
EGR1 Regulation of Vasculogenic Mimicry in the MDA-MB-231 Triple-Negative Breast Cancer Cell Line through the Upregulation of KLF4 Expression

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Vasculogenic Mimicry of Aggressive Breast Cancer Cells
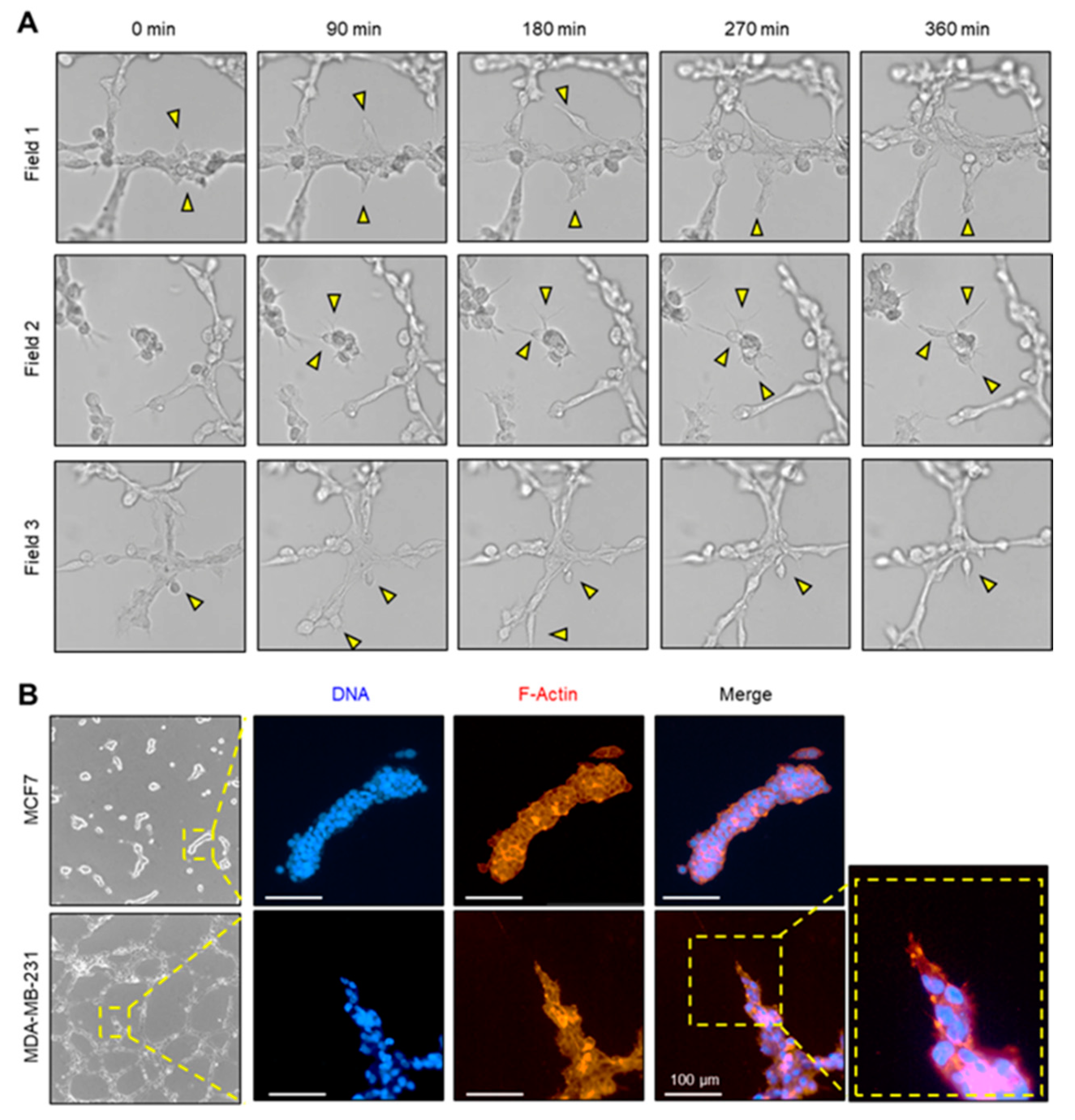
2.2. Tip-Cell-Like Behavior during VM Formation Was Observed in MDA-MB-231 Cells by Live Cell Imaging
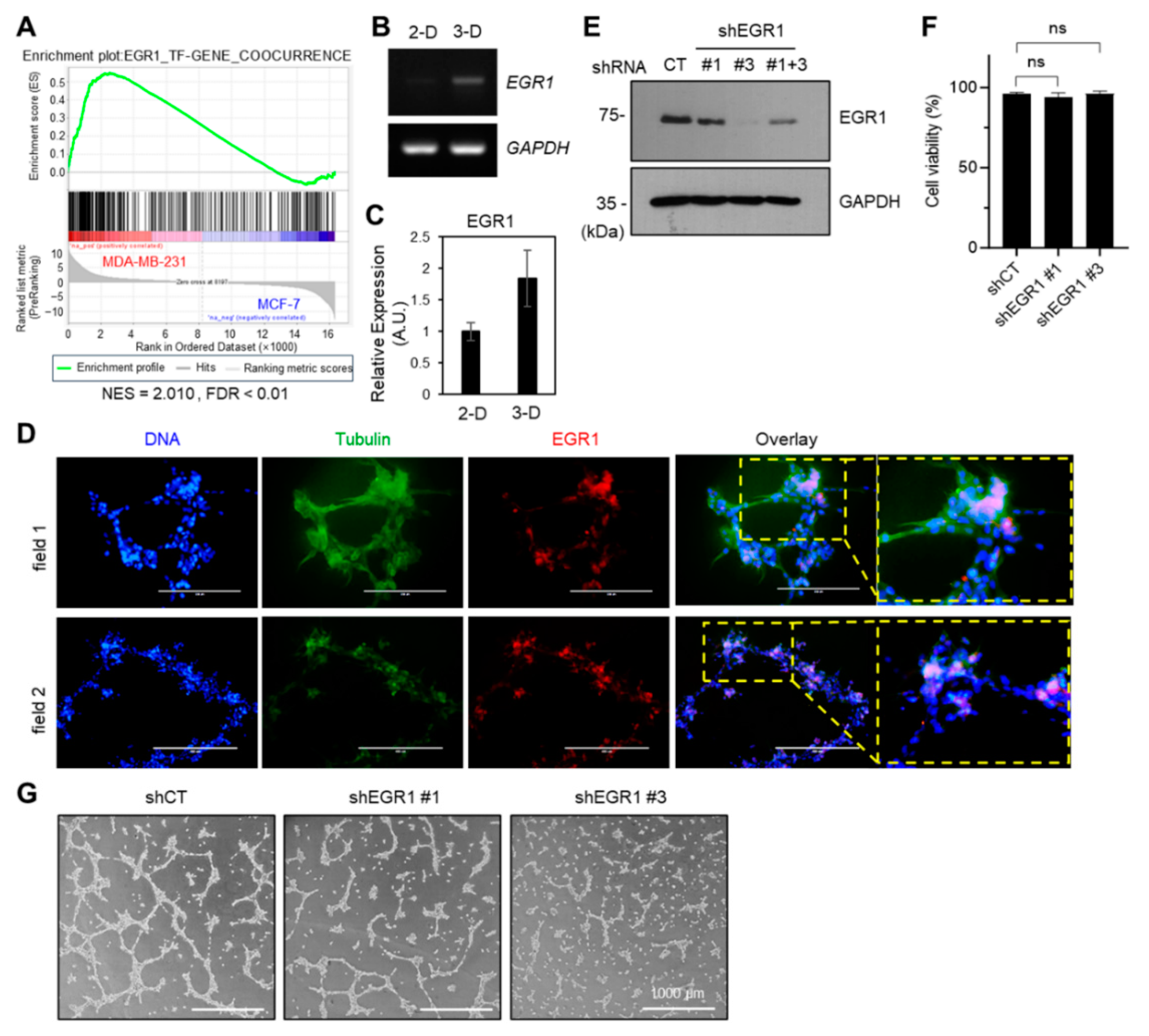
2.3. EGR1 Positively Modulated the VM Formation
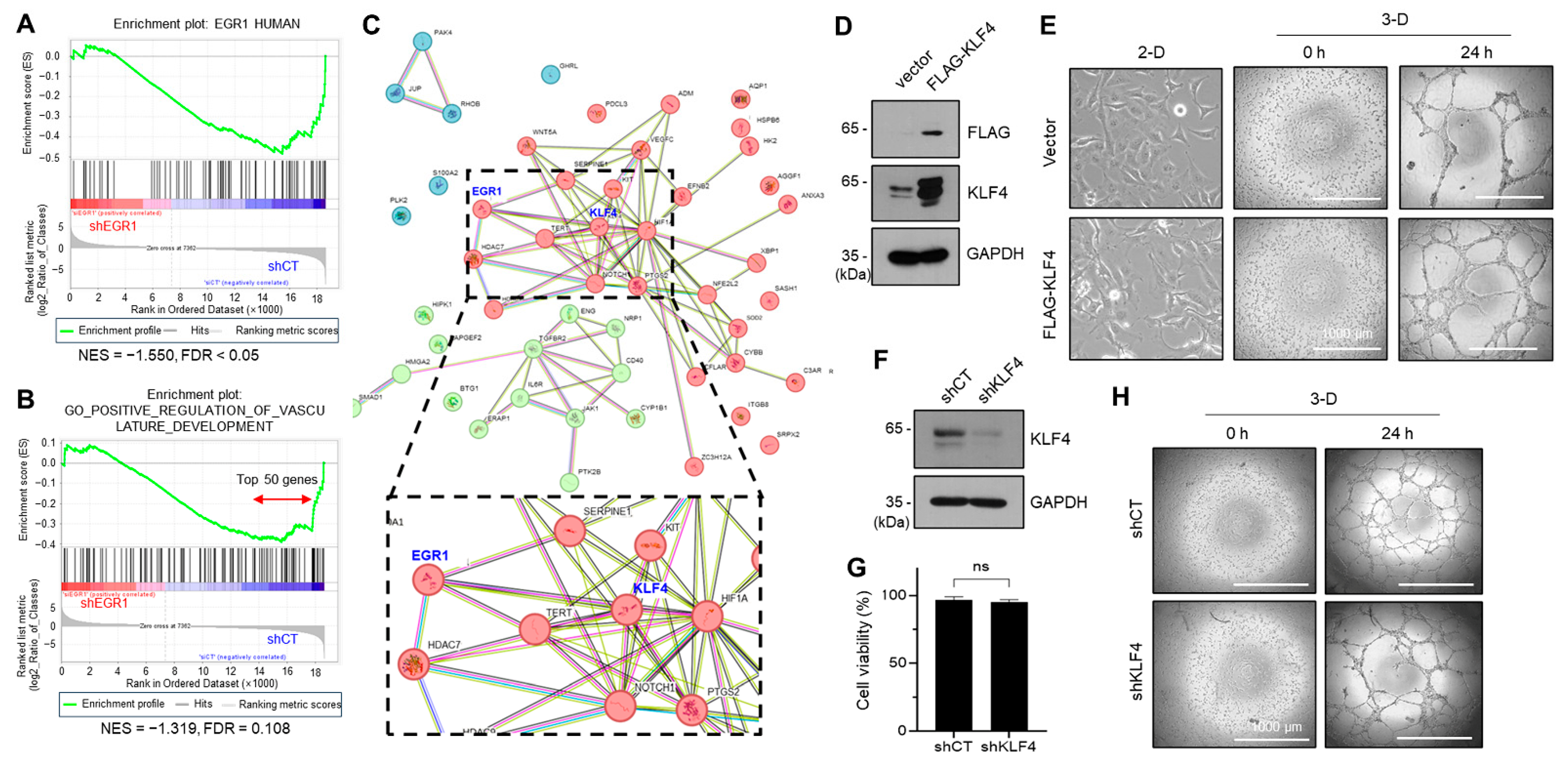
2.4. KLF4, a Yamanaka Factor, Positively Regulates the VM Formation of Aggressive Breast Cancer Cell
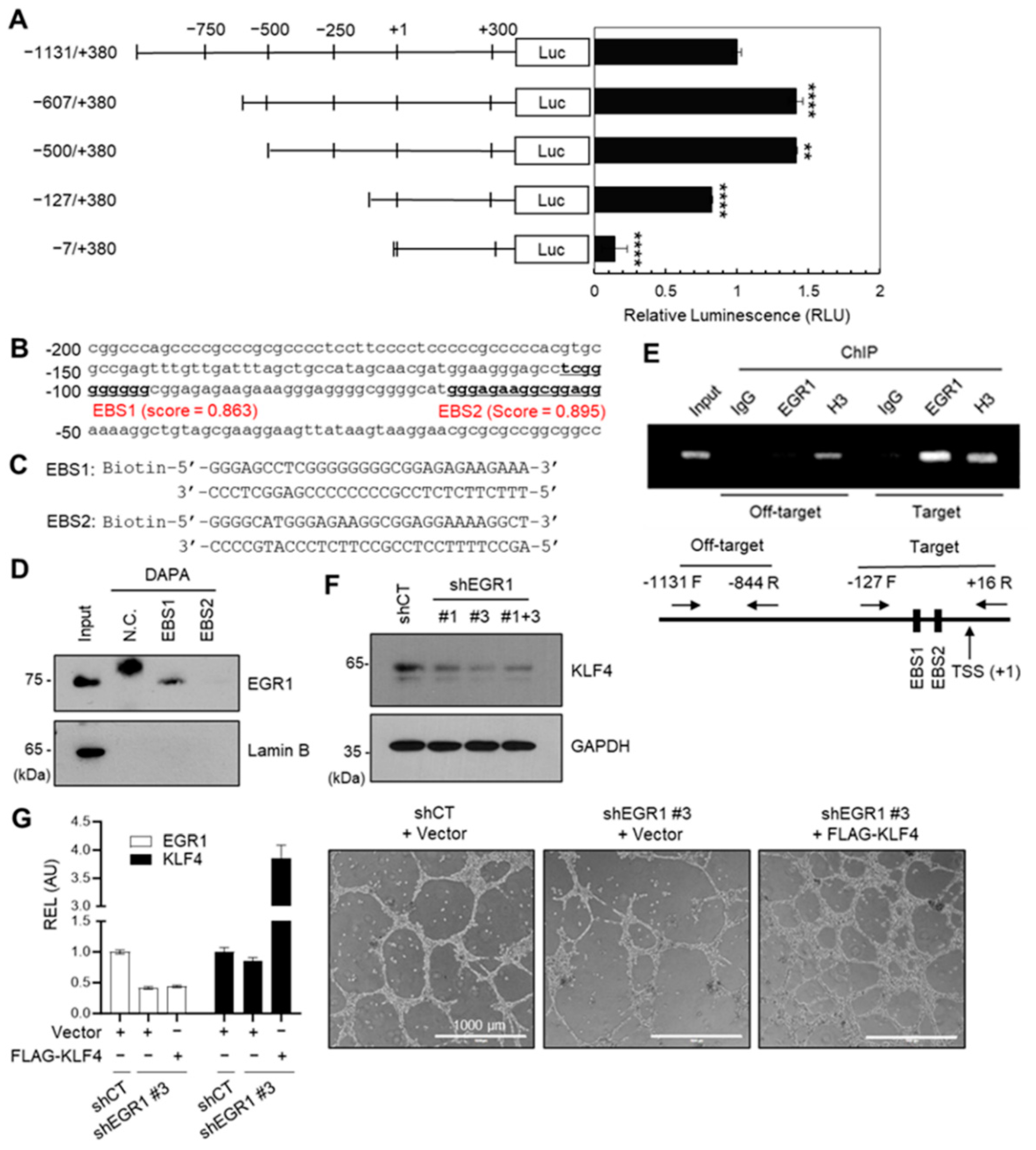
2.5. EGR1 Regulates KLF4 Expression
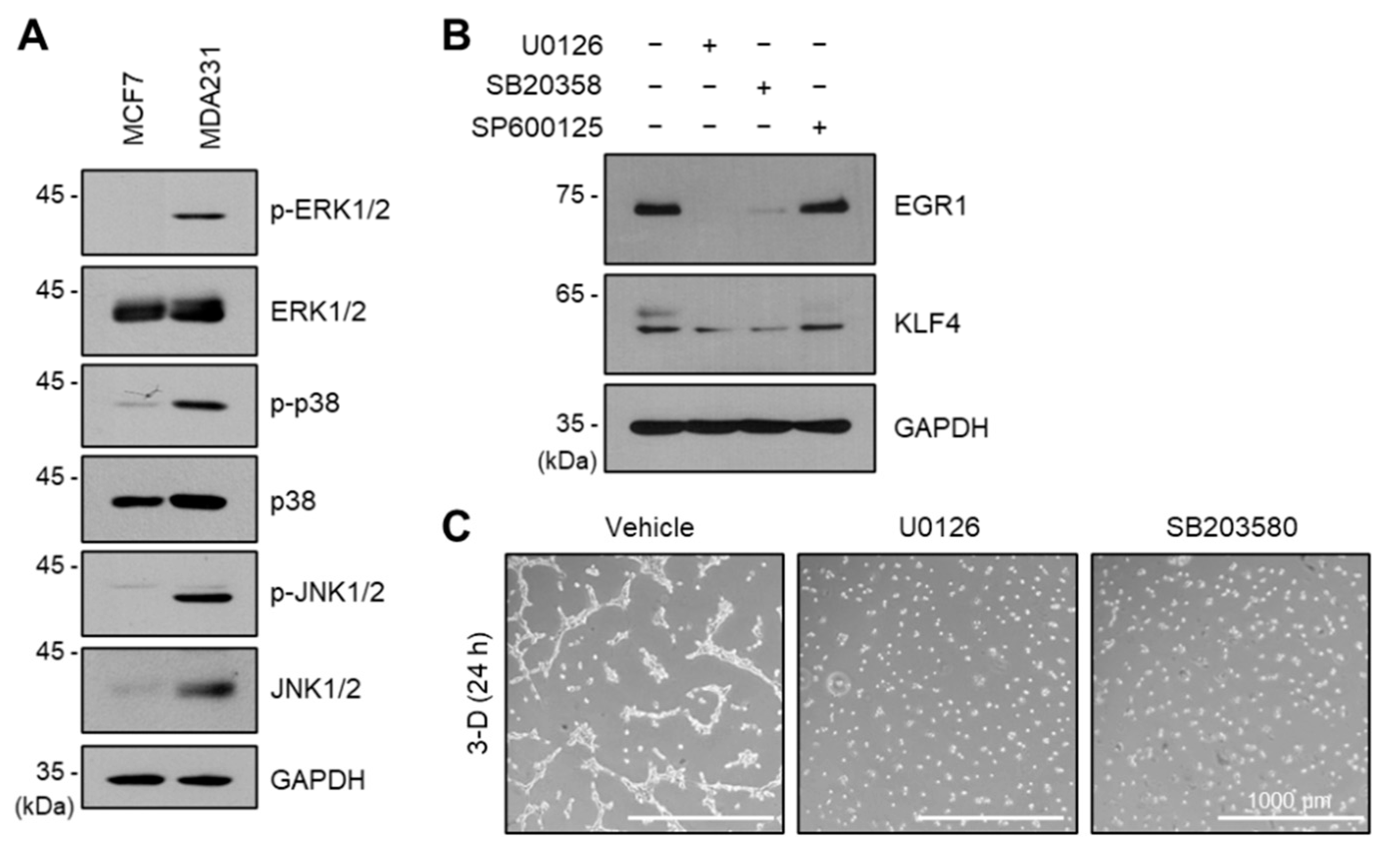
2.6. ERK and p38 Kinases Regulate EGR1 and KLF4 Expression
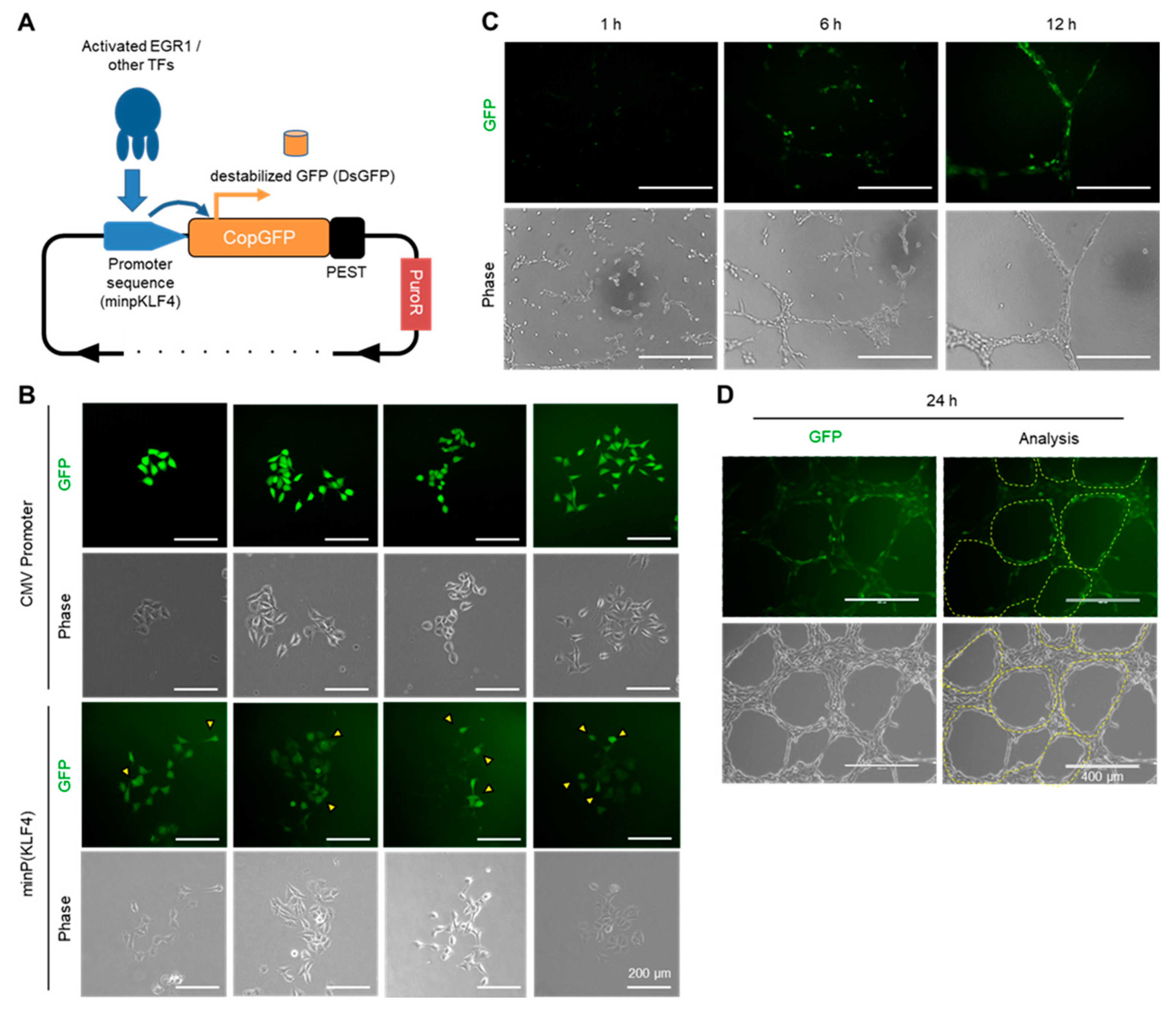
2.7. Visualizing the Activation of the EGR1-KLF4 Signaling Pathway during VM Formation Using a Fluorescent-Based Reporter System
3. Discussion
4. Materials and Methods
4.1. Reagents and Plasmids
4.2. Cell Culture
4.3. Animal Study
4.4. VM Tube Formation Assay
4.5. Transcriptome Analysis
4.6. Reverse Transcription (RT)-PCR and Quantitative Real-Time PCR (qPCR)
4.7. Generation of MDA-MB-231 Transfectants Expressing shRNA
4.8. Immunoblotting
4.9. Cell Viability Assay
4.10. KLF4 Promoter Cloning and PROMOTER-Reporter Activity Assay
4.11. DNA Affinity Precipitation Assay (DAPA)
4.12. Chromatin Immunoprecipitation (ChIP) Assay
4.13. The Fluorescent Reporter for KLF4 Promoter
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ChIP | Chromatin immunoprecipitation |
| DAPA | DNA affinity precipitation assay |
| EGR1 | Early Growth Response 1 |
| EBS | EGR1-binding sequence |
| EMT | Epithelial-to-mesenchymal transition |
| GSEA | Gene Set Enrichment Analysis |
| GEO | Gene Expression Omnibus |
| KLF4 | Kruppel-like factor 4 |
| MAPK | Mitogen-activated protein kinase |
| MSC | Mesenchymal stem cell |
| PAS | Periodic acid-Schiff |
| RT-PCR | Reverse transcriptase-PCR |
| shRNA | Short hairpin RNA |
| VM | Vasculogenic mimicry |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.; Hess, A.R.; Seftor, E.A.; Kirschmann, D.A.; Hardy, K.M.; Margaryan, N.V.; Hendrix, M.J. Tumor Cell Vasculogenic Mimicry: From Controversy to Therapeutic Promise. Am. J. Pathol. 2012, 181, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [PubMed]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A New Alternative Mechanism in Glioblastoma Vascularization: Tubular Vasculogenic Mimicry. Brain 2010, 133, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.E.; Simms, N.; Polanski, R.; et al. Vasculogenic Mimicry in Small Cell Lung Cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef]
- Wu, S.; Yu, L.; Wang, D.; Zhou, L.; Cheng, Z.; Chai, D.; Ma, L.; Tao, Y. Aberrant Expression of CD133 in Non-Small Cell Lung Cancer and Its Relationship to Vasculogenic Mimicry. BMC Cancer 2012, 12, 535. [Google Scholar] [CrossRef]
- Liu, T.J.; Sun, B.C.; Zhao, X.L.; Zhao, X.M.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.Y.; Zhao, N.; Liu, N. CD133 + Cells with Cancer Stem Cell Characteristics Associates with Vasculogenic Mimicry in Triple-Negative Breast Cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef]
- Valdivia, A.; Mingo, G.; Aldana, V.; Pinto, M.P.; Ramirez, M.; Retamal, C.; Gonzalez, A.; Nualart, F.; Corvalan, A.H.; Owen, G.I. Fact or Fiction, It Is Time for a Verdict on Vasculogenic Mimicry? Front. Oncol. 2019, 9, 680. [Google Scholar] [CrossRef]
- Liu, Q.; Qiao, L.; Liang, N.; Xie, J.; Zhang, J.; Deng, G.; Luo, H.; Zhang, J. The Relationship between Vasculogenic Mimicry and Epithelial-Mesenchymal Transitions. J. Cell. Mol. Med. 2016, 20, 1761–1769. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic Mimicry in Carcinogenesis and Clinical Applications. J. Hematol. Oncol. 2020, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Welch-Reardon, K.M.; Wu, N.; Hughes, C.C.W. A Role for Partial Endothelial-Mesenchymal Transitions in Angiogenesis? Arter. Thromb. Vasc. Biol. 2015, 35, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Faure, S.; Clere, N. Vasculogenic Mimicry, a Complex and Devious Process Favoring Tumorigenesis—Interest in Making It a Therapeutic Target. Pharmacol. Ther. 2021, 223, 107805. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Du, K.; Guan, W.; Wu, D.; Tang, H.; Wang, N.; Qi, J.; Gu, Z.; Yang, J.; Ding, J. A Novel Definition of Microvessel Density in Renal Cell Carcinoma: Angiogenesis plus Vasculogenic Mimicry. Oncol. Lett. 2020, 20, 192. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reynolds, A.R. Anti-Angiogenic Therapy for Cancer: Current Progress, Unresolved Questions and Future Directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef]
- Gashler, A.; Sukhatme, V.P. Early Growth Response Protein 1 (Egr-1): Prototype of a Zinc-Finger Family of Transcription Factors. Prog. Nucleic Acid Res. Mol. Biol. 1995, 50, 191–224. [Google Scholar]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front. Oncol. 2021, 11, 642547. [Google Scholar] [CrossRef]
- Li, L.; Ameri, A.H.; Wang, S.; Jansson, K.H.; Casey, O.M.; Yang, Q.; Beshiri, M.L.; Fang, L.; Lake, R.G.; Agarwal, S.; et al. EGR1 Regulates Angiogenic and Osteoclastogenic Factors in Prostate Cancer and Promotes Metastasis. Oncogene 2019, 38, 6241–6255. [Google Scholar] [CrossRef]
- Wu, S.Y.; Rupaimoole, R.; Shen, F.; Pradeep, S.; Pecot, C.V.; Ivan, C.; Nagaraja, A.S.; Gharpure, K.M.; Pham, E.; Hatakeyama, H.; et al. A MiR-192-EGR1-HOXB9 Regulatory Network Controls the Angiogenic Switch in Cancer. Nat. Commun. 2016, 7, 11169. [Google Scholar] [CrossRef]
- Guerquin, M.J.; Charvet, B.; Nourissat, G.; Havis, E.; Ronsin, O.; Bonnin, M.A.; Ruggiu, M.; Olivera-Martinez, I.; Robert, N.; Lu, Y.; et al. Transcription Factor EGR1 Directs Tendon Differentiation and Promotes Tendon Repair. J. Clin. Investig. 2013, 123, 3564–3576. [Google Scholar] [CrossRef]
- Jung, E.; Ou, S.; Ahn, S.S.; Yeo, H.; Lee, Y.H.; Shin, S.Y. The JNK-EGR1 Signaling Axis Promotes TNF-α-Induced Endothelial Differentiation of Human Mesenchymal Stem Cells via VEGFR2 Expression. Cell Death Differ. 2023, 30, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, G.; Chen, X.; Chen, Z.; Tan, A.Y.; Lin, A.; Zhang, C.; Torres, L.K.; Bajrami, S.; Zhang, T.; et al. Low-Dose Carbon Monoxide Suppresses Metastatic Progression of Disseminated Cancer Cells. Cancer Lett. 2022, 546, 215831. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. A Decade of Transcription Factor-Mediated Reprogramming to Pluripotency. Nat. Rev. Mol. Cell. Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Fatma, H.; Siddique, H.R. Pluripotency Inducing Yamanaka Factors: Role in Stemness and Chemoresistance of Liver Cancer. Expert Rev. Anticancer Ther. 2021, 21, 853–864. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and Reprogramming: Origins of Cancer Stem Cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Chen, H.F.; Huang, C.H.; Liu, C.J.; Hung, J.J.; Hsu, C.C.; Teng, S.C.; Wu, K.J. Twist1 Induces Endothelial Differentiation of Tumour Cells through the Jagged1-KLF4 Axis. Nat. Commun. 2014, 5, 4697. [Google Scholar] [CrossRef]
- Hollestelle, A.; Elstrodt, F.; Nagel, J.H.A.; Kallemeijn, W.W.; Schutte, M. Phosphatidylinositol-3-OH Kinase or RAS Pathway Mutations in Human Breast Cancer Cell Lines. Mol. Cancer Res. 2007, 5, 195–201. [Google Scholar] [CrossRef]
- Di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef]
- Fan, G.; Lou, L.; Song, Z.; Zhang, X.; Xiong, X.F. Targeting Mutated GTPase KRAS in Tumor Therapies. Eur. J. Med. Chem. 2021, 226, 113816. [Google Scholar] [CrossRef]
- Tang, B.; Raviv, A.; Esposito, D.; Flanders, K.C.; Daniel, C.; Nghiem, B.T.; Garfield, S.; Lim, L.; Mannan, P.; Robles, A.I.; et al. A Flexible Reporter System for Direct Observation and Isolation of Cancer Stem Cells. Stem Cell Rep. 2015, 4, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Forster, J.; Harriss-Phillips, W.; Douglass, M.; Bezak, E. A Review of the Development of Tumor Vasculature and Its Effects on the Tumor Microenvironment. Hypoxia 2017, 5, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Specialization of Tumour Vasculature. Nat. Rev. Cancer 2002, 2, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun Signal 2020, 18, 59. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Shan, L.N.; Song, Y.G.; Su, D.; Liu, Y.L.; Shi, X.B.; Lu, S.J. Early Growth Response Protein-1 Involves in Transforming Growth Factor-Β1 Induced Epithelial-Mesenchymal Transition and Inhibits Migration of Non-Small-Cell Lung Cancer Cells. Asian Pac. J. Cancer Prev. 2015, 16, 4137–4142. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, C.; Zhao, B.; Li, Z.; Li, T.; Yang, X.; Zhao, Y.; Wang, W. EGR1 Induces EMT in Pancreatic Cancer via a P300/SNAI2 Pathway. J. Transl. Med. 2023, 21, 201. [Google Scholar] [CrossRef]
- Arner, E.N.; Du, W.; Brekken, R.A. Behind the Wheel of Epithelial Plasticity in KRAS-Driven Cancers. Front. Oncol. 2019, 9, 1049. [Google Scholar] [CrossRef]
- Lizárraga-Verdugo, E.; Avendaño-Félix, M.; Bermúdez, M.; Ramos-Payán, R.; Pérez-Plasencia, C.; Aguilar-Medina, M. Cancer Stem Cells and Its Role in Angiogenesis and Vasculogenic Mimicry in Gastrointestinal Cancers. Front. Oncol. 2020, 10, 413. [Google Scholar] [CrossRef]
- Yao, X.; Ping, Y.; Bian, X. Contribution of Cancer Stem Cells to Tumor Vasculogenic Mimicry. Protein Cell 2011, 2, 266–272. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Jensen, L.J.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-Tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, E.; Lee, Y.H.; Ou, S.; Kim, T.Y.; Shin, S.Y. EGR1 Regulation of Vasculogenic Mimicry in the MDA-MB-231 Triple-Negative Breast Cancer Cell Line through the Upregulation of KLF4 Expression. Int. J. Mol. Sci. 2023, 24, 14375. https://doi.org/10.3390/ijms241814375
Jung E, Lee YH, Ou S, Kim TY, Shin SY. EGR1 Regulation of Vasculogenic Mimicry in the MDA-MB-231 Triple-Negative Breast Cancer Cell Line through the Upregulation of KLF4 Expression. International Journal of Molecular Sciences. 2023; 24(18):14375. https://doi.org/10.3390/ijms241814375
Chicago/Turabian StyleJung, Euitaek, Young Han Lee, Sukjin Ou, Tae Yoon Kim, and Soon Young Shin. 2023. "EGR1 Regulation of Vasculogenic Mimicry in the MDA-MB-231 Triple-Negative Breast Cancer Cell Line through the Upregulation of KLF4 Expression" International Journal of Molecular Sciences 24, no. 18: 14375. https://doi.org/10.3390/ijms241814375
APA StyleJung, E., Lee, Y. H., Ou, S., Kim, T. Y., & Shin, S. Y. (2023). EGR1 Regulation of Vasculogenic Mimicry in the MDA-MB-231 Triple-Negative Breast Cancer Cell Line through the Upregulation of KLF4 Expression. International Journal of Molecular Sciences, 24(18), 14375. https://doi.org/10.3390/ijms241814375