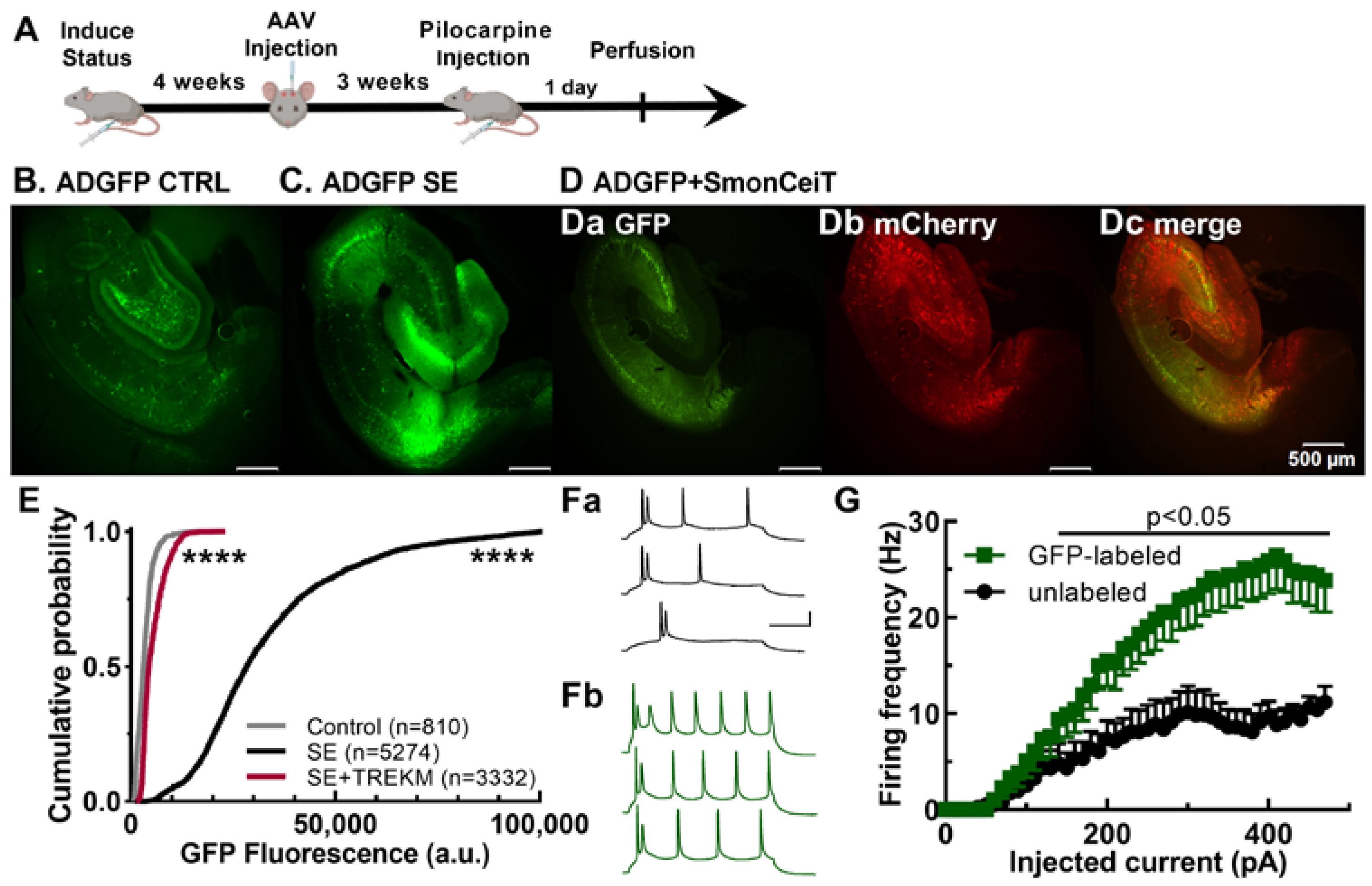
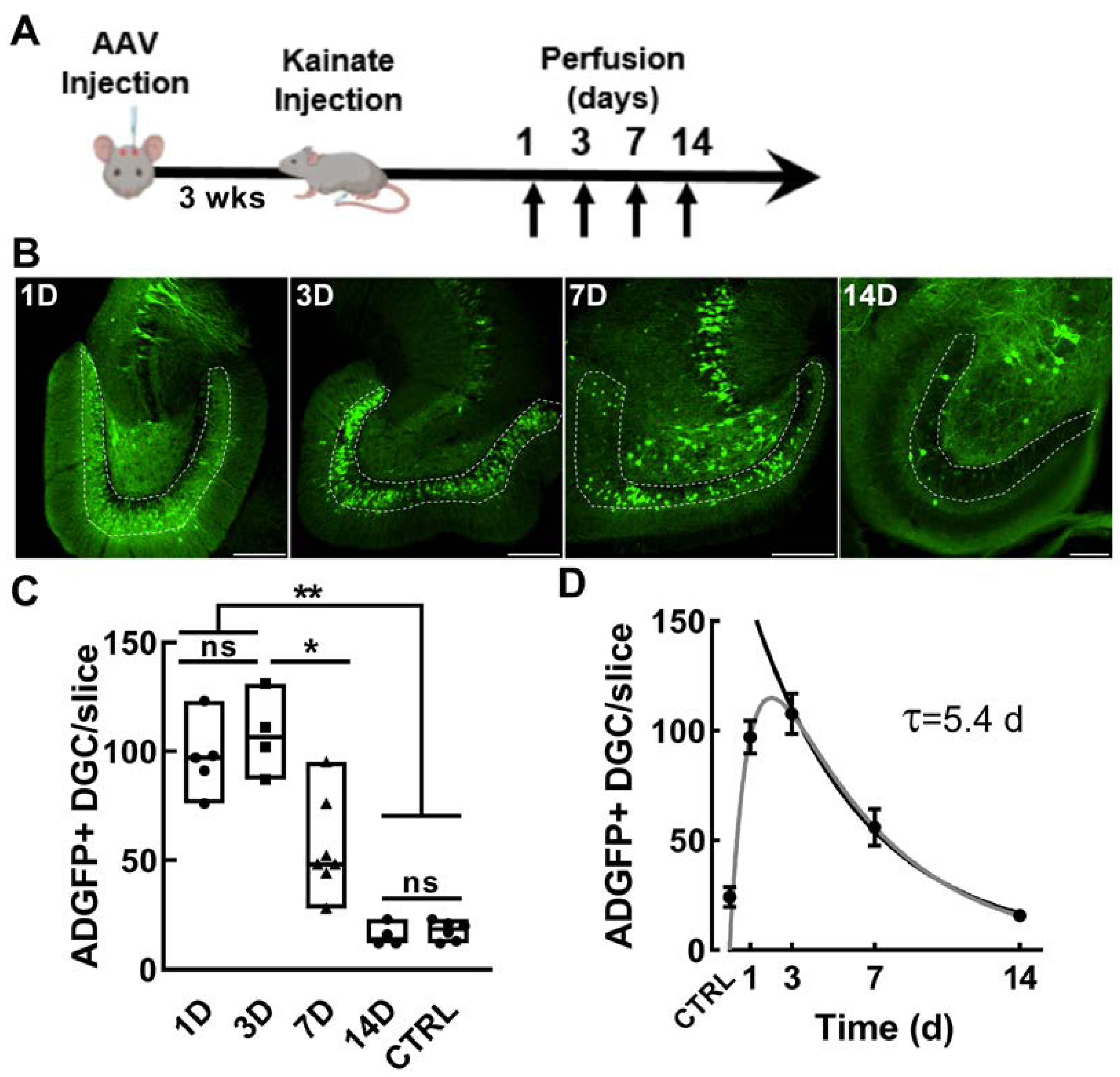
2.1. Validation of the Activity Dependence of EpiPro in Rats
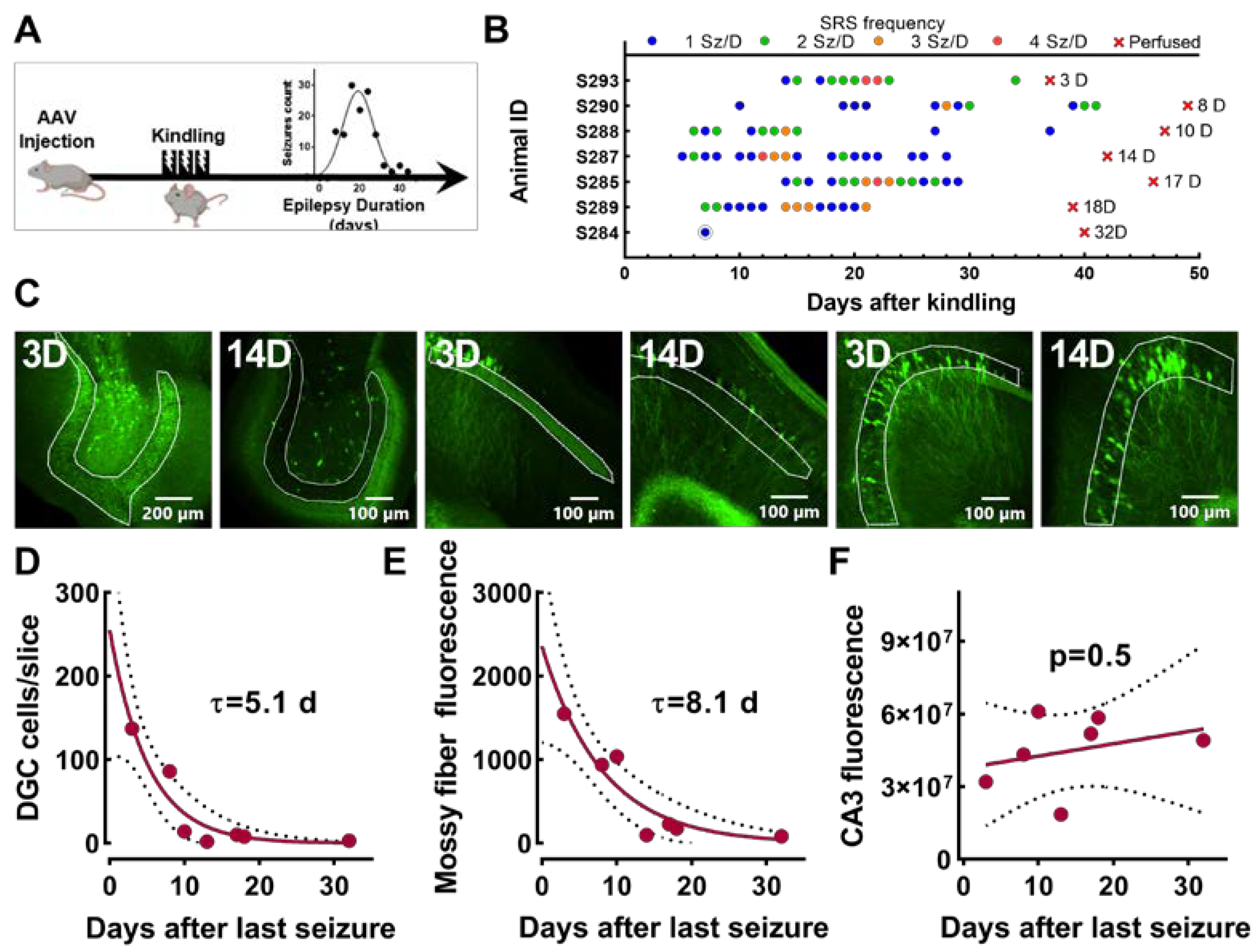
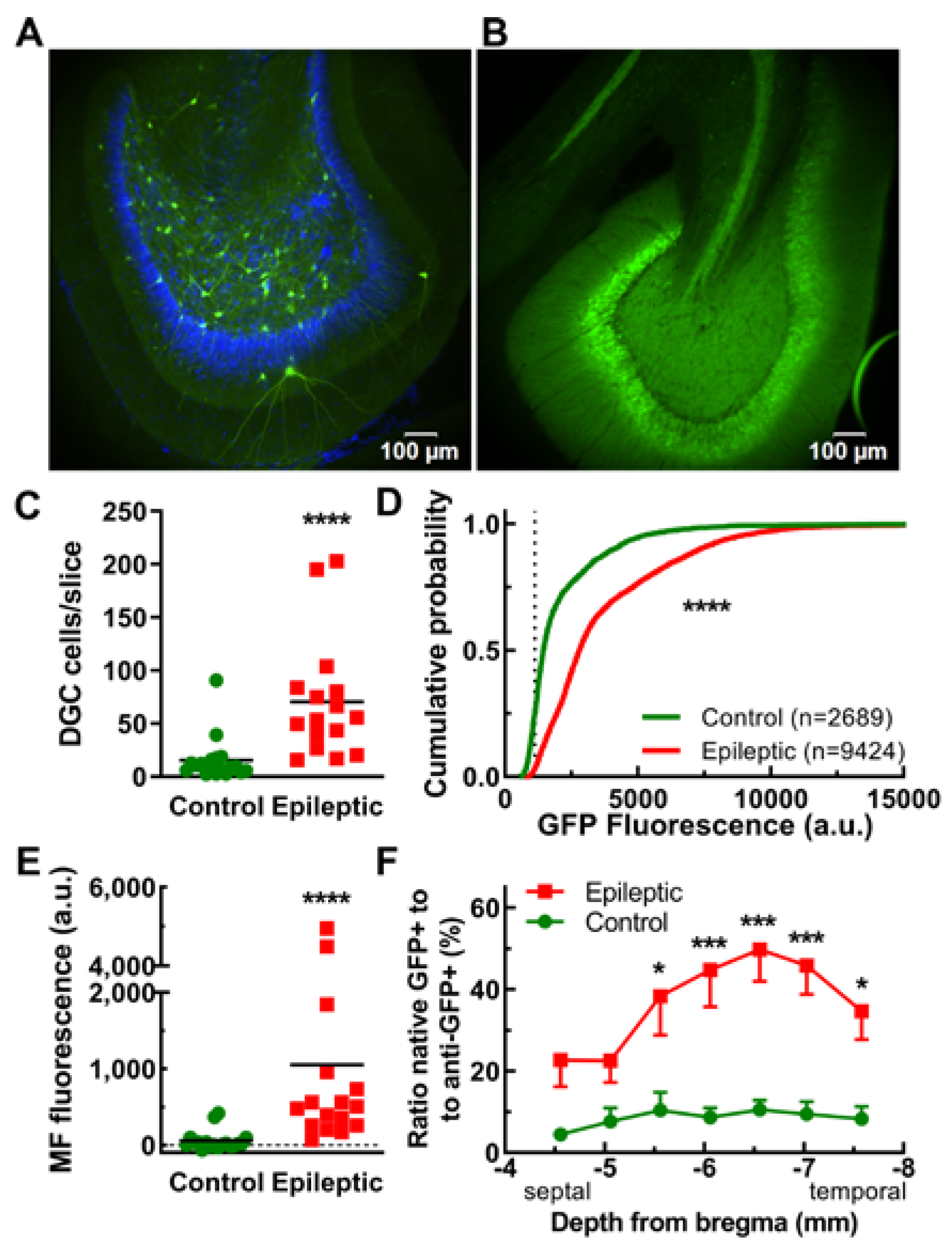
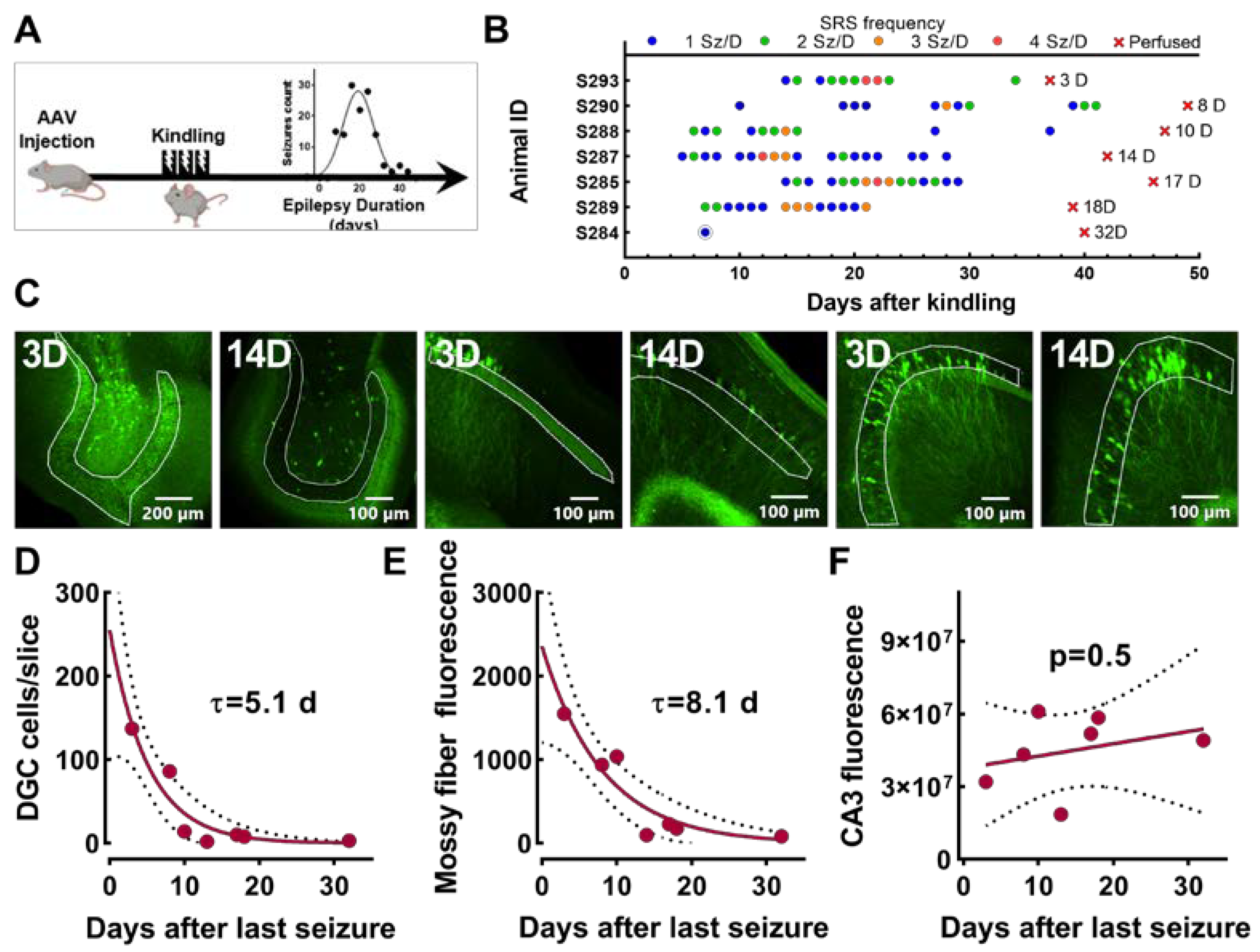
2.2. Experiments Using Epileptic Rats with Spontaneous Seizures
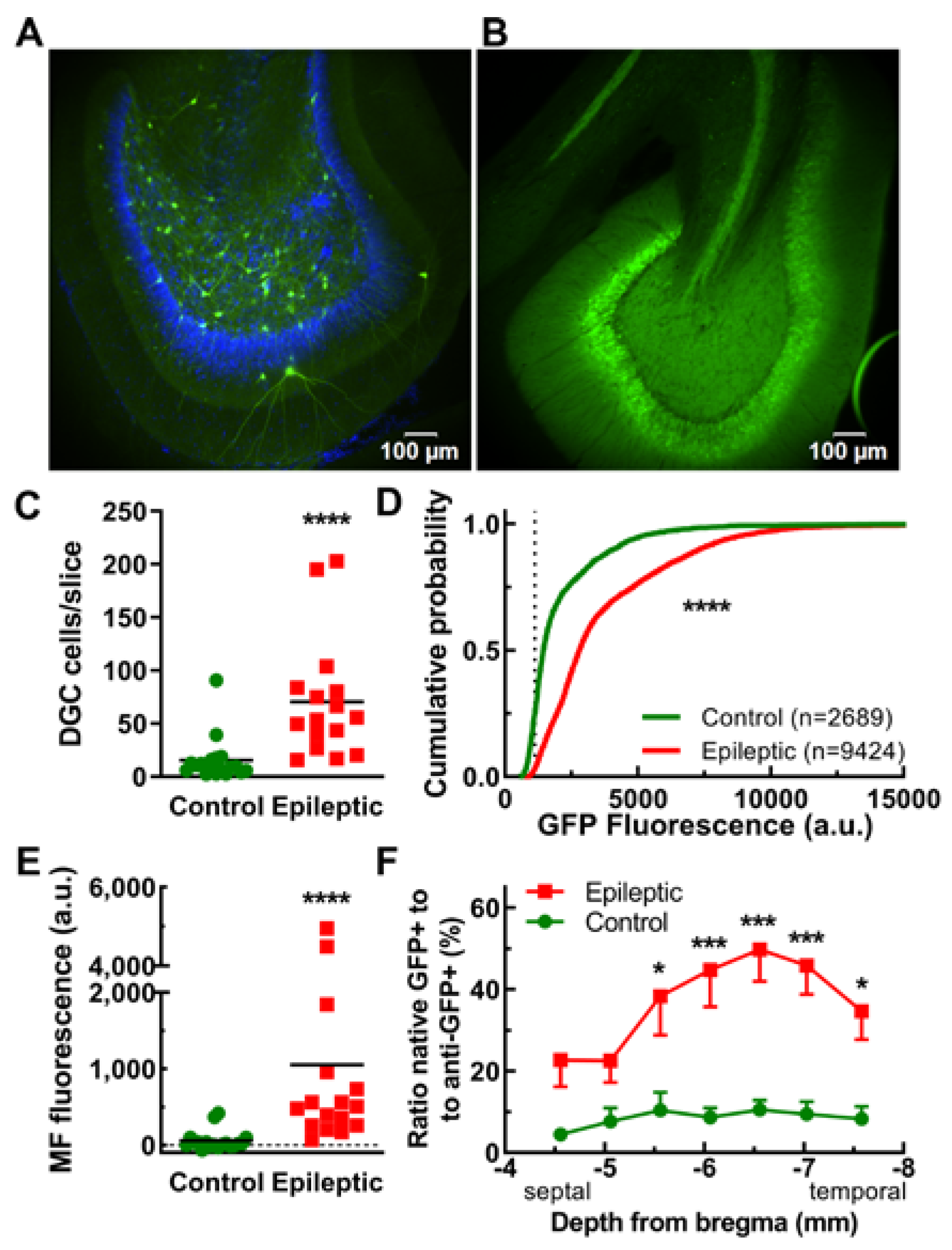
2.3. Dentate Granule Cells and Their Mossy Fiber Projections
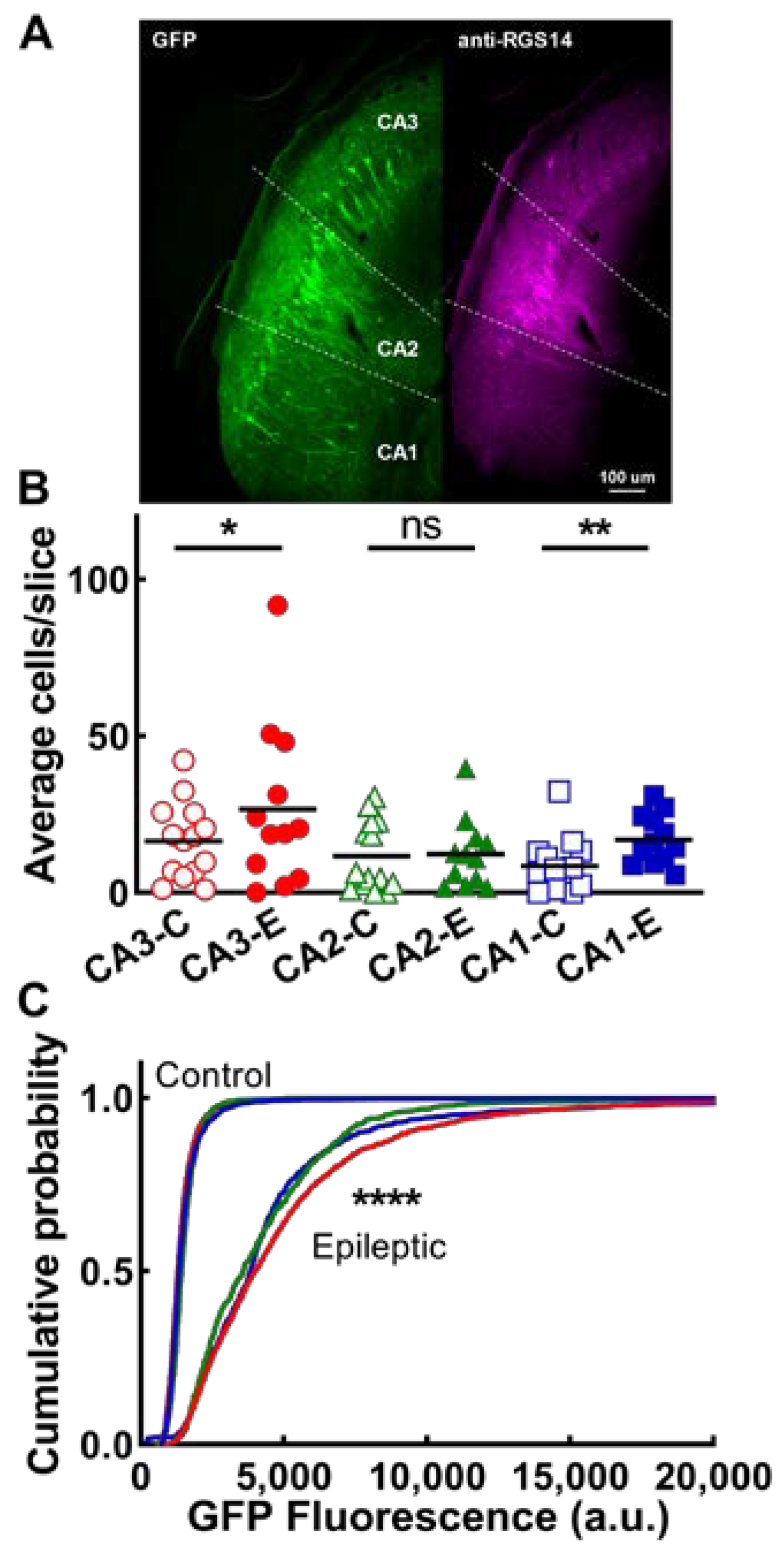
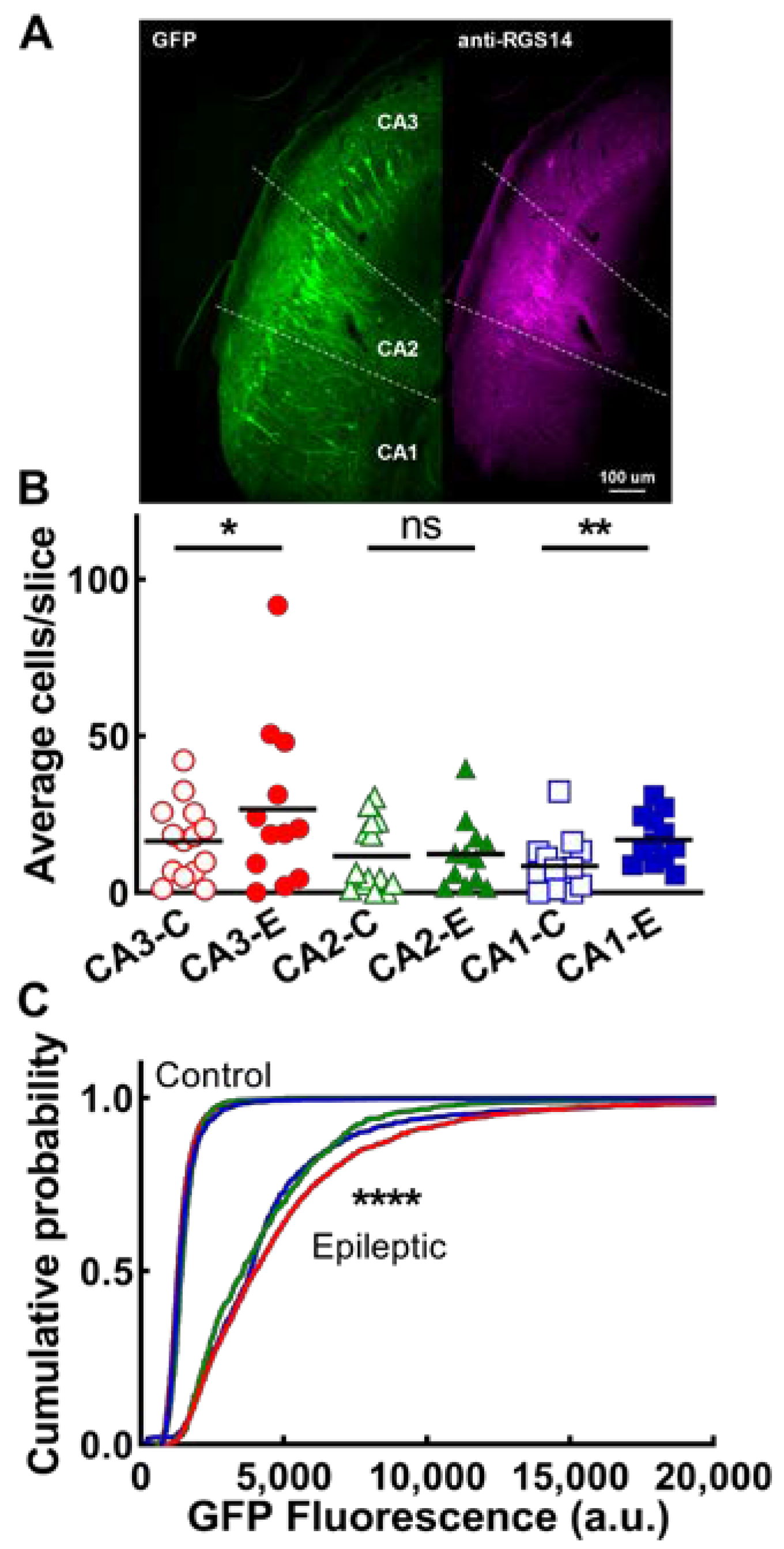
2.4. CA Neurons
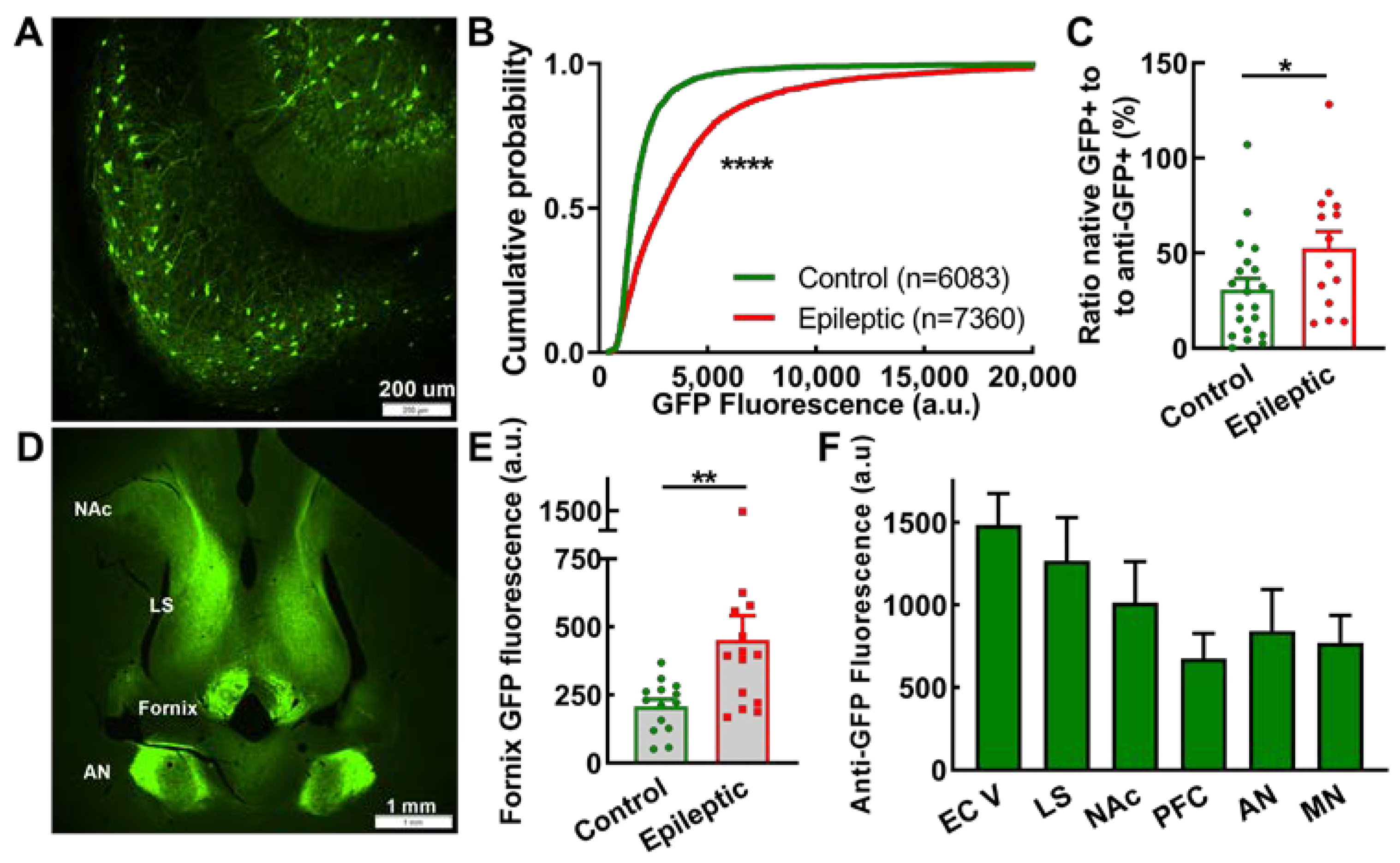
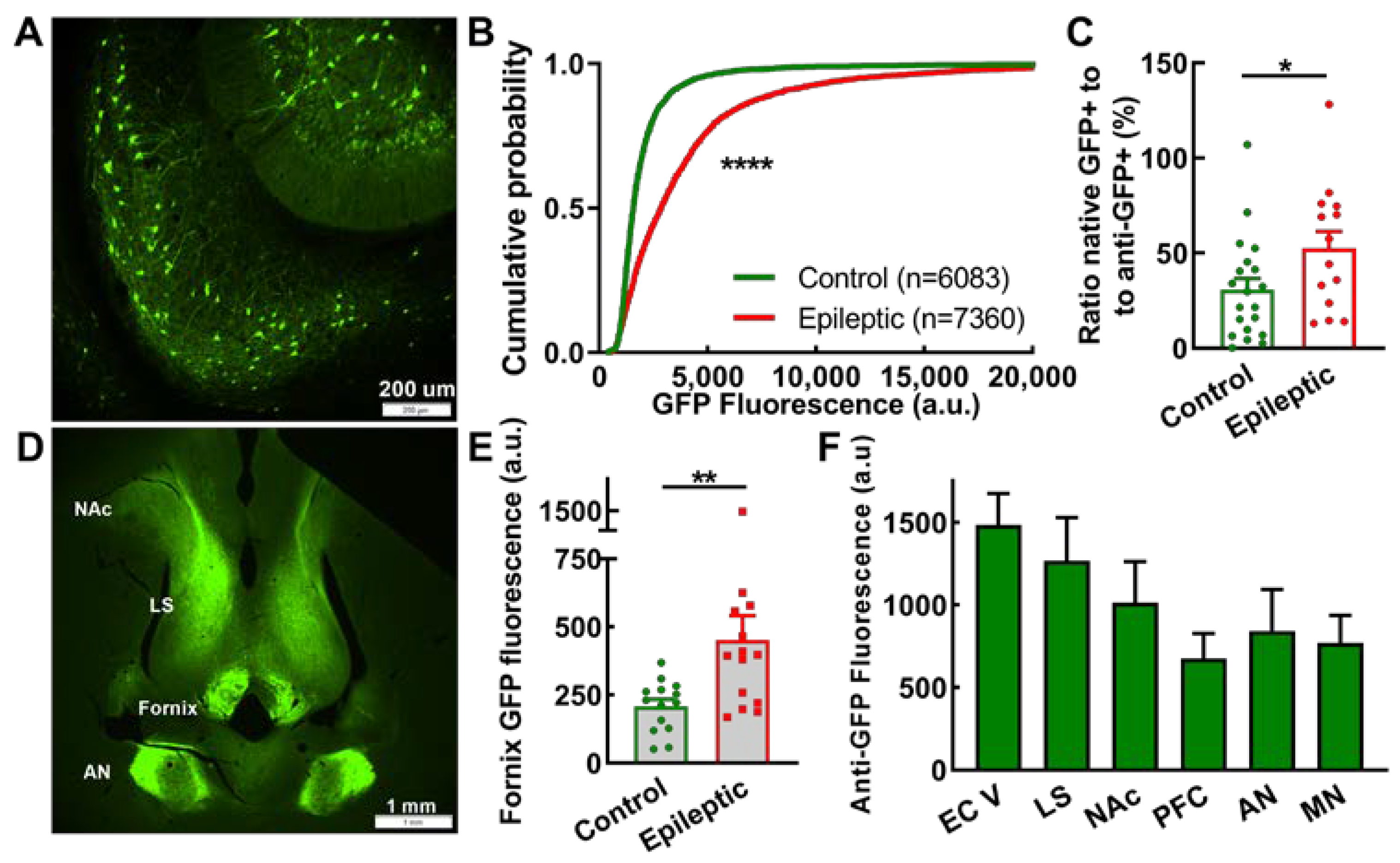
2.5. Subiculum and Projections

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal ID | AAV Injected | Seizure Status | Seizures (sz/Day) | Days after Last Seizure | Days after Inj. | DGC/ Slice | Comments on Unique Features |
|---|---|---|---|---|---|---|---|
| Experiment 1: Acute status epilepticus triggered by Li/pilocarpine treatment | |||||||
| SV234 | ADGFP + CSRH1S | control (cage) | nd | -- | 20 | 52 | |
| SV235 | ADGFP + CSRH1S | control (cage) | nd | -- | 21 | 11 | SGC, EC |
| SV236 | ADGFP + CSRH1S | control (cage) | nd | -- | 21 | 7 | SGC, P/E |
| SV229 | ADGFP | status | status | 1 | 23 | 249 | P/E |
| SV231 | ADGFP | status | status | 1 | 23 | 191 | EC, P/E |
| SV230 | ADGFP + SmonCeiT | status | status | 1 | 22 | 109 | GFP low in TREK-M-infected cells |
| SV232 | ADGFP + SmonCeiT | status | status | 1 | 22 | 65 | GFP low in TREK-M-infected cells |
| Experiment 2: Chronic TLE, AAV injected before Li/pilocarpine treatment | |||||||
| SV273 | ADGFP | control (cage) | nd | nd | 43 | 13 | SGC |
| SV249 | ADGFP | epileptic | nd | nd | 53 | 43 | Extensive HS, post-fixed |
| SV250 | ADGFP | epileptic | nd | nd | 57 | 65 | Extensive HS, P/E |
| SV265 | ADGFP | epileptic | nd | 1 | 65 | 164 | Extensive HS, EC |
| Experiment 3: Chronic TLE, AAV injected into spontaneously seizing rats weeks after Li/pilocarpine treatment | |||||||
| BV311 | ADGFP | sz unknown cause | 5 (1) | 1 | 25 | 155 | Excluded neither control nor epileptic |
| BV312 | ADGFP | control (video) | 0 | -- | 32 | 11 | SGC, OML, P/E |
| BV315 | ADGFP | control (video) | 0 | -- | 103 | nd | Used for EPhys, not imaged |
| BV316 | ADGFP | control (video) | 0 | -- | 104 | nd | Used for EPhys, not imaged |
| BV322 | ADGFP | control (video) | 0 | -- | 55 | 21 | P/E, OML |
| BV323 | ADGFP | control (video) | 0 | -- | 60 | 5 | SGC, EC |
| BV324 | ADGFP | control (video) | 0 | -- | 60 | 7 | P/E, OML |
| BV340 | ADGFP | control (video) | 0 | -- | 27 | 2 | GFP+ glia at injection site |
| BV341 | ADGFP | control (video) | 0 | -- | 27 | 9 | Highly active subicular neurons |
| BV297 | ADGFP | epileptic | 5 (0.4) | 1 | 31 | 48 | Extensive HS, P/E, EC |
| BV302 | ADGFP | epileptic | 6 (0.5) | 3 | 34 | 79 | Excluded due to >50% atrophy |
| BV304 | ADGFP | single Sz | 1 (0.1) | na | 54 | 75 | Lowest Sz. freq., no HS, GCD, EC |
| BV305A | ADGFP | epileptic | 2 (0.3) | 4 | 21 | nd | Excluded due to >50% atrophy |
| BV305B | ADGFP | epileptic | 6 (0.8) | 1 | 28 | 40 | Extensive HS, P/E, OML, EC |
| BV306B | ADGFP | epileptic | 5 (1.0) | 1 | 24 | 40 | Extensive HS, EC |
| BV307 | ADGFP | epileptic | 89 (11) | 1 | 27 | 88 | Extensive HS, P/E, OML, EC |
| BV309 | ADGFP | epileptic | 14 (2.8) | 1 | 21 | 74 | Minimal HS, P/E |
2.6. Measuring the Time Course of Activation of EpiPro after a Single Seizure
2.7. Experiment Measuring EpiPro Activity after Mice Stop Having Chronic Seizures
3. Discussion
3.1. Validation of the Activity Dependence of EpiPro
3.2. EpiPro as a Biomarker for the Breakdown of the Dentate Gate
3.3. Exploration of Epileptic Circuits
3.4. Activation and Deactivation of EpiPro Promoter Activity
3.5. Potential Utility of EpiPro as a Gene Therapy Driver
4. Materials and Methods
4.1. Experimental Design
4.2. Cloning of the AAV Targeting Vectors
4.3. Lithium/Pilocarpine Treatment and Seizure Monitoring
4.4. Stereotaxic AAV Injection
4.5. Perfusion and Brain Slicing
4.6. Imaging and Analysis
4.7. Immunohistochemistry
4.8. Brain Slice Electrophysiology
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Devinsky, O. Diagnosis and treatment of temporal lobe epilepsy. Rev. Neurol. Dis. 2004, 1, 2–9. [Google Scholar] [PubMed]
- Asadi-Pooya, A.A.; Stewart, G.R.; Abrams, D.J.; Sharan, A. Prevalence and incidence of drug-resistant mesial temporal lobe epilepsy in the United States. World Neurosurg. 2017, 99, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, T.; Dudley, R.W.; Hasan, A.; Al Jishi, A.; Al Hinai, Q.; Poulin, N.; Colnat-Coulbois, S.; Olivier, A. Memory outcome after temporal lobe epilepsy surgery: Corticoamygdalohippocampectomy versus selective amygdalohippocampectomy. J. Neurosurg. 2010, 113, 1164–1175. [Google Scholar] [CrossRef]
- Shaimardanova, A.A.; Chulpanova, D.S.; Mullagulova, A.I.; Afawi, Z.; Gamirova, R.G.; Solovyeva, V.V.; Rizvanov, A.A. Gene and cell therapy for epilepsy: A mini review. Front. Mol. Neurosci. 2022, 15, 868531. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; O’Neill, N.; Maffei, B.; Zourray, C.; Almacellas-Barbanoj, A.; Carpenter, J.C.; Jones, S.P.; Leite, M.; Turner, T.J.; Moreira, F.C.; et al. On-demand cell-autonomous gene therapy for brain circuit disorders. Science 2022, 378, 523–532. [Google Scholar] [CrossRef]
- Sorensen, A.T.; Cooper, Y.A.; Baratta, M.V.; Weng, F.J.; Zhang, Y.; Ramamoorthi, K.; Fropf, R.; LaVerriere, E.; Xue, J.; Young, A.; et al. A robust activity marking system for exploring active neuronal ensembles. Elife 2016, 5, e13918. [Google Scholar] [CrossRef]
- Beaumont, T.L.; Yao, B.; Shah, A.; Kapatos, G.; Loeb, J.A. Layer-specific CREB target gene induction in human neocortical epilepsy. J. Neurosci. 2012, 32, 14389–14401. [Google Scholar] [CrossRef]
- Rakhade, S.N.; Shah, A.K.; Agarwal, R.; Yao, B.; Asano, E.; Loeb, J.A. Activity-dependent gene expression correlates with interictal spiking in human neocortical epilepsy. Epilepsia 2007, 48 (Suppl. 5), 86–95. [Google Scholar] [CrossRef]
- Xiang, G.; Pan, L.; Xing, W.; Zhang, L.; Huang, L.; Yu, J.; Zhang, R.; Wu, J.; Cheng, J.; Zhou, Y. Identification of activity-dependent gene expression profiles reveals specific subsets of genes induced by different routes of Ca2+ entry in cultured rat cortical neurons. J. Cell. Physiol. 2007, 212, 126–136. [Google Scholar] [CrossRef]
- Stanley, S.A.; Gagner, J.E.; Damanpour, S.; Yoshida, M.; Dordick, J.S.; Friedman, J.M. Radio-wave heating of iron oxide nanoparticles can regulate plasma glucose in mice. Science 2012, 336, 604–608. [Google Scholar] [CrossRef]
- Roberts, D.S.; Raol, Y.H.; Bandyopadhyay, S.; Lund, I.V.; Budreck, E.C.; Passini, M.A.; Wolfe, J.H.; Brooks-Kayal, A.R.; Russek, S.J. Egr3 stimulation of GABRA4 promoter activity as a mechanism for seizure-induced up-regulation of GABAA receptor a4 subunit expression. Proc. Natl. Acad. Sci. USA 2005, 102, 11894–11899. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Okuno, H.; Nonaka, M.; Adachi-Morishima, A.; Kyo, N.; Okamura, M.; Takemoto-Kimura, S.; Worley, P.F.; Bito, H. Synaptic activity-responsive element in the Arc/Arg3.1 promoter essential for synapse-to-nucleus signaling in activated neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 316–321. [Google Scholar] [CrossRef]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.L.; Dayton, R.D.; Tatom, J.B.; Henderson, K.M.; Henning, P.P. AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: Effects of serotype, promoter and purification method. Mol. Ther. 2008, 16, 89–96. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M.; Fu, H.; Monahan, P.E.; Toulson, C.E.; Naik, P.; Samulski, R.J. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003, 10, 2112–2118. [Google Scholar] [CrossRef]
- Shi, Y.; Abe, C.; Holloway, B.B.; Shu, S.; Kumar, N.N.; Weaver, J.L.; Sen, J.; Perez-Reyes, E.; Stornetta, R.L.; Guyenet, P.G.; et al. Nalcn Is a “leak” sodium channel that regulates excitability of brainstem chemosensory neurons and breathing. J. Neurosci. 2016, 36, 8174–8187. [Google Scholar] [CrossRef]
- Lonser, R.R.; Sarntinoranont, M.; Morrison, P.F.; Oldfield, E.H. Convection-enhanced delivery to the central nervous system. J. Neurosurg. 2015, 122, 697–706. [Google Scholar] [CrossRef]
- Gradinaru, V.; Zhang, F.; Ramakrishnan, C.; Mattis, J.; Prakash, R.; Diester, I.; Goshen, I.; Thompson, K.R.; Deisseroth, K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell 2010, 141, 154–165. [Google Scholar] [CrossRef]
- Kawashima, T.; Kitamura, K.; Suzuki, K.; Nonaka, M.; Kamijo, S.; Takemoto-Kimura, S.; Kano, M.; Okuno, H.; Ohki, K.; Bito, H. Functional labeling of neurons and their projections using the synthetic activity-dependent promoter E-SARE. Nat. Methods 2013, 10, 889–895. [Google Scholar] [CrossRef]
- Corish, P.; Tyler-Smith, C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999, 12, 1035–1040. [Google Scholar] [CrossRef]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D. The dentate gyrus as a filter or gate: A look back and a look ahead. Prog. Brain Res. 2007, 163, 601–613. [Google Scholar] [CrossRef]
- Dengler, C.G.; Coulter, D.A. Normal and epilepsy-associated pathologic function of the dentate gyrus. Prog. Brain Res. 2016, 226, 155–178. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Buckmaster, P.S. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J. Neurosci. 2003, 23, 2440–2452. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.; Gawda, A.; Ravichandran, P.; McGrew, B.; Nylund, E.; Kang, J.; Burke, C.; Vitko, I.; Scott, M.; Williamson, J.; et al. Characterization of kindled VGAT-Cre mice as a new animal model of temporal lobe epilepsy. Epilepsia 2020, 61, 2277–2288. [Google Scholar] [CrossRef]
- Gibbs-Shelton, S.; Benderoth, J.; Gaykema, R.P.; Straub, J.; Okojie, K.A.; Uweru, J.O.; Lentferink, D.H.; Rajbanshi, B.; Cowan, M.N.; Patel, B.; et al. Microglia play beneficial roles in multiple experimental seizure models. Glia 2023, 71, 1699–1714. [Google Scholar] [CrossRef]
- Miyakawa, H.; Woo, S.K.; Dahl, S.C.; Handler, J.S.; Kwon, H.M. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc. Natl. Acad. Sci. USA 1999, 96, 2538–2542. [Google Scholar] [CrossRef]
- Loew, R.; Heinz, N.; Hampf, M.; Bujard, H.; Gossen, M. Improved Tet-responsive promoters with minimized background expression. BMC Biotechnol. 2010, 10, 81. [Google Scholar] [CrossRef]
- Ma, D.; Zerangue, N.; Lin, Y.F.; Collins, A.; Yu, M.; Jan, Y.N.; Jan, L.Y. Role of ER export signals in controlling surface potassium channel numbers. Science 2001, 291, 316–319. [Google Scholar] [CrossRef]
- Dey, D.; Eckle, V.-S.; Vitko, I.; Sullivan, K.A.; Lasiecka, Z.M.; Winckler, B.; Stornetta, R.L.; Williamson, J.M.; Kapur, J.; Perez-Reyes, E. A potassium leak channel silences hyperactive neurons and ameliorates status epilepticus. Epilepsia 2014, 55, 203–213. [Google Scholar] [CrossRef]
- Lim, J.A.; Moon, J.; Kim, T.J.; Jun, J.S.; Park, B.; Byun, J.I.; Sunwoo, J.S.; Park, K.I.; Lee, S.T.; Jung, K.H.; et al. Clustering of spontaneous recurrent seizures separated by long seizure-free periods: An extended video-EEG monitoring study of a pilocarpine mouse model. PLoS ONE 2018, 13, e0194552. [Google Scholar] [CrossRef]
- Sullivan, K.A.; Vitko, I.; Blair, K.; Gaykema, R.P.; Failor, M.J.; San Pietro, J.M.; Dey, D.; Williamson, J.M.; Stornetta, R.L.; Kapur, J.; et al. Drug-Inducible Gene Therapy Effectively Reduces Spontaneous Seizures in Kindled Rats but Creates Off-Target Side Effects in Inhibitory Neurons. Int. J. Mol. Sci. 2023, 24, 11347. [Google Scholar] [CrossRef]
- Barker, B.S.; Nigam, A.; Ottolini, M.; Gaykema, R.P.; Hargus, N.J.; Patel, M.K. Pro-excitatory alterations in sodium channel activity facilitate subiculum neuron hyperexcitability in temporal lobe epilepsy. Neurobiol. Dis. 2017, 108, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Simons, S.B.; Heldt, S.A.; Zhao, M.; Schroeder, J.P.; Vellano, C.P.; Cowan, D.P.; Ramineni, S.; Yates, C.K.; Feng, Y.; et al. RGS14 is a natural suppressor of both synaptic plasticity in CA2 neurons and hippocampal-based learning and memory. Proc. Natl. Acad. Sci. USA 2010, 107, 16994–16998. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.; Morris, R.; Amaral, D.; Bliss, T.; O’Keefe, J. The Hippocampus Book; Oxford University Press: Oxford, UK; New York, NY, USA, 2007; p. 832. [Google Scholar]
- Aggleton, J.P.; Christiansen, K. The subiculum: The heart of the extended hippocampal system. Prog. Brain Res. 2015, 219, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, I.; Bower, M.R.; Leyva, F.; Buckmaster, P.S. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J. Neurosci. 2013, 33, 11100–11115. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Pearce, P.S.; Friedman, D.; Lafrancois, J.J.; Iyengar, S.S.; Fenton, A.A.; Maclusky, N.J.; Scharfman, H.E. Spike-wave discharges in adult Sprague-Dawley rats and their implications for animal models of temporal lobe epilepsy. Epilepsy Behav. 2014, 32, 121–131. [Google Scholar] [CrossRef]
- Lothman, E.W.; Collins, R.C. Kainic acid induced limbic seizures: Metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res. 1981, 218, 299–318. [Google Scholar] [CrossRef]
- West, A.E.; Greenberg, M.E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 2011, 3, a005744. [Google Scholar] [CrossRef]
- Delorenzo, R.J.; Sun, D.A.; Deshpande, L.S. Cellular mechanisms underlying acquired epilepsy: The calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol. Ther. 2005, 105, 229–266. [Google Scholar] [CrossRef]
- Xia, Z.; Refsdal, C.D.; Merchant, K.M.; Dorsa, D.M.; Storm, D.R. Distribution of mRNA for the calmodulin-sensitive adenylate cyclase in rat brain: Expression in areas associated with learning and memory. Neuron 1991, 6, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Kugler, S.; Kilic, E.; Bahr, M. Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 2003, 10, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Dittgen, T.; Nimmerjahn, A.; Komai, S.; Licznerski, P.; Waters, J.; Margrie, T.W.; Helmchen, F.; Denk, W.; Brecht, M.; Osten, P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 18206–18211. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Yaari, Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat. Rev. Neurosci. 2008, 9, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S.; Bumanglag, A.V.; Schwarcz, R.; Frotscher, M. Abnormal dentate gyrus network circuitry in temporal lobe epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; Bethesda: Rockville, MD, USA, 2012. [Google Scholar]
- Weinberg, M.S.; Blake, B.L.; Samulski, R.J.; McCown, T.J. The influence of epileptic neuropathology and prior peripheral immunity on CNS transduction by rAAV2 and rAAV5. Gene Ther. 2011, 18, 961–968. [Google Scholar] [CrossRef]
- Peltola, J.; Colon, A.J.; Pimentel, J.; Coenen, V.A.; Gil-Nagel, A.; Gonçalves Ferreira, A.; Lehtimäki, K.; Ryvlin, P.; Taylor, R.S.; Ackermans, L.; et al. Deep Brain Stimulation of the Anterior Nucleus of the Thalamus in Drug-Resistant Epilepsy in the MORE Multicenter Patient Registry. Neurology 2023, 100, e1852–e1865. [Google Scholar] [CrossRef]
- Peng, Z.; Houser, C.R. Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J. Neurosci. 2005, 25, 7210–7220. [Google Scholar] [CrossRef]
- Reimsnider, S.; Manfredsson, F.P.; Muzyczka, N.; Mandel, R.J. Time course of transgene expression after intrastriatal pseudotyped rAAV2/1, rAAV2/2, rAAV2/5, and rAAV2/8 transduction in the rat. Mol. Ther. 2007, 15, 1504–1511. [Google Scholar] [CrossRef]
- Manfredsson, F.P.; Burger, C.; Rising, A.C.; Zuobi-Hasona, K.; Sullivan, L.F.; Lewin, A.S.; Huang, J.; Piercefield, E.; Muzyczka, N.; Mandel, R.J. Tight long-term dynamic doxycycline responsive nigrostriatal GDNF using a single rAAV vector. Mol. Ther. 2009, 17, 1857–1867. [Google Scholar] [CrossRef]
- Costantini, I.; Ghobril, J.P.; Di Giovanna, A.P.; Allegra Mascaro, A.L.; Silvestri, L.; Mullenbroich, M.C.; Onofri, L.; Conti, V.; Vanzi, F.; Sacconi, L.; et al. A versatile clearing agent for multi-modal brain imaging. Sci. Rep. 2015, 5, 9808. [Google Scholar] [CrossRef]
- Scharfman, H.E.; Myers, C.E. Hilar mossy cells of the dentate gyrus: A historical perspective. Front. Neural Circuits 2012, 6, 106. [Google Scholar] [CrossRef]
- Volz, F.; Bock, H.H.; Gierthmuehlen, M.; Zentner, J.; Haas, C.A.; Freiman, T.M. Stereologic estimation of hippocampal GluR2/3- and calretinin-immunoreactive hilar neurons (presumptive mossy cells) in two mouse models of temporal lobe epilepsy. Epilepsia 2011, 52, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Seress, L.; Ábrahám, H.; Czéh, B.; Fuchs, E.; Léránth, C. Calretinin expression in hilar mossy cells of the hippocampal dentate gyrus of nonhuman primates and humans. Hippocampus 2008, 18, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F.; Buzsáki, G. Interneurons of the hippocampus. Hippocampus 1996, 6, 347–470. [Google Scholar] [CrossRef]
- Straub, J.; Vitko, I.; Gaykema, R.P.; Perez-Reyes, E. Preparation and Implantation of Electrodes for Electrically Kindling VGAT-Cre Mice to Generate a Model for Temporal Lobe Epilepsy. J. Vis. Exp. 2021, 174, e62929. [Google Scholar] [CrossRef]
- Park, J.; Yu, Y.P.; Zhou, C.-Y.; Li, K.-W.; Wang, D.; Chang, E.; Kim, D.-S.; Vo, B.; Zhang, X.; Gong, N.; et al. Central Mechanisms Mediating Thrombospondin-4-induced Pain States. J. Biol. Chem. 2016, 291, 13335–13348. [Google Scholar] [CrossRef] [PubMed]
| Animal ID | AAV Injected | Seizure Status | Sz/Day | Perf. after Last Sz | Perf. after inj. | DGC/Slice |
|---|---|---|---|---|---|---|
| Controls | ||||||
| G054 | ADGFP | control | na | na | 18 | 7 |
| G055 | ADGFP | control | na | na | 19 | 15 |
| G086 | ADGFP | control | na | na | 69 | 23 |
| G092 | ADGFP | control | na | na | 64 | 6 |
| S179 | ADGFP + CSRH1S | control | na | na | 34 | 25 |
| S180 | ADGFP + CSRH1S | control | na | na | 34 | 41 |
| S181 | ADGFP + CSRH1S | control | na | na | 33 | 31 |
| S183 | ADGFP + CSRH1S | control | na | na | 33 | 25 |
| S184 | ADGFP + CSRH1S | control | na | na | 32 | 45 |
| Experiment 1: Chronic epilepsy triggered by electrical kindling | ||||||
| G051 | ADGFP | K + SRS | 2.3 | 1 | 54 | 73 |
| G053 | ADGFP | K + SRS | 2.6 | 1 | 43 | 30 |
| G084 | ADGFP | K + SRS | 1.1 | 0 | 71 | 65 |
| G091 | ADGFP | K + SRS | 0.2 | 3 | 59 | 109 |
| G093 | ADGFP | K + SRS | nd | 2 | 56 | 55 |
| G095 | ADGFP | K + SRS | 3.4 | 1 | 55 | 64 |
| Experiment 2: Time course after single kainate-induced seizure | ||||||
| S136 | ADGFP + CSRH1S | 10 mg KA | na | 1 | 22 | 110 |
| S138 | ADGFP + CSRH1S | 15 mg KA | na | 1 | 21 | 94 |
| S143 | ADGFP + CSRH1S | 10 mg KA | na | 1 | 18 | 76 |
| S122 | ADGFP + CSRH1S | 17 mg KA | na | 3 | 66 | 87 |
| S123 | ADGFP + CSRH1S | 10 mg KA | na | 3 | 65 | 116 |
| S127 | ADGFP + CSRH1S | 15 mg KA | na | 3 | 62 | 111 |
| S137 | ADGFP + CSRH1S | 10 mg KA | na | 7 | 27 | 85 |
| S139 | ADGFP + CSRH1S | 10 mg KA | na | 7 | 26 | 50 |
| S140 | ADGFP + CSRH1S | 10 mg KA | na | 7 | 26 | 46 |
| S141 | ADGFP + CSRH1S | 10 mg KA | na | 7 | 25 | 28 |
| S142 | ADGFP + CSRH1S | 10 mg KA | na | 14 | 31 | 14 |
| S144 | ADGFP + CSRH1S | 10 mg KA | na | 14 | 31 | 23 |
| S145 | ADGFP + CSRH1S | 15 mg KA | na | 14 | 28 | 12 |
| Experiment 3: Time course after epilepsy ends | ||||||
| S293 | ADGFP + CSRH1S | K + SRS | 1.10 | 3 | 42 | 137 |
| S290 | ADGFP + CSRH1S | K + SRS | 0.52 | 8 | 54 | 86 |
| S288 | ADGFP + CSRH1S | K + SRS | 0.52 | 10 | 54 | 14 |
| S287 | ADGFP + CSRH1S | K + SRS | 1.17 | 13 | 47 | 2 |
| S285 | ADGFP + CSRH1S | K + SRS | 1.87 | 17 | 54 | 10 |
| S289 | ADGFP + CSRH1S | K + SRS | 1.79 | 18 | 47 | 8 |
| S284 | ADGFP + CSRH1S | K + SRS | na | 32 | 47 | 3 |
| S291 | ADGFP + CSRH1S | K + SRS | na | na | 55 | 9 |
| S292 | ADGFP + CSRH1S | K + No SRS | na | na | 44 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burke, C.T.; Vitko, I.; Straub, J.; Nylund, E.O.; Gawda, A.; Blair, K.; Sullivan, K.A.; Ergun, L.; Ottolini, M.; Patel, M.K.; et al. EpiPro, a Novel, Synthetic, Activity-Regulated Promoter That Targets Hyperactive Neurons in Epilepsy for Gene Therapy Applications. Int. J. Mol. Sci. 2023, 24, 14467. https://doi.org/10.3390/ijms241914467
Burke CT, Vitko I, Straub J, Nylund EO, Gawda A, Blair K, Sullivan KA, Ergun L, Ottolini M, Patel MK, et al. EpiPro, a Novel, Synthetic, Activity-Regulated Promoter That Targets Hyperactive Neurons in Epilepsy for Gene Therapy Applications. International Journal of Molecular Sciences. 2023; 24(19):14467. https://doi.org/10.3390/ijms241914467
Chicago/Turabian StyleBurke, Cassidy T., Iuliia Vitko, Justyna Straub, Elsa O. Nylund, Agnieszka Gawda, Kathryn Blair, Kyle A. Sullivan, Lara Ergun, Matteo Ottolini, Manoj K. Patel, and et al. 2023. "EpiPro, a Novel, Synthetic, Activity-Regulated Promoter That Targets Hyperactive Neurons in Epilepsy for Gene Therapy Applications" International Journal of Molecular Sciences 24, no. 19: 14467. https://doi.org/10.3390/ijms241914467
APA StyleBurke, C. T., Vitko, I., Straub, J., Nylund, E. O., Gawda, A., Blair, K., Sullivan, K. A., Ergun, L., Ottolini, M., Patel, M. K., & Perez-Reyes, E. (2023). EpiPro, a Novel, Synthetic, Activity-Regulated Promoter That Targets Hyperactive Neurons in Epilepsy for Gene Therapy Applications. International Journal of Molecular Sciences, 24(19), 14467. https://doi.org/10.3390/ijms241914467