

SAFoldNet: A Novel Tool for Discovering and Aligning Three-Dimensional Protein Structures Based on a Neural Network

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
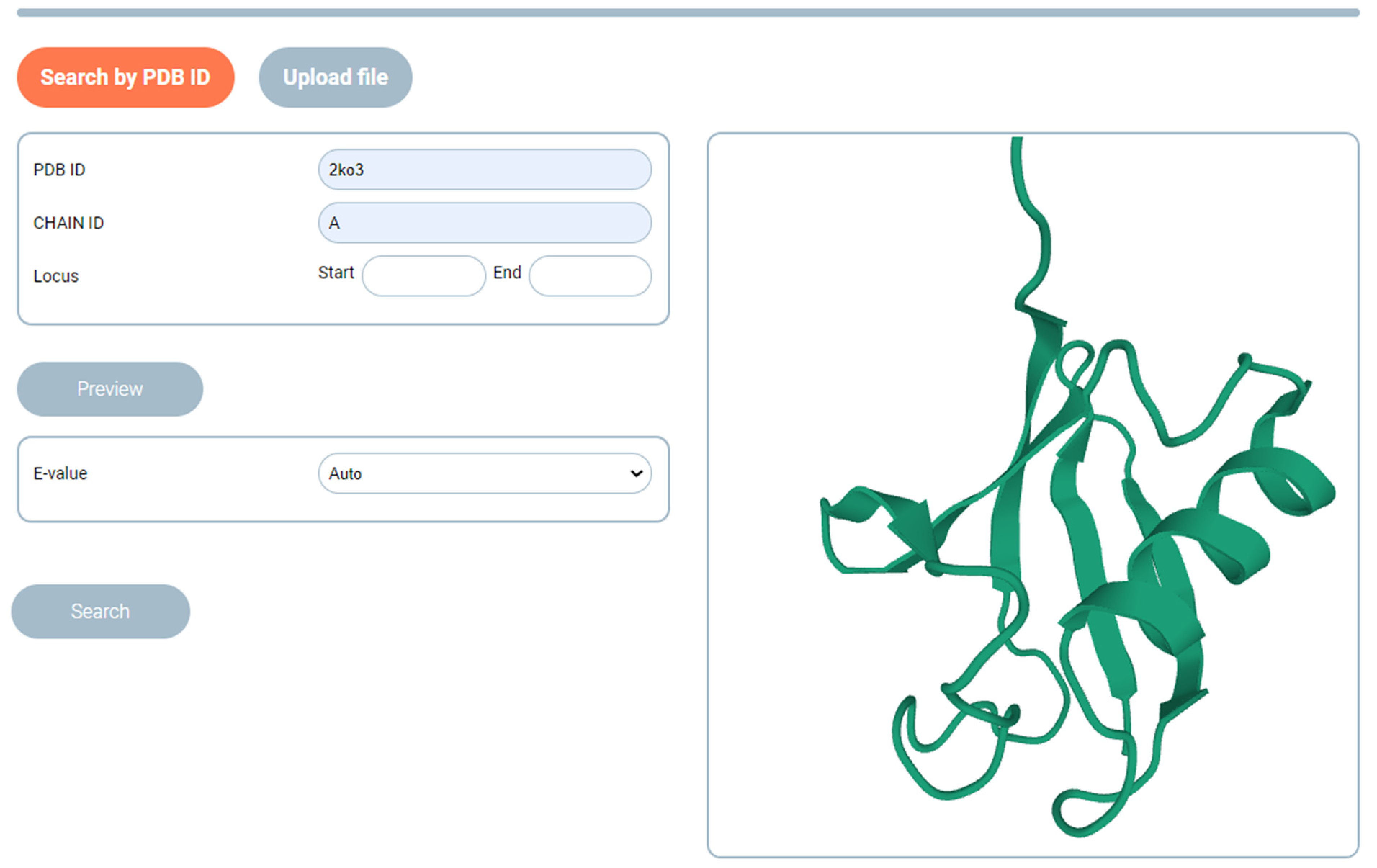
2.1. SAFoldNet
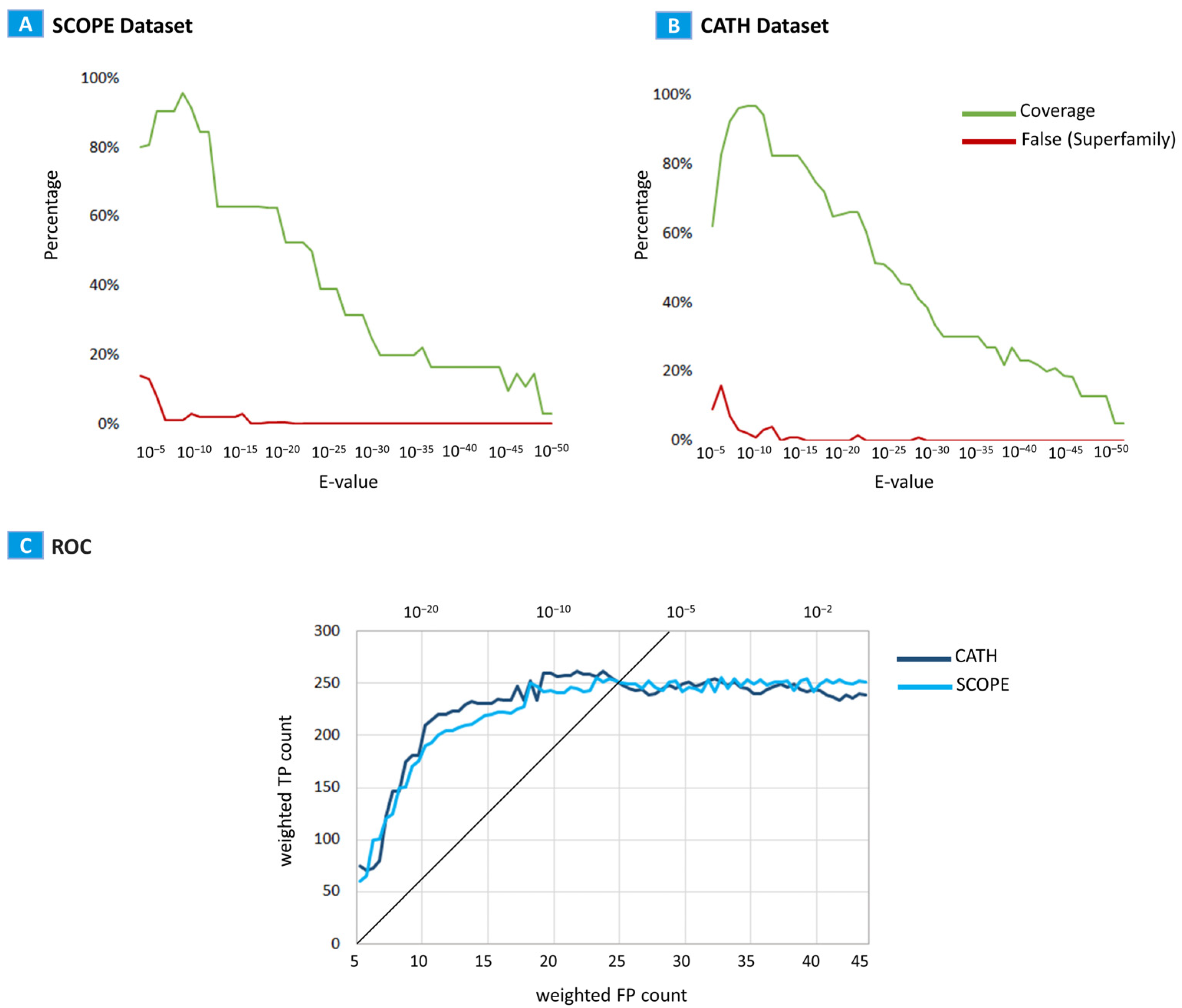
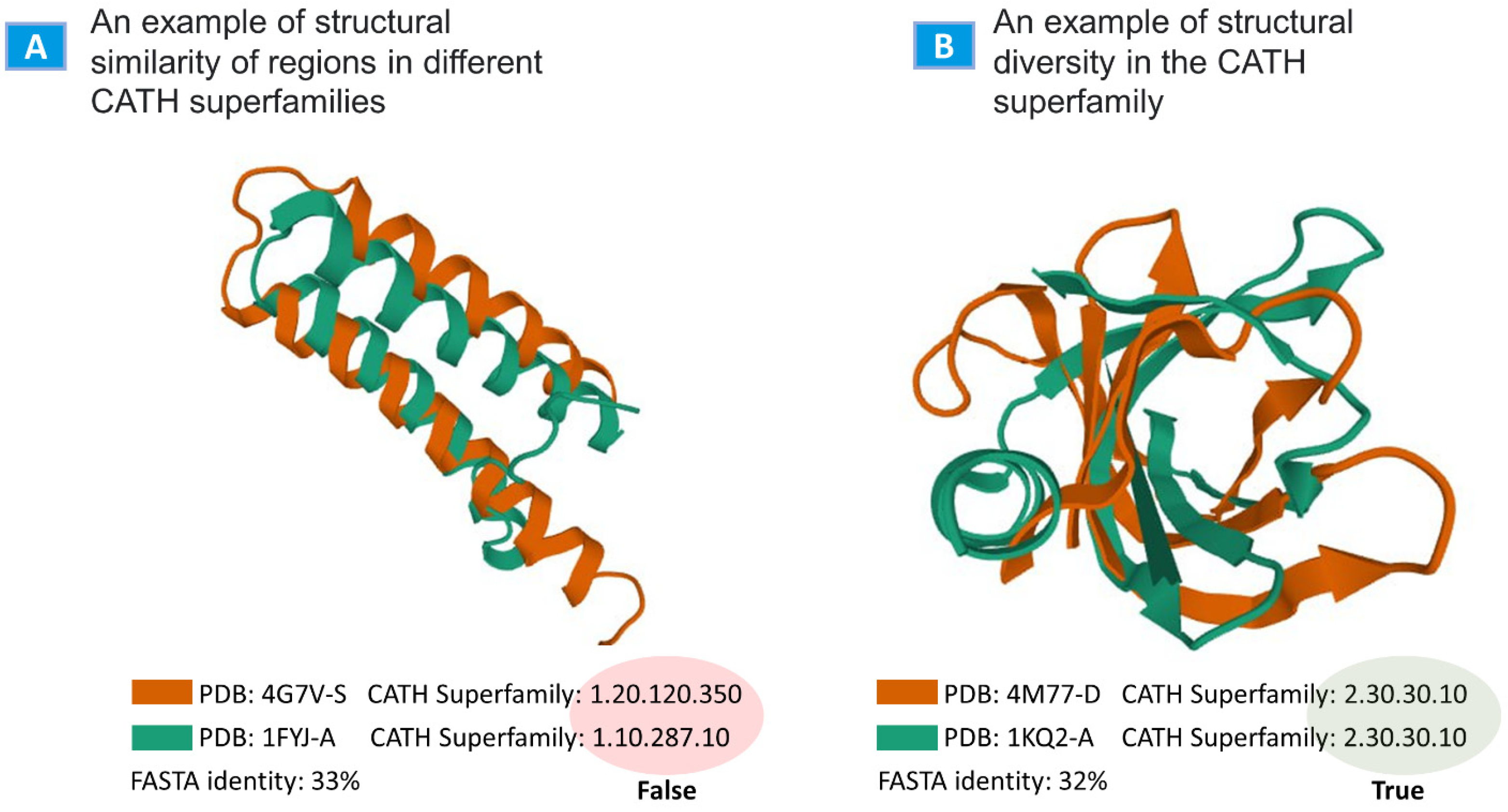
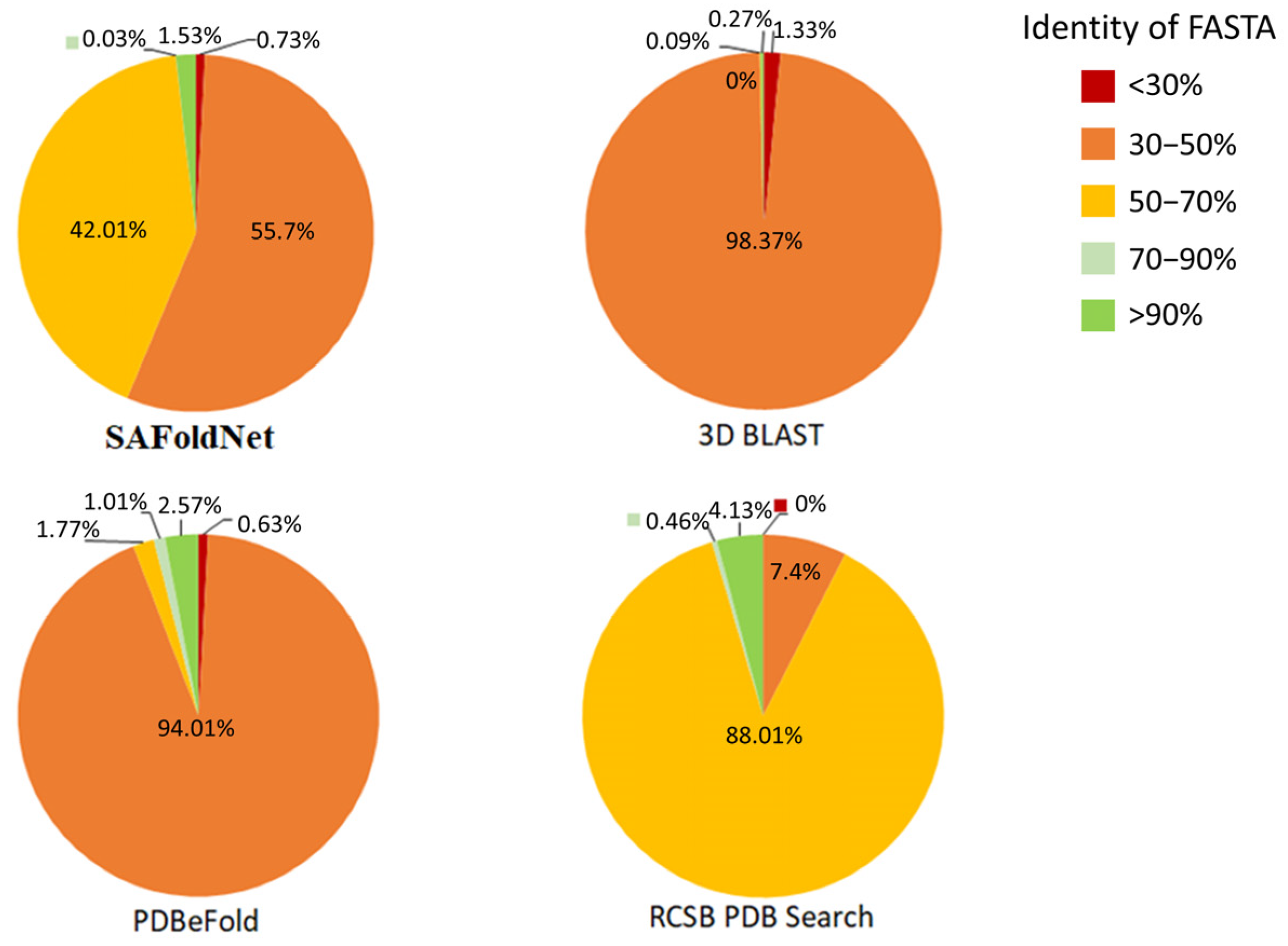
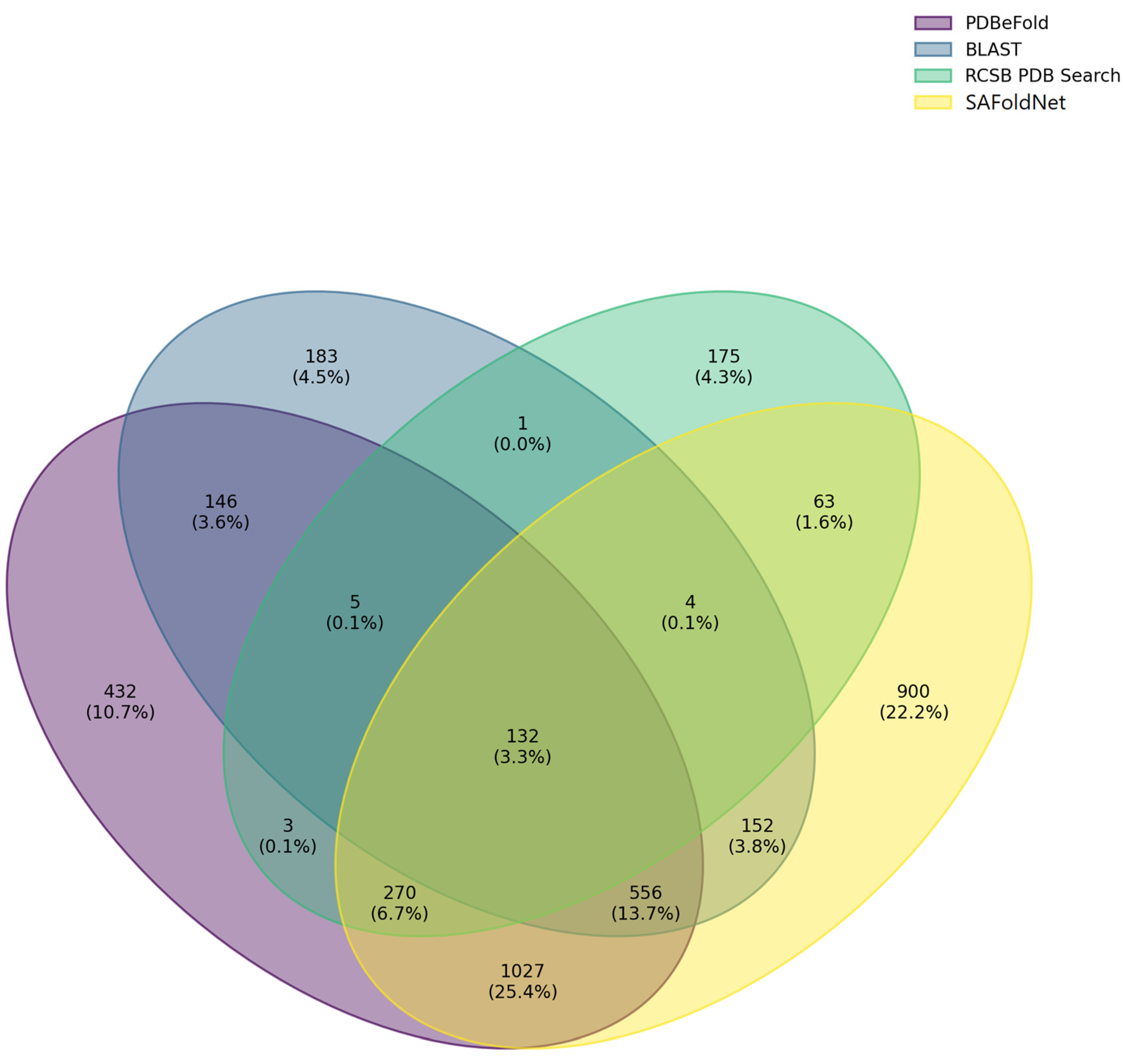
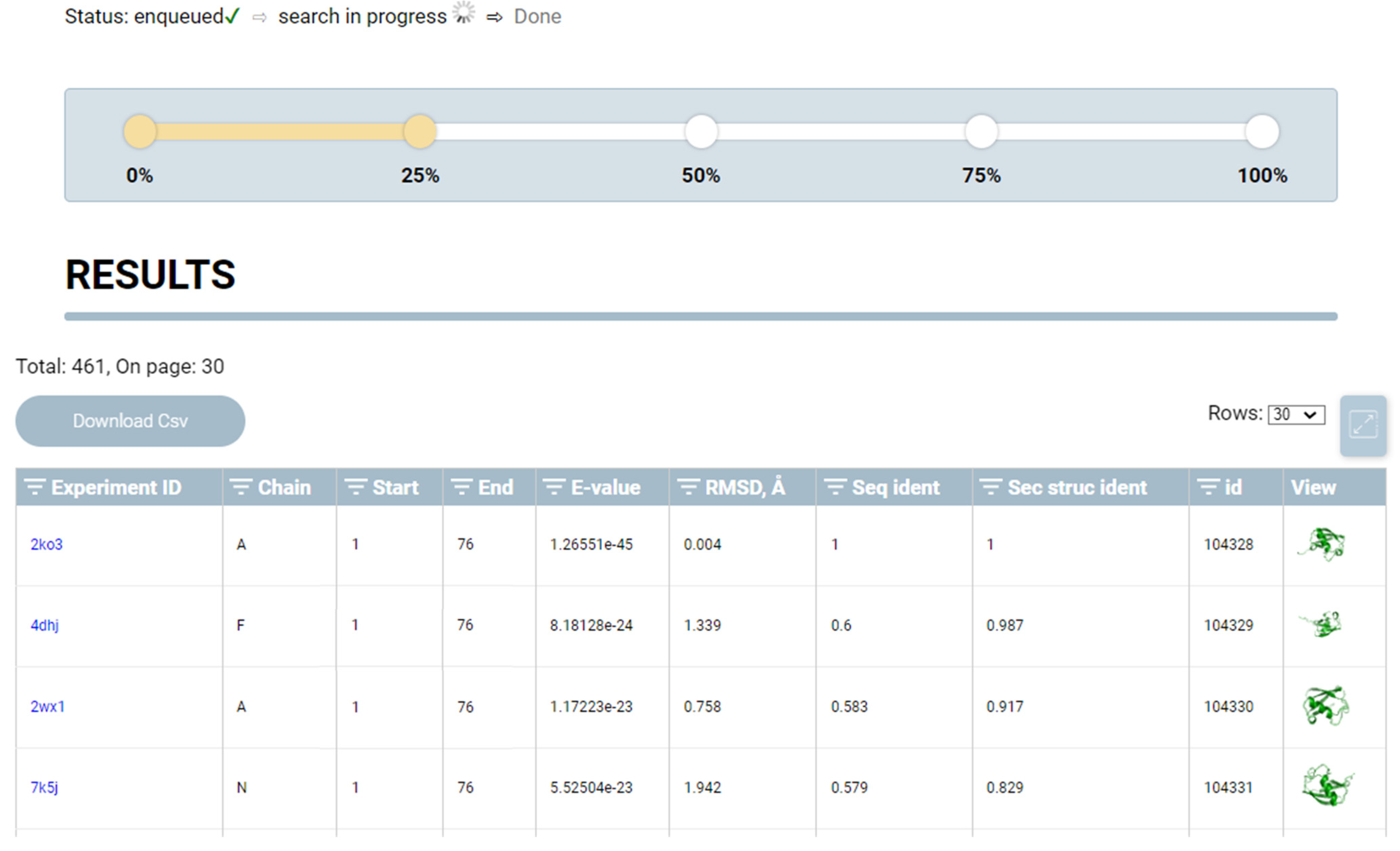
2.2. Evaluation
3. Discussion
4. Materials and Methods
4.1. SA
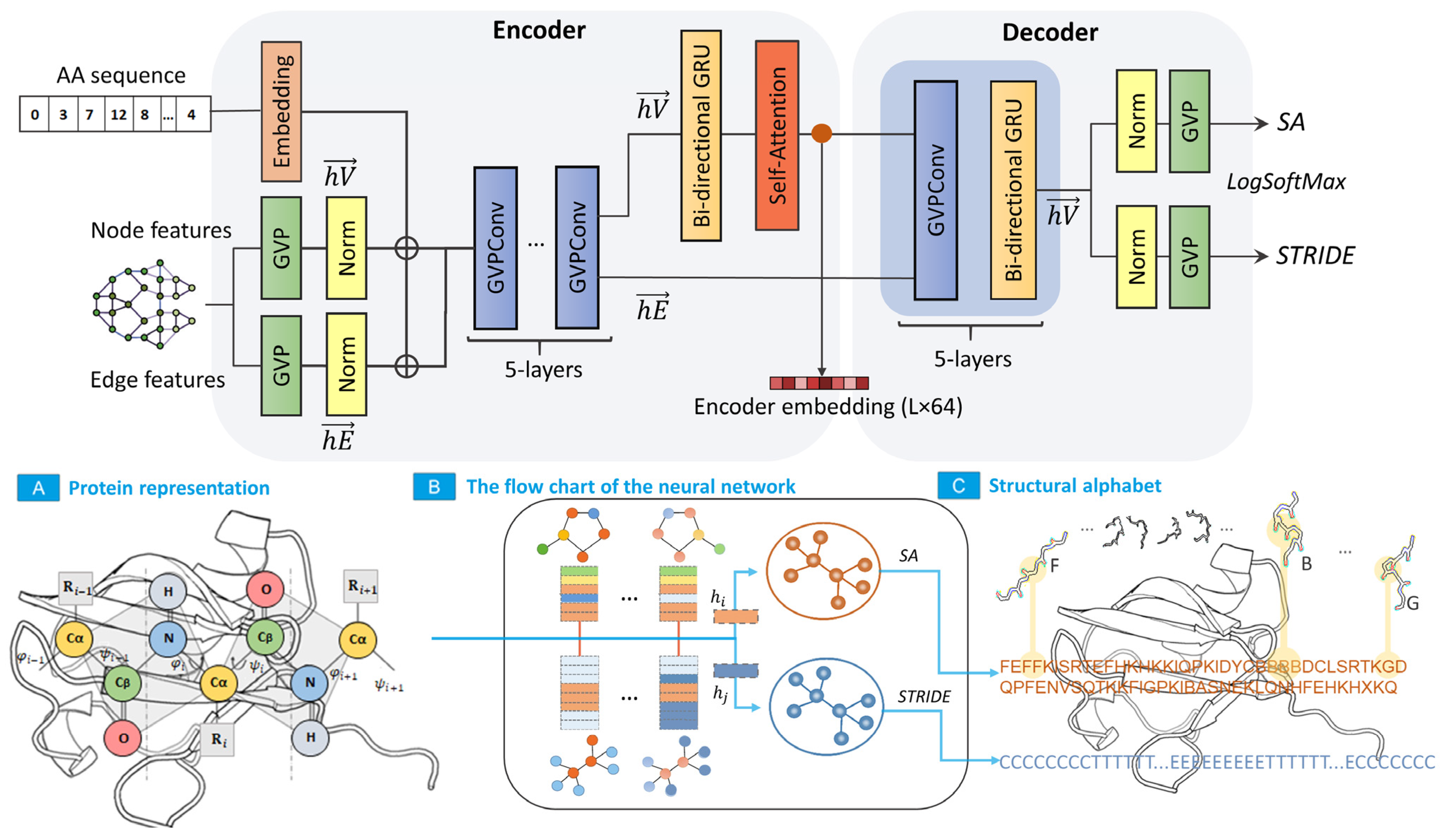
4.2. Neural Network Architecture
4.3. Neural Network Flow
- From the array of coordinates, a graph is formed, each vertex of which corresponds to the Cα-atom of the primary protein chain, connected by edges with Cα-atoms (KNN graph, k = 32). Each edge and vertex of the graph contain scalar and vector features that describe the orientation of the amino acid residue in three-dimensional Euclidean space.
- Signs of the graph vertex comprise torsion angles, single vectors that determine the direction of the previous and subsequent C and Cα atoms of the primary chain, single vectors that determine the direction of the side chain (direction to the Cβ-atom), and codes of the amino acid sequence of the protein.
- The graph edge features comprise a single vector that defines the direction of the edge, distance between graph vertices encoded using Gaussian radial basis functions, and edge position code encoded using a positional sinusoidal encoder.
- All graph features are input into the network, which comprises two interconnected blocks: an encoder, which generates a hidden feature representation, and a decoder, which converts the hidden feature representation into SA elements. A negative log likelihood loss function is used to calculate the network loss function.
4.4. Datasets
4.5. Network Training
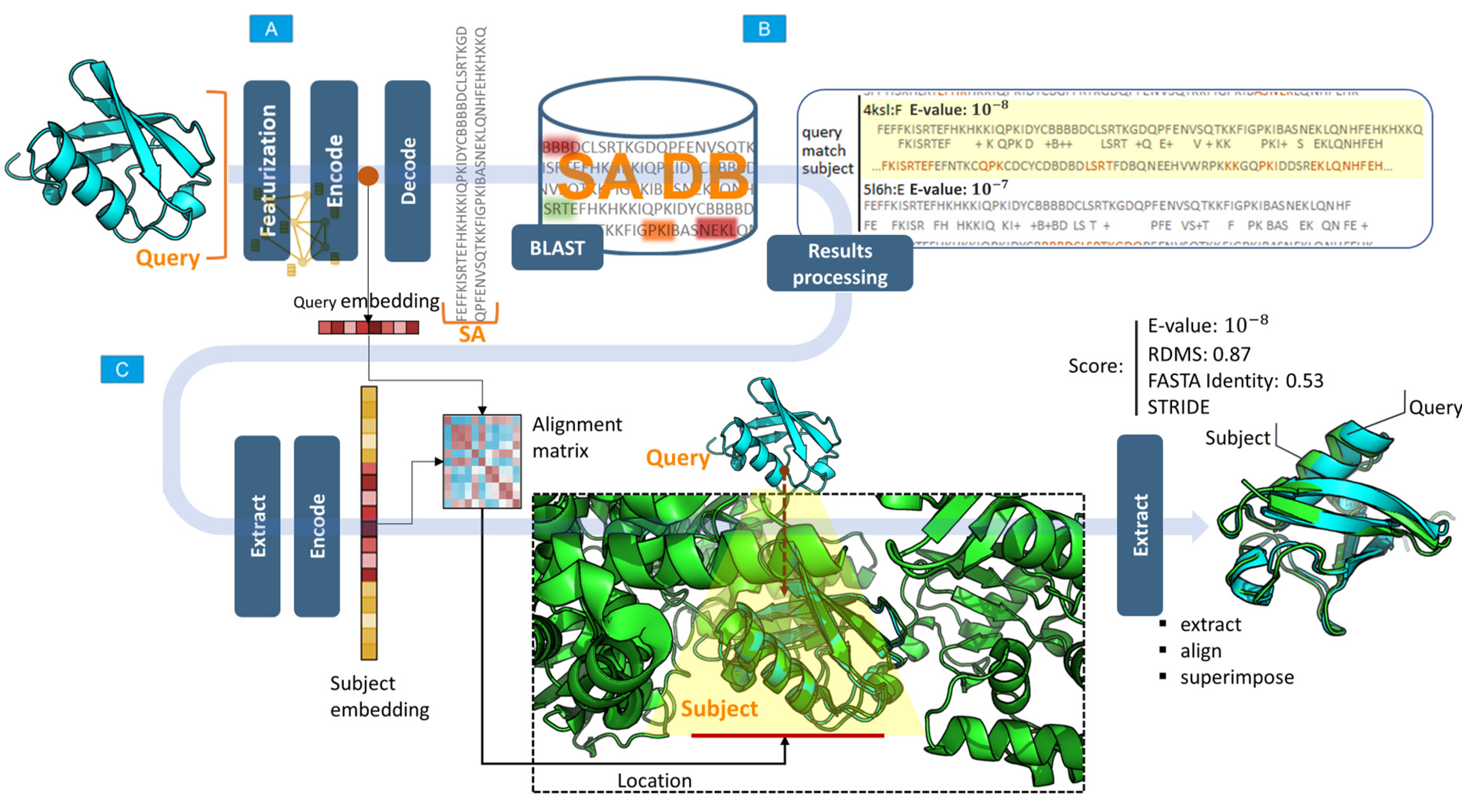
4.6. Algorithm for Searching for Structures with Similar Conformations
- From the requested structure submitted to the input of the network, the features are extracted, and a structure graph (featurization) is created.
- The network encoder block generates a vector of hidden feature representation (embedding) for the requested structure. The input for the search can be either a full chain (as deposited in the PDB) or a fragment.
- The block decoder network (decoder) generates a sequence of SA for the requested structure.
- Using the programing interface provided by the BLAST package, areas of similarity between sequences of the SA of the requested structure in the SA database are determined, and the statistical significance of similarity (E-value) is calculated. At the output, an array is formed comprising the indices of PDB files, elements of the identified areas, and value of the statistical significance index. E-value is a parameter describing the number of matches that can be “expected” or detected by chance when searching or browsing the sequence database. This value decreases exponentially as the number of matches increases. Essentially, the E-value describes random background noise. For instance, an E-value of 1 may imply that, in a SA database, one would detect a match simply by chance by looking at the content of the database. The lower the E-value or the closer it is to zero, the more “significant” the match. The E-value can also be used as a convenient metric to create a threshold of significance in structural similarity searches and in search result reports. The methodology for calculating this indicator is presented in detail elsewhere [17].
- Next, the BLAST search results are sequentially processed, and each protein from the search results containing elements of the requested sequence is passed through the network encoder block (encoder) to obtain a latent representation vector that aggregates the geometric characteristics of the protein. This vector, together with the vector (embeddings) of the desired structure, is used to determine the exact location of the structure in the protein from the search results (this is relevant if a protein fragment is input and it is necessary to determine its exact location in the search results). The location is determined using a matrix of pairwise distances between embeddings, and the region with the smallest distance determines the location. This approach allows for accurately determining the position of a structure in the composition of large proteins.
- After the location is determined, the desired and detected structures are superimposed using the Kabsh algorithm [18], and the RMSD between the Cα atoms of the structures is calculated.
- At the output, a report is generated containing the detected structures, dropped and superimposed on the original structure, and indicators:
- -
- Statistical significance (E-value);
- -
- RMSD of atomic positions;
- -
- Structural coordinates within the protein;
- -
- FASTA matches (Sequence identity);
- -
- Secondary structure identity.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lobo, I. Basic Local Alignment Search Tool (BLAST). Nat. Educ. 2008, 1, 215. [Google Scholar]
- Course Materials for BIO/CSE 597F, Fall 2001. Available online: https://globin.bx.psu.edu/courses/fall2001/ (accessed on 22 February 2023).
- Mak, L.; Grandison, S.; Morris, R.J. An Extension of Spherical Harmonics to Region-Based Rotationally Invariant Descriptors for Molecular Shape Description and Comparison. J. Mol. Graph. Model. 2008, 26, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Guzenko, D.; Burley, S.K.; Duarte, J.M. Real Time Structural Search of the Protein Data Bank. PLoS Comput. Biol. 2020, 16, e1007970. [Google Scholar] [CrossRef]
- Mavridis, L.; Ritchie, D.W. 3D-Blast: 3D Protein Structure Alignment, Comparison, and Classification Using Spherical Polar Fourier Correlations. Biocomputing 2010, 281–292. [Google Scholar] [CrossRef]
- Holder, A.; Simon, J.; Strauser, J.; Taylor, J.; Shibberu, Y. Dynamic Programming Used to Align Protein Structures with a Spectrum Is Robust. Biology 2013, 2, 1296–1310. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein Disorder Prediction: Implications for Structural Proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.; Harel, D.; Wherland, S.; Sussman, J.L. A 3D Building Blocks Approach to Analyzing and Predicting Structure of Proteins. Proteins: Struct. Funct. Bioinform. 1989, 5, 355–373. [Google Scholar] [CrossRef] [PubMed]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noã«L, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein Flexibility in the Light of Structural Alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Brevern, A.G.; Hazout, S.A. Hybrid Protein Model (HPM): A Method to Compact Protein 3D-structure Information and Physicochemical Properties. In Proceedings of the Seventh International Symposium on String Processing Information Retrieval, A Curuña, Spain, 27–29 September 2000; p. 49. [Google Scholar]
- Yang, J.-M.; Tung, C.-H. Protein Structure Database Search and Evolutionary Classification. Nucleic Acids Res. 2006, 34, 3646–3659. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A Structural Classification of Proteins Database for the Investigation of Sequences and Structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Validation Dataset for SAFoldNet. 2023. Available online: https://figshare.com/s/cf5014ecd95dcac38584 (accessed on 11 September 2023).
- Evaluation Ruleset for SCOP 1.61 Benchmarks. Available online: https://supfam.org/SUPERFAMILY/ruleset.html (accessed on 11 September 2023).
- Petrovsky, D.V.; Rudnev, V.R.; Nikolsky, K.S.; Kulikova, L.I.; Malsagova, K.M.; Kopylov, A.T.; Kaysheva, A.L. PSSNet—An Accurate Super-Secondary Structure for Protein Segmentation. Int. J. Mol. Sci. 2022, 23, 14813. [Google Scholar] [CrossRef] [PubMed]
- Heinig, M.; Frishman, D. STRIDE: A Web Server for Secondary Structure Assignment from Known Atomic Coordinates of Proteins. Nucleic Acids Res. 2004, 32 (Suppl. 2), W500–W502. [Google Scholar] [CrossRef] [PubMed]
- The Statistics of Sequence Similarity Scores. Available online: https://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html (accessed on 21 February 2023).
- LucianoSphere. The Definitive Procedure for Aligning Two Sets of 3D Points with the Kabsch Algorithm. Medium. Available online: https://towardsdatascience.com/the-definitive-procedure-for-aligning-two-sets-of-3d-points-with-the-kabsch-algorithm-a7ec2126c87e (accessed on 21 February 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Description | CATH Family | Service | Total | Accuracy of CATH Superfamily Assignment | Percent Coverage |
|---|---|---|---|---|---|---|
| 2ko3 | Ubiquitin | Beta-grasp (ubiquitin-like) 3.10.20 | RCSB PDB Search | 619 | 619 | 100% |
| 3D-BLAST | 1034 | 629 | 60.8% | |||
| PDBeFold | 1999 | 1768 | 88.4% | |||
| SAFoldNet | 2084 | 1970 | 94.5% | |||
| 2ocs | Pdz3 domain | Pdz3 domain 2.30.42 | RCSB PDB Search | 23 | 23 | 100% |
| 3D-BLAST | 500 | 477 | 95.4% | |||
| PDBeFold | 688 | 655 | 95.3% | |||
| SAFoldNet | 1000 | 935 | 93.5% | |||
| 1tit | Immunoglobulin | Immunoglobulin-like 2.60.40 | RCSB PDB Search | 13 | 13 | 100% |
| 3D-BLAST | 123 | 86 | 68.2% | |||
| PDBeFold | 198 | 148 | 74.7% | |||
| SAFoldNet | 237 | 199 | 83.9% |
| Total | Training Set (60%) | Test Set (30%) | Validation Set (10%) |
|---|---|---|---|
| 88,440 | 53,064 | 26,532 | 8844 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrovskiy, D.V.; Nikolsky, K.S.; Rudnev, V.R.; Kulikova, L.I.; Butkova, T.V.; Malsagova, K.A.; Kopylov, A.T.; Kaysheva, A.L. SAFoldNet: A Novel Tool for Discovering and Aligning Three-Dimensional Protein Structures Based on a Neural Network. Int. J. Mol. Sci. 2023, 24, 14439. https://doi.org/10.3390/ijms241914439
Petrovskiy DV, Nikolsky KS, Rudnev VR, Kulikova LI, Butkova TV, Malsagova KA, Kopylov AT, Kaysheva AL. SAFoldNet: A Novel Tool for Discovering and Aligning Three-Dimensional Protein Structures Based on a Neural Network. International Journal of Molecular Sciences. 2023; 24(19):14439. https://doi.org/10.3390/ijms241914439
Chicago/Turabian StylePetrovskiy, Denis V., Kirill S. Nikolsky, Vladimir R. Rudnev, Liudmila I. Kulikova, Tatiana V. Butkova, Kristina A. Malsagova, Arthur T. Kopylov, and Anna L. Kaysheva. 2023. "SAFoldNet: A Novel Tool for Discovering and Aligning Three-Dimensional Protein Structures Based on a Neural Network" International Journal of Molecular Sciences 24, no. 19: 14439. https://doi.org/10.3390/ijms241914439
APA StylePetrovskiy, D. V., Nikolsky, K. S., Rudnev, V. R., Kulikova, L. I., Butkova, T. V., Malsagova, K. A., Kopylov, A. T., & Kaysheva, A. L. (2023). SAFoldNet: A Novel Tool for Discovering and Aligning Three-Dimensional Protein Structures Based on a Neural Network. International Journal of Molecular Sciences, 24(19), 14439. https://doi.org/10.3390/ijms241914439