
Uncovering NOTCH1 as a Promising Target in the Treatment of MLL-Rearranged Leukemia

 ,
,
Abstract
:1. Introduction
2. Results
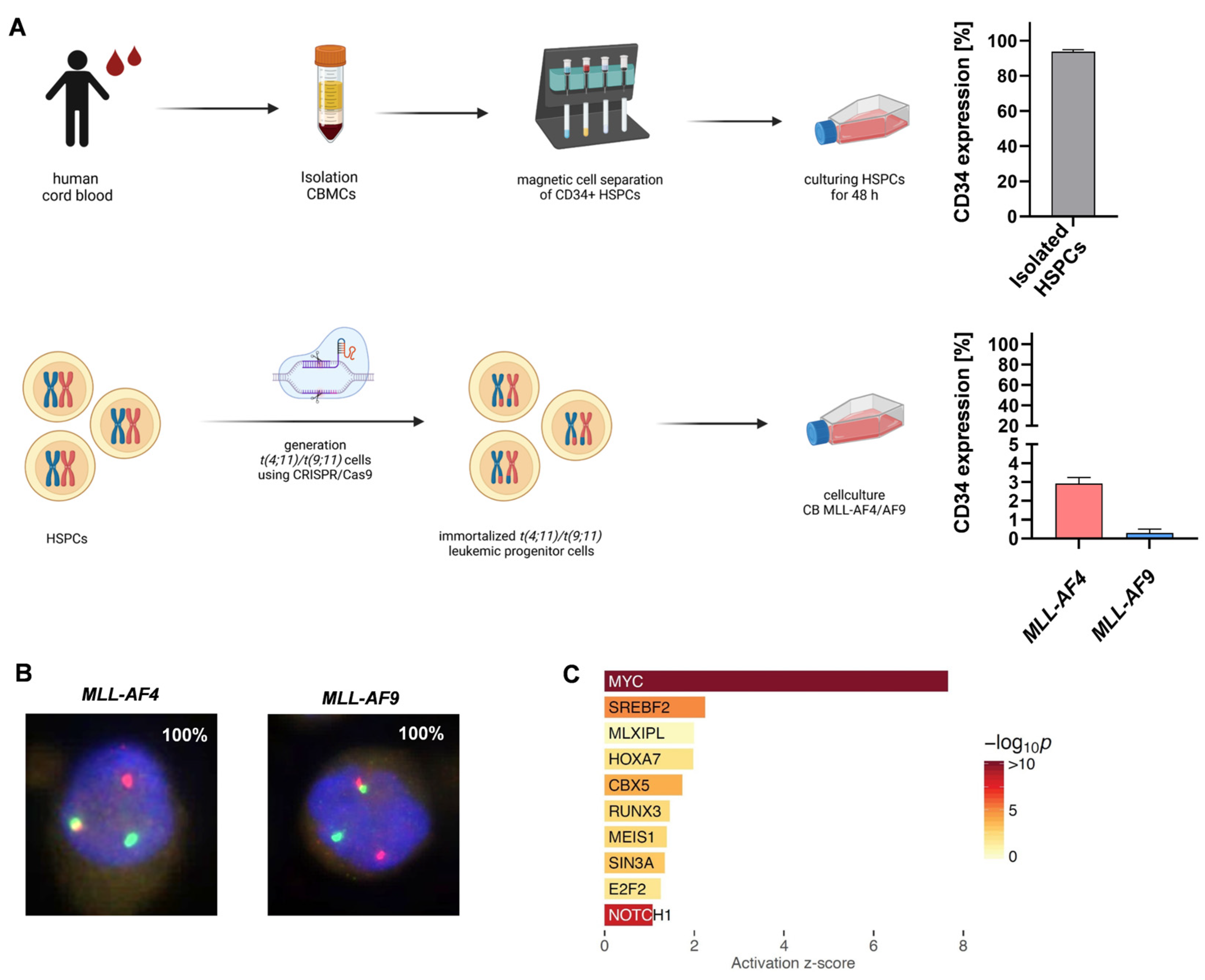
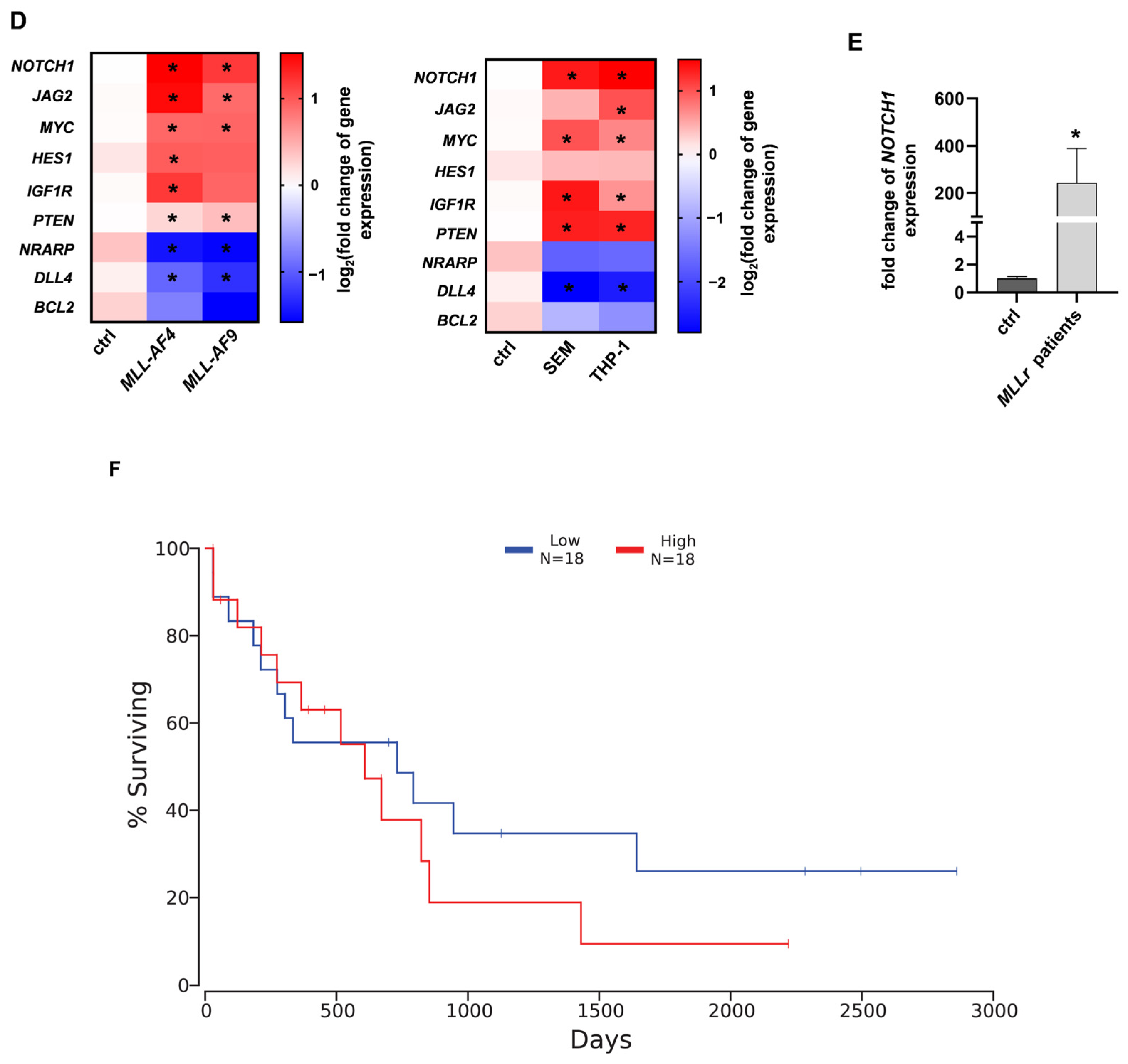
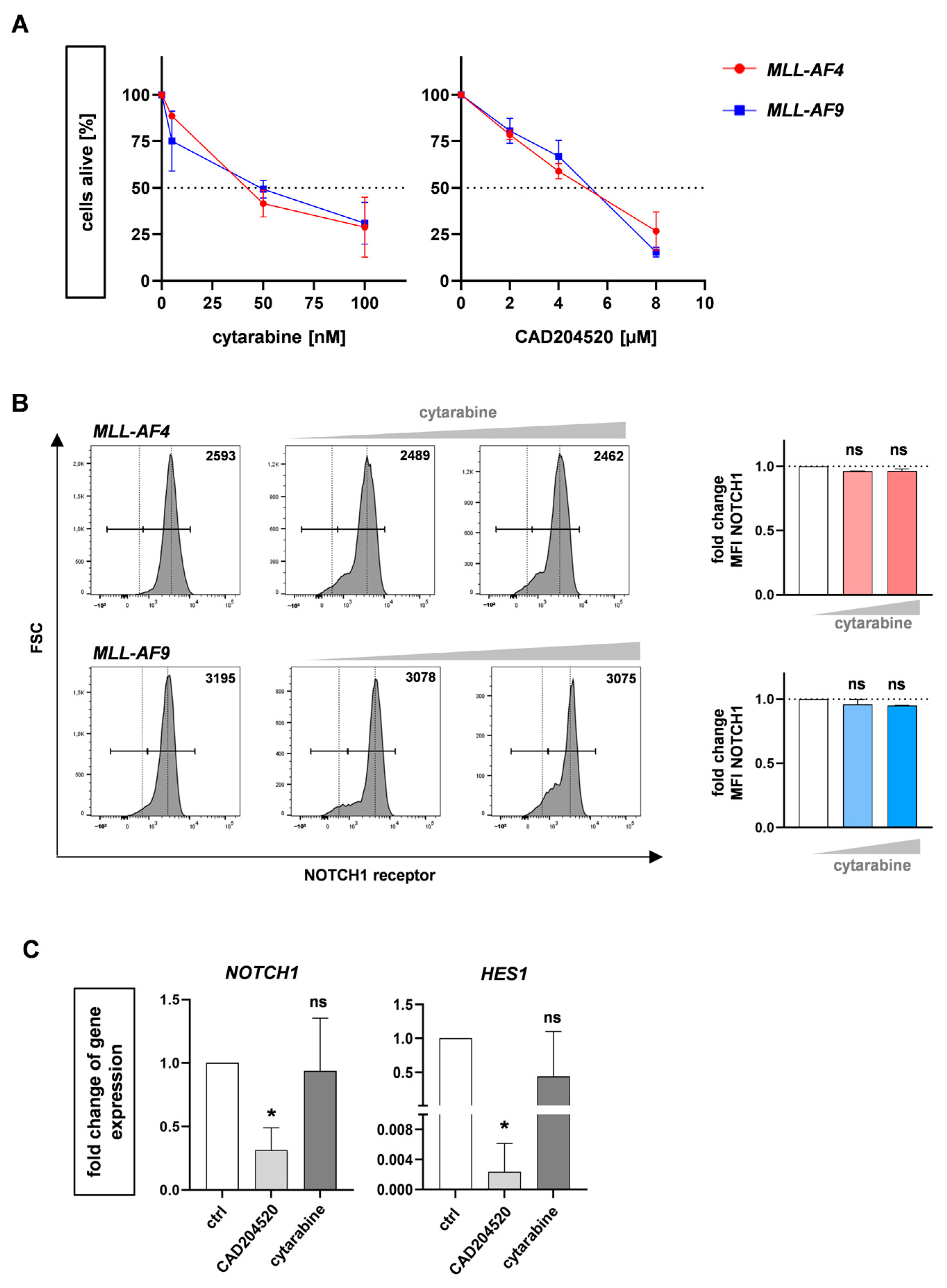
2.1. NOTCH1 as a Potential Target in MLL-AF4/-AF9-Rearranged Leukemia
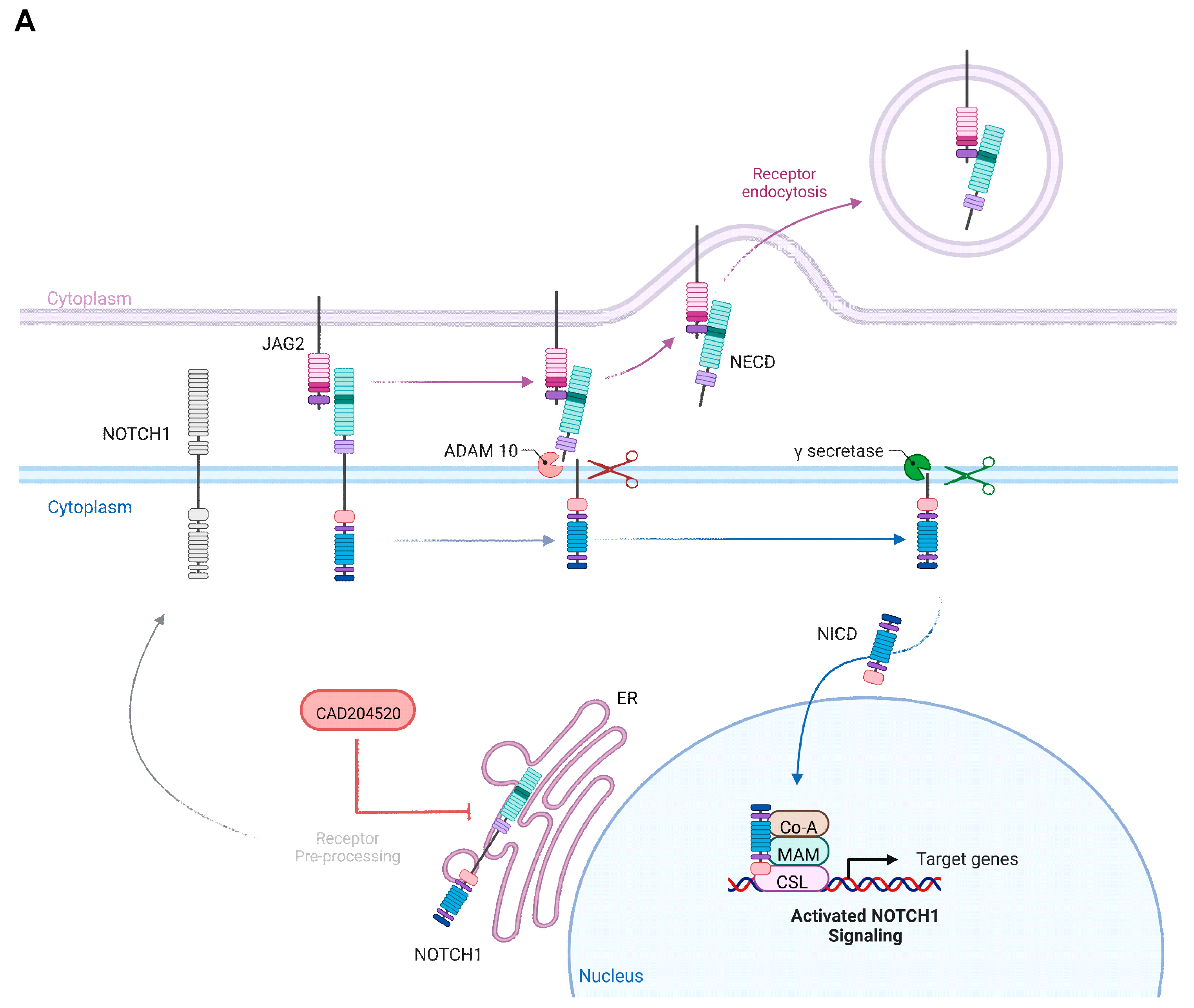
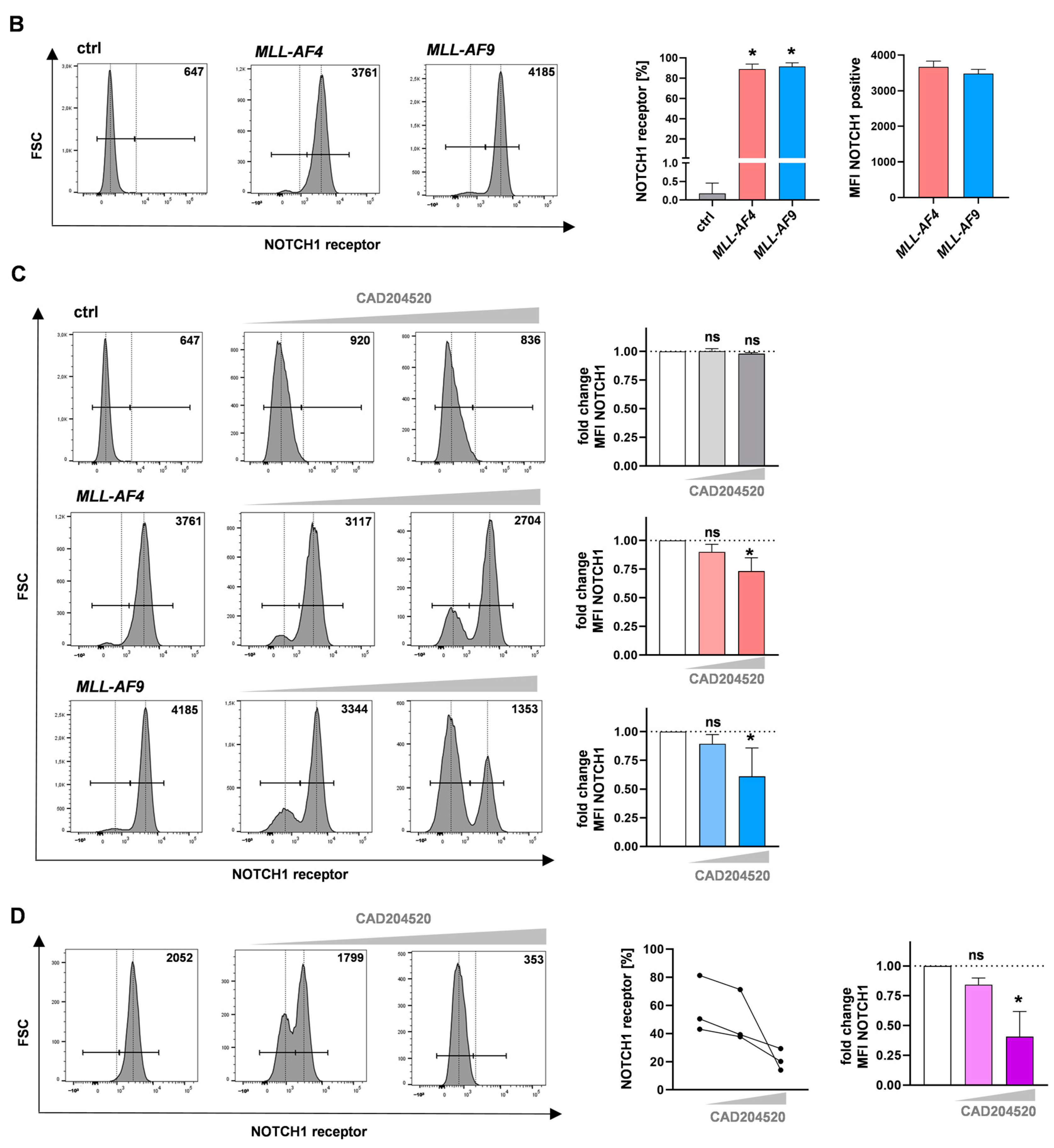
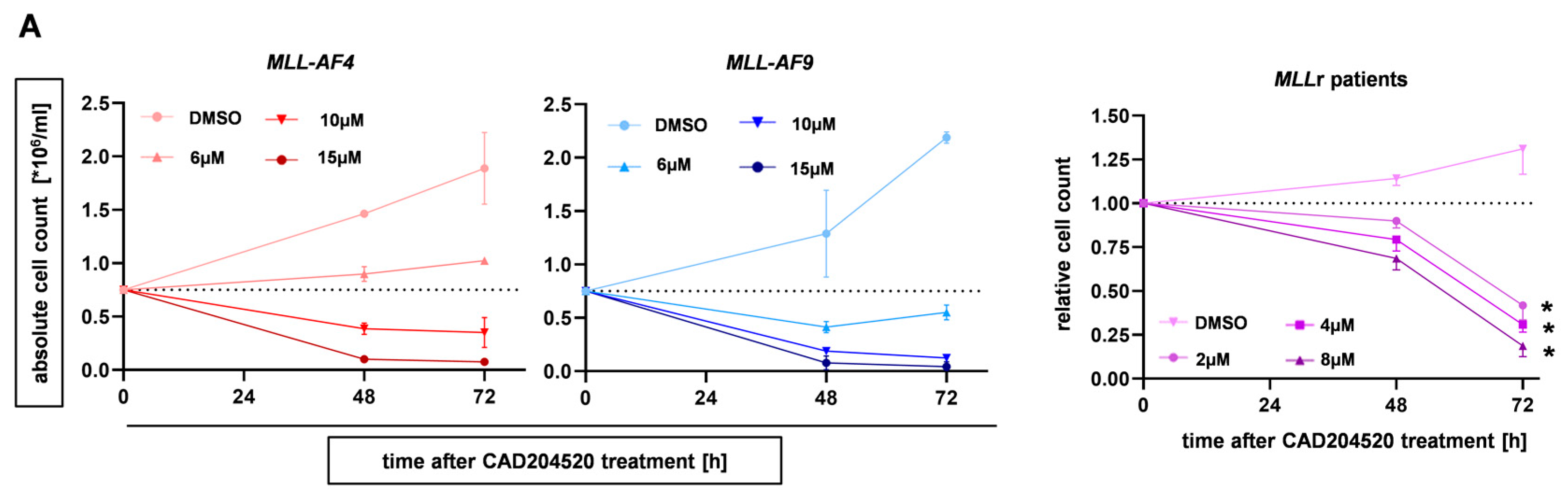
2.2. The SERCA Inhibitor CAD204520 Specifically Affects the NOTCH1 Pathway
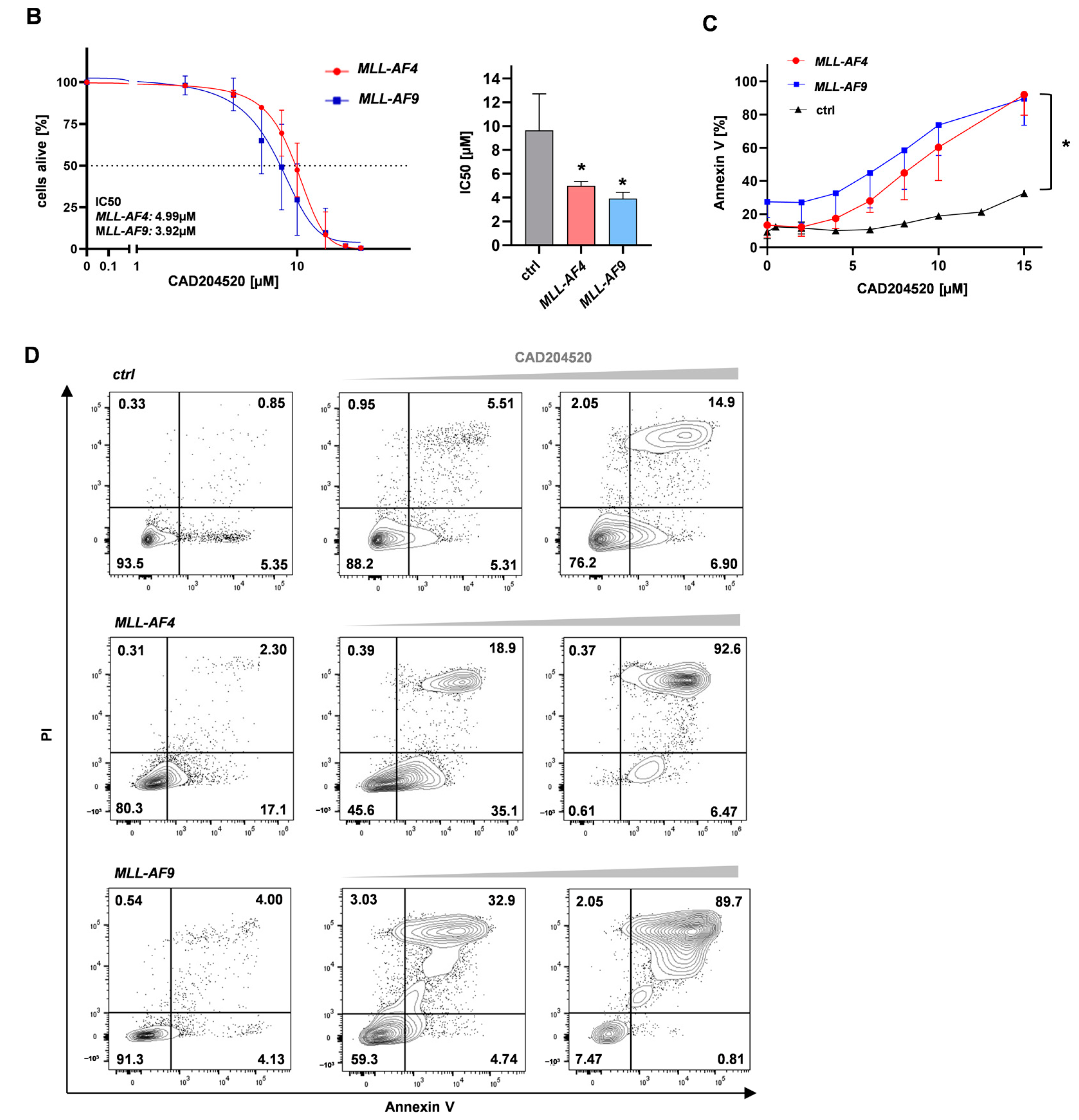
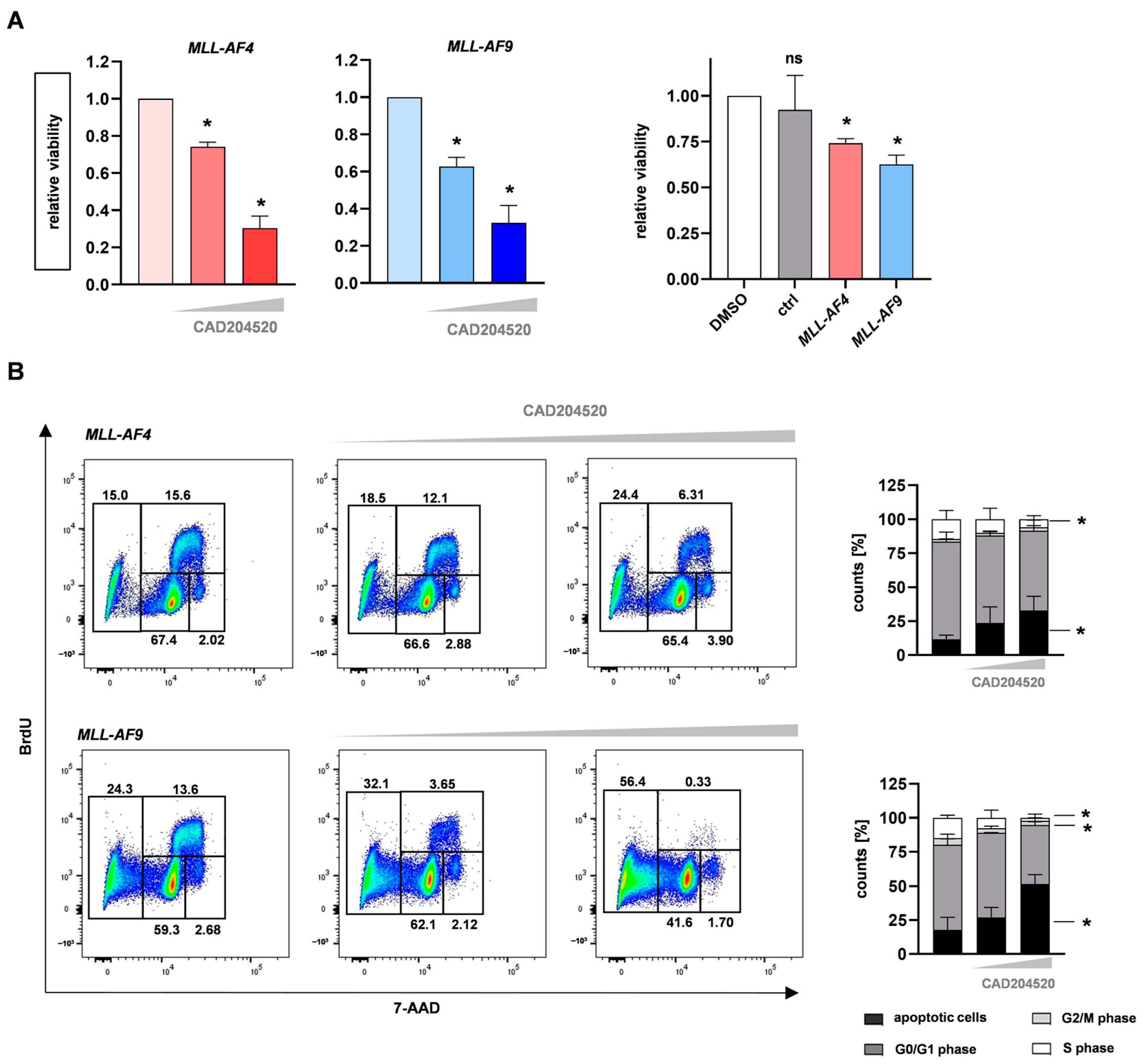
2.3. CAD204520 Induces Apoptosis and Cell Cycle Interruption Leading to Reduced Proliferation
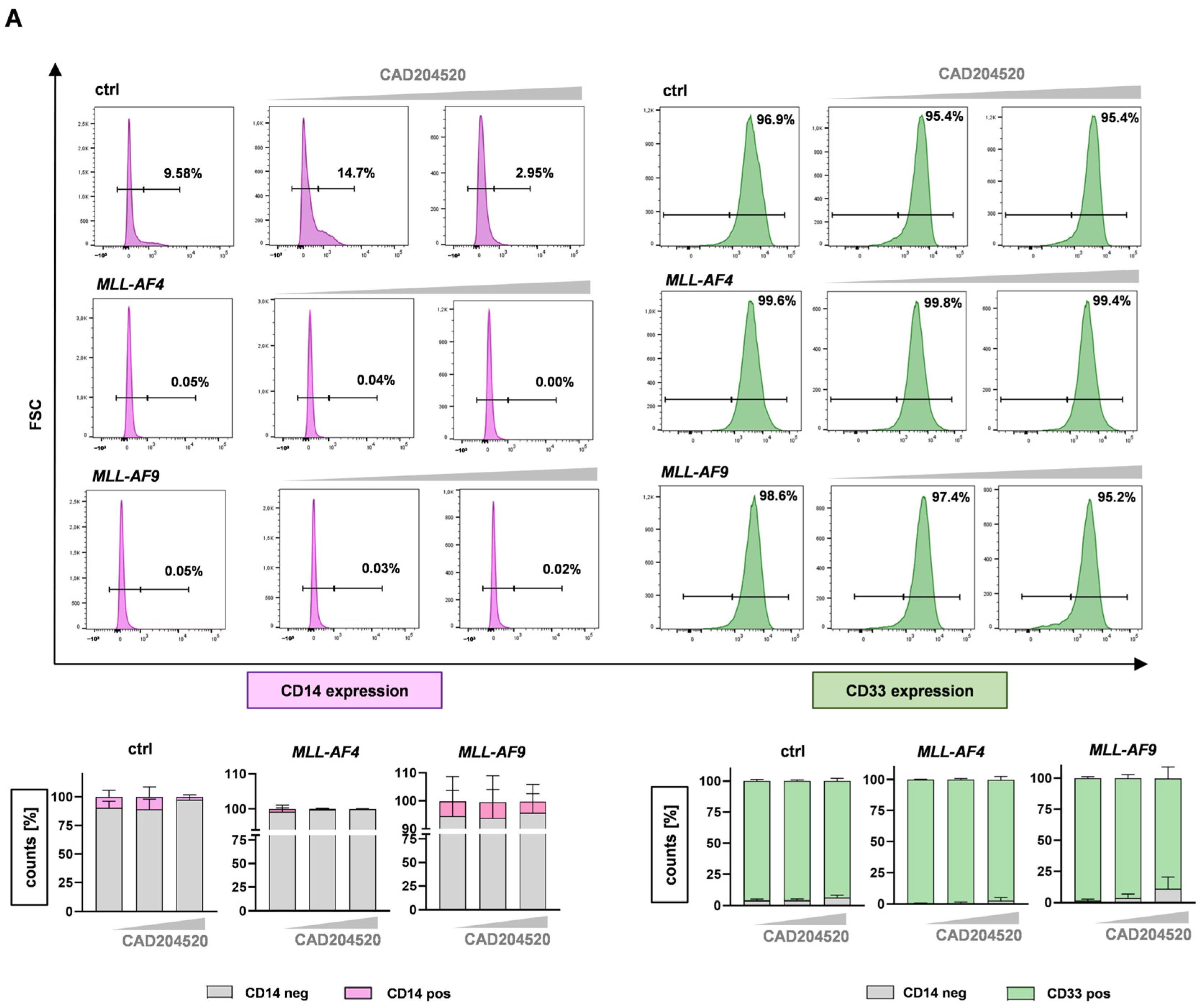
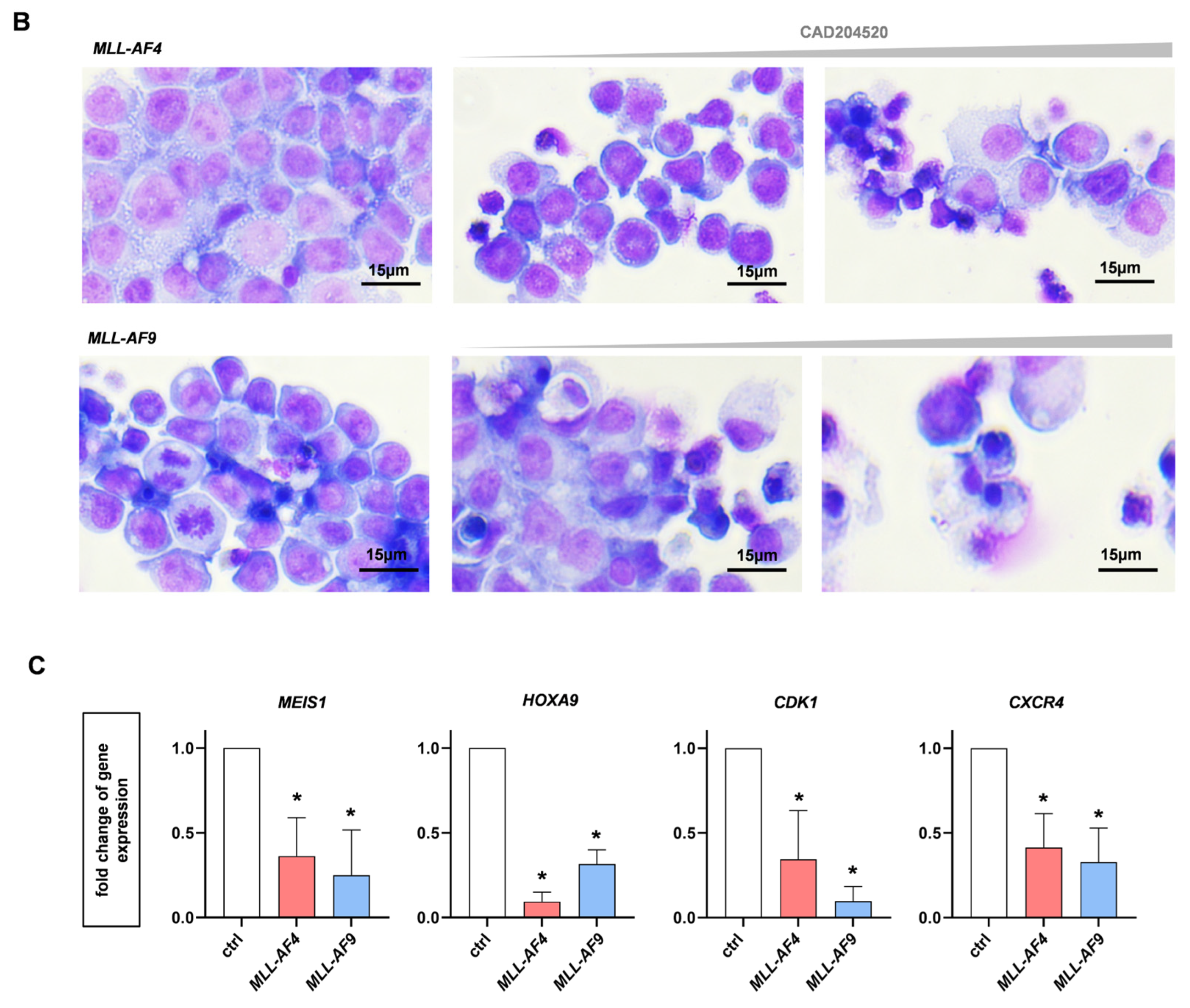
2.4. Anti-Leukemic Effects of CAD204520 Are Linked to MLL Pathway Inhibition and Not to Cell Maturation
3. Discussion
4. Materials and Methods
4.1. Human CRISPR/Cas9-MLLr Model
4.2. RNA Sequencing and Gene Expression Analysis
4.3. Cell Lines and Patient Samples
4.4. Reverse Transcription Quantitative PCR (RT-qPCR)
4.5. Inhibitor Treatment Assay
4.6. Microscopy-Based Determination of Cell Counts
4.7. Cell Viability Assay
4.8. Cell Cycle and Apoptosis Analysis
4.9. May–Gruenwald–Giemsa Staining
4.10. Flow Cytometry
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef] [PubMed]
- Tsakaneli, A.; Williams, O. Drug Repurposing for Targeting Acute Leukemia with KMT2A (MLL)—Gene Rearrangements. Front. Pharmacol. 2021, 12, 741413. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.; Kari, V.; Raghavan, S.C. Chromosomal translocations in cancer. Biochim. Biophys. Acta 2008, 1786, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Larghero, P.; Lopes, B.A.; Burmeister, T.; Gröger, D.; Sutton, R.; Venn, N.C.; Cazzaniga, G.; Abascal, L.C.; Tsaur, G.; et al. The KMT2A recombinome of acute leukemias in 2023. Leukemia 2023, 37, 988–1005. [Google Scholar] [CrossRef]
- Gaussmann, A.; Wenger, T.; Eberle, I.; Bursen, A.; Bracharz, S.; Herr, I.; Dingermann, T.; Marschalek, R. Combined effects of the two reciprocal t(4;11) fusion proteins MLL·AF4 and AF4·MLL confer resistance to apoptosis, cell cycling capacity and growth transformation. Oncogene 2007, 26, 3352–3363. [Google Scholar] [CrossRef] [PubMed]
- Secker, K.-A.; Bloechl, B.; Keppeler, H.; Duerr-Stoerzer, S.; Schmid, H.; Schneidawind, D.; Jeong, J.; Hentrich, T.; Schulze-Hentrich, J.M.; Schneidawind, C. MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis. Cancers 2020, 12, 1342. [Google Scholar] [CrossRef]
- Chiarella, E.; Aloisio, A.; Scicchitano, S.; Todoerti, K.; Cosentino, E.G.; Lico, D.; Neri, A.; Amodio, N.; Bond, H.M.; Mesuraca, M. ZNF521 Enhances MLL-AF9-Dependent Hematopoietic Stem Cell Transformation in Acute Myeloid Leukemias by Altering the Gene Expression Landscape. Int. J. Mol. Sci. 2021, 22, 10814. [Google Scholar] [CrossRef]
- Secker, K.-A.; Keppeler, H.; Duerr-Stoerzer, S.; Schmid, H.; Schneidawind, D.; Hentrich, T.; Schulze-Hentrich, J.M.; Mankel, B.; Fend, F.; Schneidawind, C. Inhibition of DOT1L and PRMT5 promote synergistic anti-tumor activity in a human MLL leukemia model induced by CRISPR/Cas9. Oncogene 2019, 38, 7181–7195. [Google Scholar] [CrossRef]
- Secker, K.-A.; Bruns, L.; Keppeler, H.; Jeong, J.; Hentrich, T.; Schulze-Hentrich, J.M.; Mankel, B.; Fend, F.; Schneidawind, D.; Schneidawind, C. Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations. Cancers 2020, 12, 1487. [Google Scholar] [CrossRef]
- Fitzel, R.; Secker-Grob, K.-A.; Keppeler, H.; Korkmaz, F.; Schairer, R.; Erkner, E.; Schneidawind, D.; Lengerke, C.; Hentrich, T.; Schulze-Hentrich, J.M.; et al. Targeting MYC in combination with epigenetic regulators induces synergistic anti-leukemic effects in MLLr leukemia and simultaneously improves immunity. Neoplasia 2023, 41, 100902. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, J.; Ji, C. The emerging roles of Notch signaling in leukemia and stem cells. Biomark. Res. 2013, 1, 23. [Google Scholar] [CrossRef]
- Zhang, H.; Liang, J.; Lu, T.; Li, M.; Shan, G.; Bi, G.; Zhao, M.; Jin, X.; Wang, Q.; Chen, Z.; et al. AGRN promotes lung adenocarcinoma progression by activating Notch signaling pathway and acts as a therapeutic target. Pharmacol. Res. 2023, 194, 106819. [Google Scholar] [CrossRef] [PubMed]
- Aifantis, I.; Raetz, E.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Han, Y.; Chen, R.; Jiang, Y.; Chen, C.; Wang, X.; Zhou, J.; Xu, Q.; Jiang, S.; Zhang, S.; et al. MicroRNA-143 acts as a tumor suppressor through Musashi-2/DLL1/Notch1 and Musashi-2/Snail1/MMPs axes in acute myeloid leukemia. J. Transl. Med. 2023, 21, 309. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Pui, J.C.; Allman, D.; Xu, L.; DeRocco, S.; Karnell, F.G.; Bakkour, S.; Lee, J.Y.; Kadesch, T.; Hardy, R.R.; Aster, J.C.; et al. Notch1 Expression in Early Lymphopoiesis Influences B versus T Lineage Determination. Immunity 1999, 11, 299–308. [Google Scholar] [CrossRef]
- McCarter, A.C.; Wang, Q.; Chiang, M. Notch in Leukemia. Adv. Exp. Med. Biol. 2018, 1066, 355–394. [Google Scholar]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Carrieri, F.A.; Murray, P.J.; Ditsova, D.; Ferris, M.A.; Davies, P.; Dale, J.K. CDK 1 and CDK 2 regulate NICD 1 turnover and the periodicity of the segmentation clock. EMBO Rep. 2019, 20, e46436. [Google Scholar] [CrossRef]
- Klinakis, A.; Lobry, C.; Abdel-Wahab, O.; Oh, P.; Haeno, H.; Buonamici, S.; van De Walle, I.; Cathelin, S.; Trimarchi, T.; Araldi, E.; et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 2011, 473, 230–233. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Almeida, A.; Partyka, K.A.; Lu, Y.; Schwan, J.V.; Lambert, K.; Rogers, M.; Robinson, W.A.; Robinson, S.E.; Shellman, Y.G. Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 2016, 7, 84594–84607. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I Pharmacologic and Pharmacodynamic Study of the Gamma Secretase (Notch) Inhibitor MK-0752 in Adult Patients With Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary Genomic Screens Identify SERCA as a Therapeutic Target in NOTCH1 Mutated Cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef]
- Marchesini, M.; Gherli, A.; Montanaro, A.; Patrizi, L.; Sorrentino, C.; Pagliaro, L.; Rompietti, C.; Kitara, S.; Heit, S.; Olesen, C.E.; et al. Blockade of Oncogenic NOTCH1 with the SERCA Inhibitor CAD204520 in T Cell Acute Lymphoblastic Leukemia. Cell Chem. Biol. 2020, 27, 678–697.e13. [Google Scholar] [CrossRef]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Yang, X.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef]
- Pinto, I.; Duque, M.; Gonçalves, J.; Akkapeddi, P.; Oliveira, M.L.; Cabrita, R.; Yunes, J.A.; Durum, S.K.; Barata, J.T.; Fragoso, R. NRARP displays either pro- or anti-tumoral roles in T-cell acute lymphoblastic leukemia depending on Notch and Wnt signaling. Oncogene 2020, 39, 975–986. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; Boer, M.L.D.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef]
- Porter, L.A.; Donoghue, D.J. Cyclin B1 and CDK1: Nuclear localization and upstream regulators. Prog. Cell Cycle Res. 2003, 5, 335–347. [Google Scholar] [PubMed]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Tsaouli, G.; Ferretti, E.; Bellavia, D.; Vacca, A.; Felli, M.P. Notch/CXCR4 Partnership in Acute Lymphoblastic Leukemia Progression. J. Immunol. Res. 2019, 2019, 5601396. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Buendía, G.A.R.; Fournier, N.; Liu, Y.; Armand, F.; Hamelin, R.; Pavlou, M.; Radtke, F. Resistance mechanism to Notch inhibition and combination therapy in human T-cell acute lymphoblastic leukemia. Blood Adv. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.U.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Zhang, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laurenti, L.; Bruscaggin, A.; Cerri, M.; et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012, 119, 521–529. [Google Scholar] [CrossRef]
- D’altri, T.; Gonzalez, J.; Aifantis, I.; Espinosa, L.; Bigas, A. Hes1 expression and CYLD repression are essential events downstream of Notch1 in T-cell leukemia. Cell Cycle 2011, 10, 1031–1036. [Google Scholar] [CrossRef]
- Espinosa, L.; Cathelin, S.; D’Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Ingle’s-Esteve, J.; Nomdedeu, J.; Bigas, A.; et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell 2010, 18, 268–281. [Google Scholar] [CrossRef]
- Kato, T.; Sakata-Yanagimoto, M.; Nishikii, H.; Ueno, M.; Miyake, Y.; Yokoyama, Y.; Asabe, Y.; Kamada, Y.; Muto, H.; Obara, N.; et al. Hes1 suppresses acute myeloid leukemia development through FLT3 repression. Leukemia 2015, 29, 576–585. [Google Scholar] [CrossRef]
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.-C.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J. Exp. Med. 2011, 208, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Riccio, O.; Van Gijn, M.E.; Bezdek, A.C.; Pellegrinet, L.; Van Es, J.H.; Zimber-Strobl, U.; Strobl, L.J.; Honjo, T.; Clevers, H.; Radtke, F. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27 Kip1 and p57 Kip2. EMBO Rep. 2008, 9, 377–383. [Google Scholar] [CrossRef]
- Roti, G.; Qi, J.; Kitara, S.; Sanchez-Martin, M.; Conway, A.S.; Varca, A.C.; Su, A.; Wu, L.; Kung, A.L.; Ferrando, A.A.; et al. Leukemia-specific delivery of mutant NOTCH1 targeted therapy. J. Exp. Med. 2017, 215, 197–216. [Google Scholar] [CrossRef]
- Tavor, S.; Petit, I.; Porozov, S.; Avigdor, A.; Dar, A.; Leider-Trejo, L.; Shemtov, N.; Deutsch, V.; Naparstek, E.; Nagler, A.; et al. CXCR4 Regulates Migration and Development of Human Acute Myelogenous Leukemia Stem Cells in Transplanted NOD/SCID Mice. Cancer Res. 2004, 64, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Jesus, C.D.C.D.; Lasgi, C.; Quang, C.T.; Ghysdael, J. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.-Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [PubMed]
- Aref, S.; Rizk, R.; El Agder, M.; Fakhry, W.; El Zafarany, M.; Sabry, M. NOTCH-1 Gene Mutations Influence Survival in Acute Myeloid Leukemia Patients. Asian Pac. J. Cancer Prev. 2020, 21, 1987–1992. [Google Scholar] [CrossRef]
- Kamga, P.T.; Bassi, G.; Cassaro, A.; Midolo, M.; Di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef]
- Takam Kamga, P.; Collo, G.D.; Resci, F.; Bazzoni, R.; Mercuri, A.; Quaglia, F.M.; Tanasi, I.; Krampera, M. Notch Signaling Molecules as Prognostic Biomarkers for Acute Myeloid Leukemia. Cancers 2019, 11, 1958. [Google Scholar] [CrossRef]
- Carrillo-Tornel, S.; Chen-Liang, T.H.; Zurdo, M.; Puiggros, A.; Gómez-Llonín, A.; García-Malo, M.D.; Jerez, A. NOTCH1 mutation in chronic lymphocytic leukaemia is associated with an enhanced cell cycle G1/S transition and specific cyclin overexpression: Preclinical ground for targeted inhibition. Br. J. Haematol. 2023, 201, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, W.; Basyal, M.; Chang, K.H.; Ostermann, L.; Burks, J.K.; Andreeff, M. FLT3 inhibitors upregulate CXCR4 and E-selectin ligands via ERK suppression in AML cells and CXCR4/E-selectin inhibition enhances anti-leukemia efficacy of FLT3-targeted therapy in AML. Leukemia 2023, 37, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 September 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Stam, R.W.; Schneider, P.; Hagelstein, J.A.P.; Van Der Linden, M.H.; Stumpel, D.J.P.M.; De Menezes, R.X.; De Lorenzo, P.; Valsecchi, M.; Maria, G.; Pieters, R. Gene expression profiling–based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010, 115, 2835–2844. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Buechele, C.; Breese, E.H.; Schneidawind, D.; Lin, C.-H.; Jeong, J.; Duque-Afonso, J.; Wong, S.H.K.; Smith, K.S.; Negrin, R.S.; Porteus, M.; et al. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood 2015, 126, 1683–1694. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence 5′-3′ | Reverse Primer Sequence 5′-3′ |
|---|---|---|
| NOTCH1 | GCCTGGACAAGATCAATGAGTTC | TCCACATCGTACTGGCACAGA |
| JAG2 | GACAACGATACCACCCCGAAT | CATGCGACACTCGCTCGAT |
| MYC | CCTGGTGCTCCATGAGGAGAC | CAGACTCTGACCTTTTGCCAGG |
| HES1 | GGACATTCTGGAAATGACAGTGAA | CCCAGCACACTTGGGTCTG |
| IGF1R | GCCCCTCGGGCTTCAT | ACCTTCACAAGGGATGCAGTACA |
| PTEN | GGGAAGTAAGGACCAGAGACAAAAA | AGCGCCTCTGACTGGGAATA |
| NRARP | GCTGCACCAGTCGGTCATC | CCGAACTTGACCAGCAGCTT |
| DLL4 | CCCTGGCAATGTACTTGTGATG | GAGTGGTGGGTGCAGTAGTTGAG |
| BCL2 | CCTGTGGATGACTGAGTACCTGAAC | CAGCCAGGAGAAATCAAACAGA |
| CDK1 | CCATTGACTAACTATGGAAGATTATACCA | TGTCTACCCTTATACACAACTCCATAGG |
| CXCR4 | TGGAGGGGATCAGTATATACACTTCA | TCATAGTCCCCTGAGCCCATT |
| MEIS1 | TGGCCACACGTCACACAGT | TTTGTCCTTATCAGGGTCATCATC |
| HOXA9 | ATGAGAGCGGCGGAGACA | CGCGCATGAAGCCAGTT |
| 18S | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, J.; Erkner, E.; Fitzel, R.; Radszuweit, P.; Keppeler, H.; Korkmaz, F.; Roti, G.; Lengerke, C.; Schneidawind, D.; Schneidawind, C. Uncovering NOTCH1 as a Promising Target in the Treatment of MLL-Rearranged Leukemia. Int. J. Mol. Sci. 2023, 24, 14466. https://doi.org/10.3390/ijms241914466
Fischer J, Erkner E, Fitzel R, Radszuweit P, Keppeler H, Korkmaz F, Roti G, Lengerke C, Schneidawind D, Schneidawind C. Uncovering NOTCH1 as a Promising Target in the Treatment of MLL-Rearranged Leukemia. International Journal of Molecular Sciences. 2023; 24(19):14466. https://doi.org/10.3390/ijms241914466
Chicago/Turabian StyleFischer, Jacqueline, Estelle Erkner, Rahel Fitzel, Pia Radszuweit, Hildegard Keppeler, Fulya Korkmaz, Giovanni Roti, Claudia Lengerke, Dominik Schneidawind, and Corina Schneidawind. 2023. "Uncovering NOTCH1 as a Promising Target in the Treatment of MLL-Rearranged Leukemia" International Journal of Molecular Sciences 24, no. 19: 14466. https://doi.org/10.3390/ijms241914466
APA StyleFischer, J., Erkner, E., Fitzel, R., Radszuweit, P., Keppeler, H., Korkmaz, F., Roti, G., Lengerke, C., Schneidawind, D., & Schneidawind, C. (2023). Uncovering NOTCH1 as a Promising Target in the Treatment of MLL-Rearranged Leukemia. International Journal of Molecular Sciences, 24(19), 14466. https://doi.org/10.3390/ijms241914466