
A Benchmark Study of Protein–Fragment Complex Structure Calculations with NMR2



Abstract
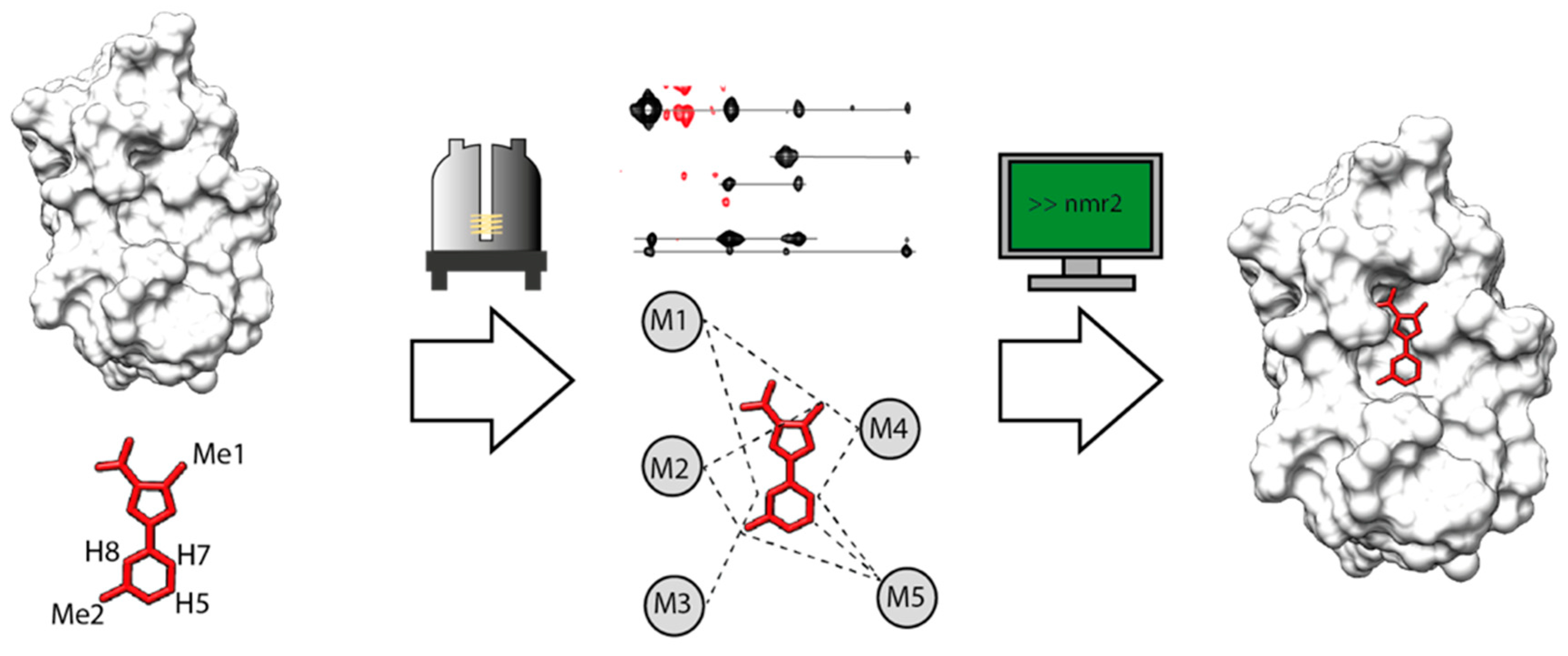
:1. Introduction
2. Results
2.1. NMR2 Performances with Exact Distances
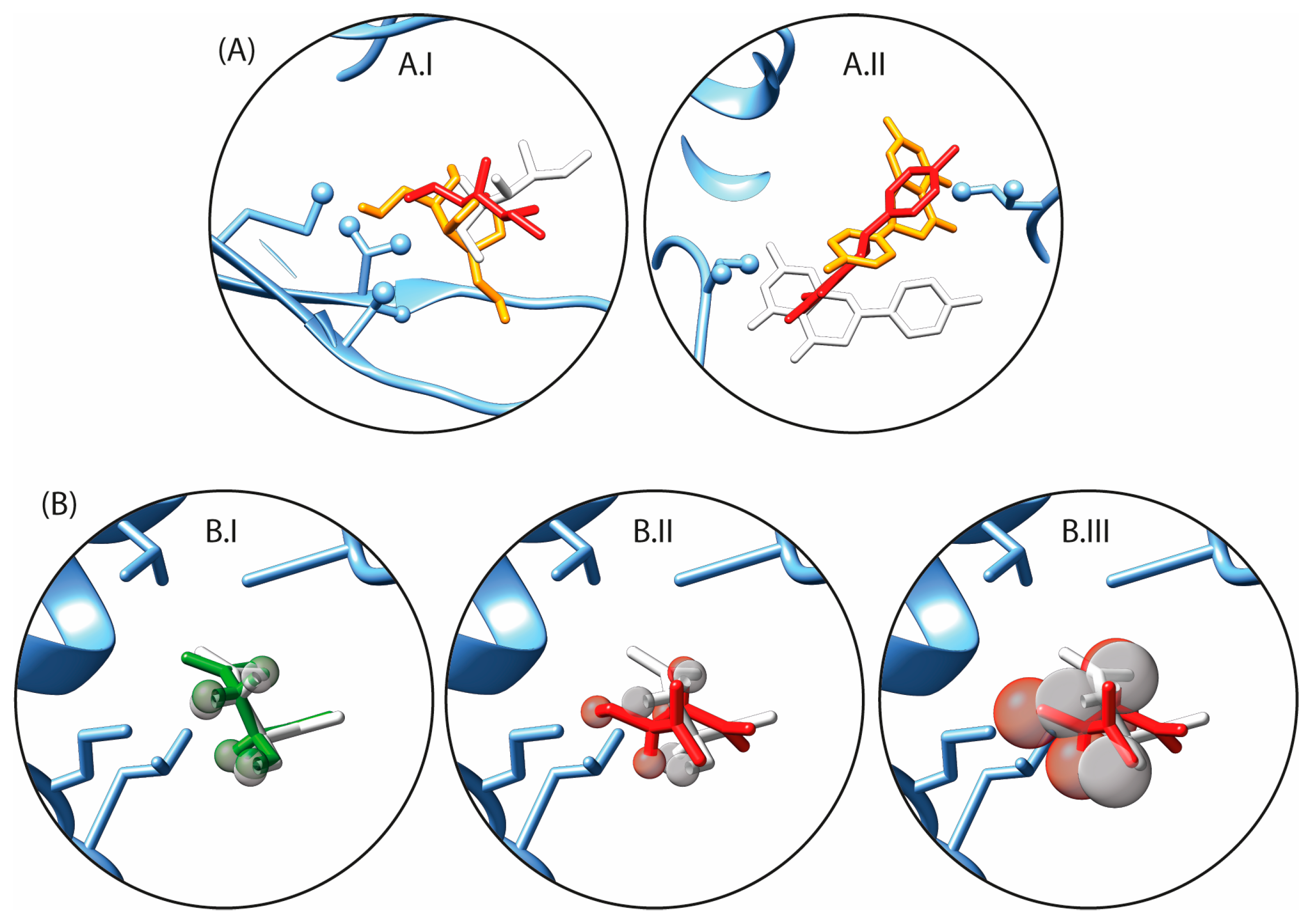
2.2. Conditions on the Binding Site Topology
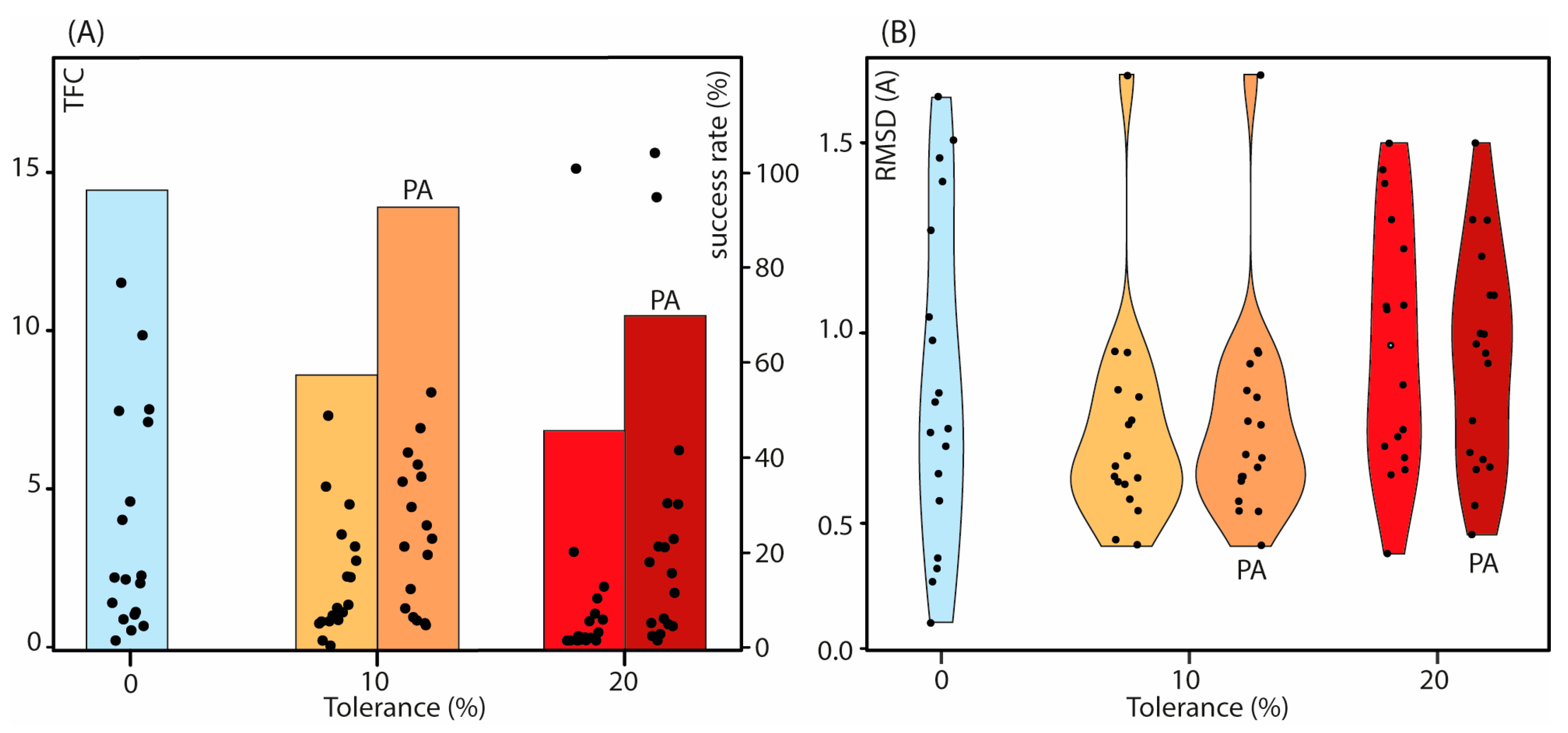
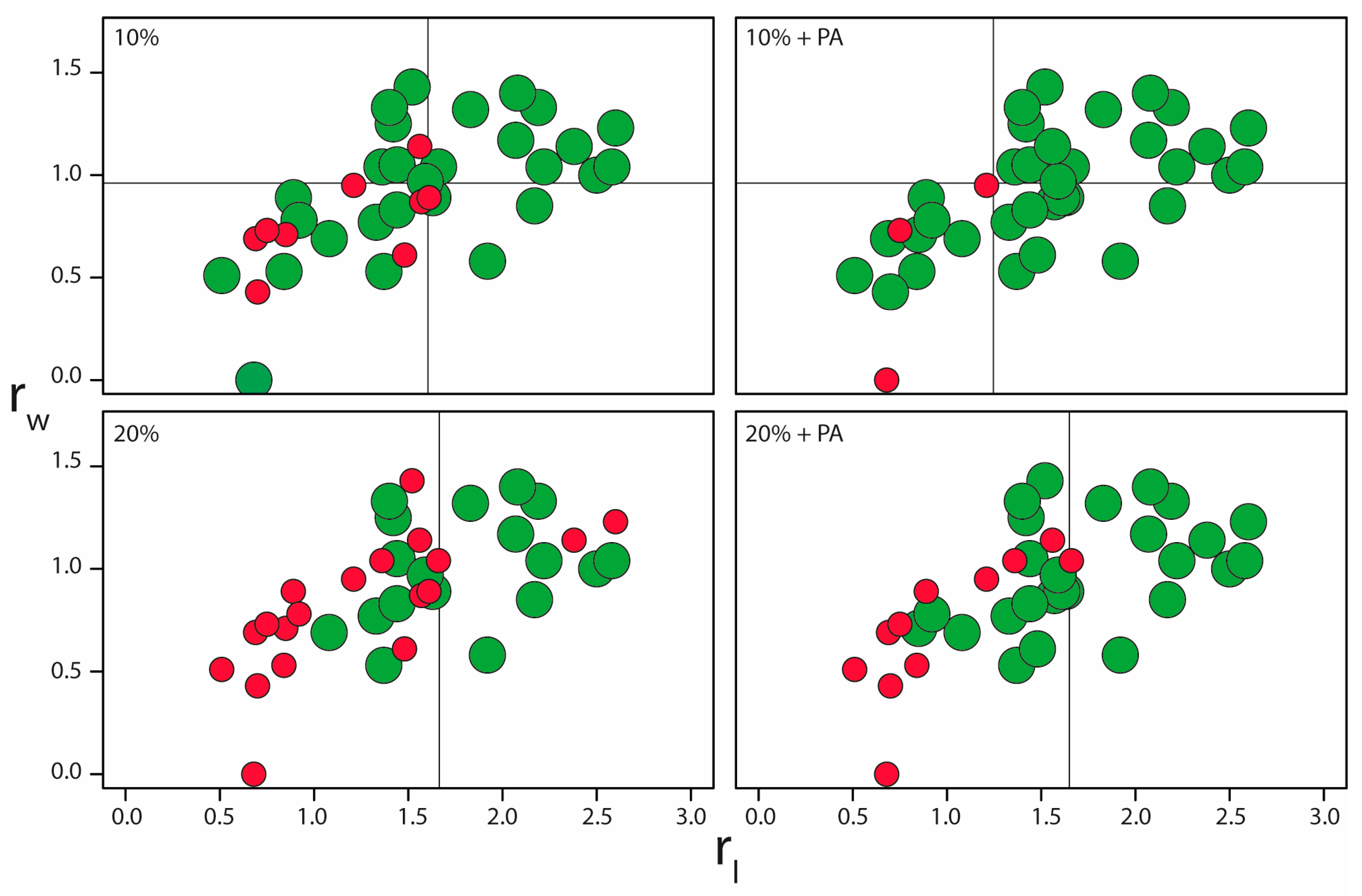
2.3. Impact of the Distance Restraints Tolerance and Protein Partial Assignment on the NMR2 Convergence
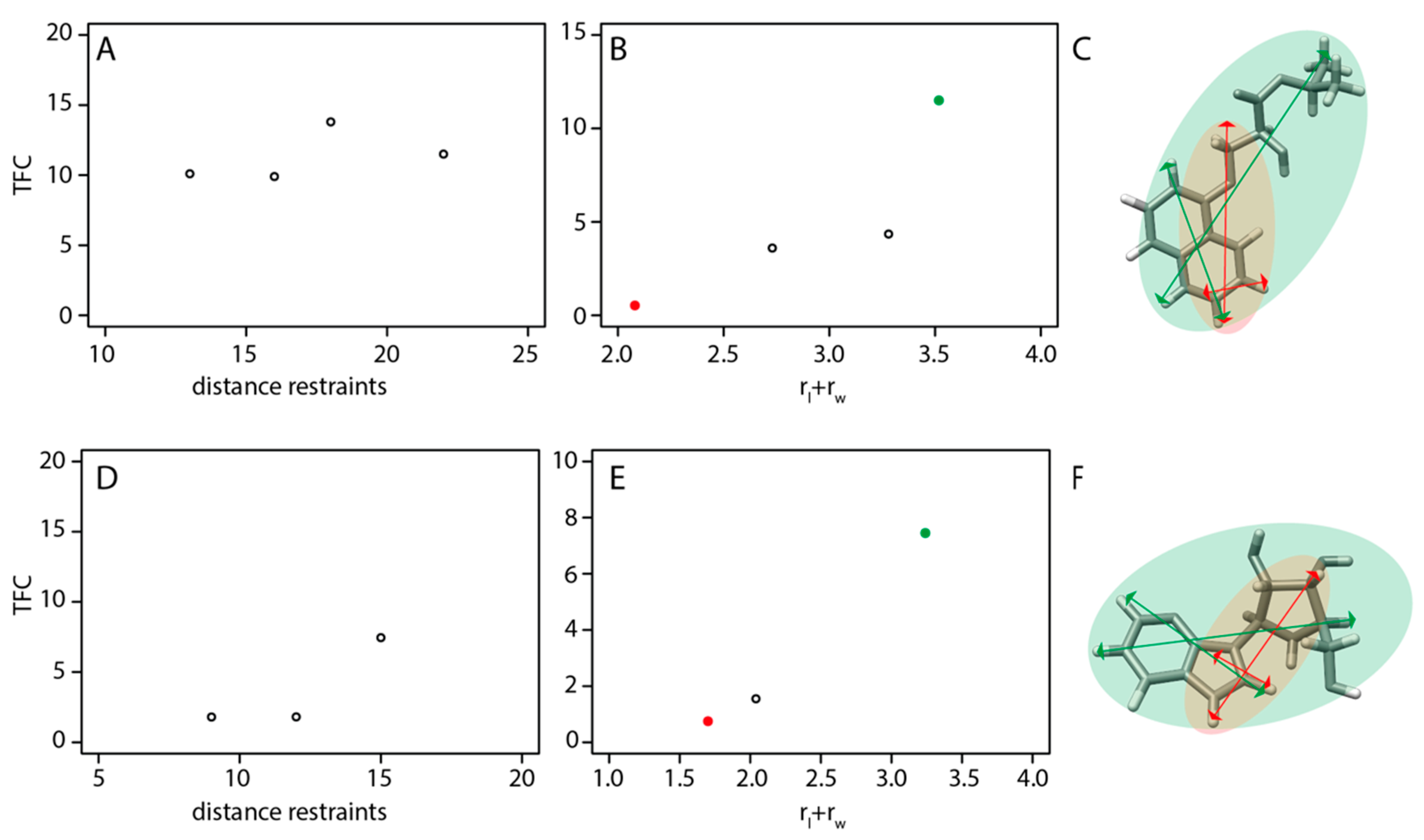
2.4. Distance Restraints Network Topology Is Critical for a Successful NMR2 Structure Calculation
2.5. Computational Time
3. Discussion
4. Material and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fesik, S.W.; Hajduk, P.J.; Sheppard, G.; Olejniczak, E.T.; Nettesheim, D.G.; Meadows, R.P.; Marcotte, P.A.; Shuker, S.B.; Steinman, D.H.; Carrera, G.M.; et al. Discovery of potent nonpeptide inhibitors of matrix metalloproteinases using SAR by NMR. Abstr. Pap. Am. Chem. Soc. 1997, 214, 109-Medi. [Google Scholar]
- Hajduk, P.J.; Sheppard, G.; Nettesheim, D.G.; Olejniczak, E.T.; Shuker, S.B.; Meadows, R.P.; Steinman, D.H.; Carrera, G.M.; Marcotte, P.A.; Severin, J.; et al. Discovery of potent nonpeptide inhibitors of stromelysin using SAR by NMR. J. Am. Chem. Soc. 1997, 119, 5818–5827. [Google Scholar] [CrossRef]
- Schonauer, E.; Kany, A.M.; Haupenthal, J.; Husecken, K.; Hoppe, I.J.; Voos, K.; Yahiaoui, S.; Elsasser, B.; Ducho, C.; Brandstetter, H.; et al. Discovery of a Potent Inhibitor Class with High Selectivity toward Clostridial Collagenases. J. Am. Chem. Soc. 2017, 139, 12696–12703. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Greer, J. A decade of fragment-based drug design: Strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Norton, R.S.; Scanlon, M.J. The ways and means of fragment-based drug design. Pharmacol. Ther. 2016, 167, 28–37. [Google Scholar] [CrossRef]
- Gorse, A.D. Diversity in medicinal chemistry space. Curr. Top. Med. Chem. 2006, 6, 3–18. [Google Scholar] [CrossRef]
- Reymond, J.L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef]
- Fink, T.; Bruggesser, H.; Reymond, J.L. Virtual exploration of the small-molecule chemical universe below 160 Daltons. Angew. Chem. Int. Ed. 2005, 44, 1504–1508. [Google Scholar] [CrossRef]
- Mattos, C.; Ringe, D. Locating and characterizing binding sites on proteins. Nat. Biotechnol. 1996, 14, 595–599. [Google Scholar] [CrossRef]
- Geschwindner, S.; Olsson, L.L.; Albert, J.S.; Deinum, J.; Edwards, P.D.; de Beer, T.; Folmer, R.H. Discovery of a novel warhead against β-secretase through fragment-based lead generation. J. Med. Chem. 2007, 50, 5903–5911. [Google Scholar] [CrossRef]
- Ahn, S.; Kahsai, A.W.; Pani, B.; Wang, Q.T.; Zhao, S.; Wall, A.L.; Strachan, R.T.; Staus, D.P.; Wingler, L.M.; Sun, L.D.; et al. Allosteric “β-blocker” isolated from a DNA-encoded small molecule library. Proc. Natl. Acad. Sci. USA 2017, 114, 1708–1713. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Verdonk, M.L.; Rees, D.C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Bauman, J.D.; Arnold, E. Advantages of crystallographic fragment screening: Functional and mechanistic insights from a powerful platform for efficient drug discovery. Prog. Biophys. Mol. Biol. 2014, 116, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M.; Correy, G.J.; Gahbauer, S.; Fearon, D.; Wu, T.; Díaz, R.E.; Young, I.D.; Martins, L.C.; Smith, D.H.; Schulze-Gahmen, U.; et al. Fragment Binding to the Nsp3 Macrodomain of SARS-CoV-2 Identified Through Crystallographic Screening and Computational Docking. bioRxiv 2020. [Google Scholar] [CrossRef]
- Potter, A.; Oldfield, V.; Nunns, C.; Fromont, C.; Ray, S.; Northfield, C.J.; Bryant, C.J.; Scrace, S.F.; Robinson, D.; Matossova, N.; et al. Discovery of cell-active phenyl-imidazole Pin1 inhibitors by structure-guided fragment evolution. Bioorg. Med. Chem. Lett. 2010, 20, 6483–6488. [Google Scholar] [CrossRef]
- Walti, M.A.; Riek, R.; Orts, J. Fast NMR-Based Determination of the 3D Structure of the Binding Site of Protein-Ligand Complexes with Weak Affinity Binders. Angew. Chem. Int. Ed. 2017, 56, 5208–5211. [Google Scholar] [CrossRef]
- Torres, F.; Ghosh, D.; Strotz, D.; Chi, C.N.; Davis, B.; Orts, J. Protein–fragment complex structures derived by NMR molecular replacement. RSC Med. Chem. 2020, 11, 591–596. [Google Scholar] [CrossRef]
- Favia, A.D.; Bottegoni, G.; Nobeli, I.; Bisignano, P.; Cavalli, A. SERAPhiC: A benchmark for in silico fragment-based drug design. J. Chem. Inf. Model. 2011, 51, 2882–2896. [Google Scholar] [CrossRef]
- Ren, X.D.; Shi, Y.S.; Zhang, Y.; Liu, B.; Zhang, L.H.; Peng, Y.B.; Zeng, R. Novel Consensus Docking Strategy to Improve Ligand Pose Prediction. J. Chem. Inf. Model. 2018, 58, 1662–1668. [Google Scholar] [CrossRef]
- Morrone, J.A.; Weber, J.K.; Huynh, T.; Luo, H.; Cornell, W.D. Combining Docking Pose Rank and Structure with Deep Learning Improves Protein-Ligand Binding Mode Prediction over a Baseline Docking Approach. J. Chem. Inf. Model. 2020, 60, 4170–4179. [Google Scholar] [CrossRef] [PubMed]
- Ammar, A.; Cavill, R.; Evelo, C.; Willighagen, E. PSnpBind-ML: Predicting the effect of binding site mutations on protein-ligand binding affinity. J. Cheminform. 2023, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Strotz, D.; Orts, J.; Chi, C.N.; Riek, R.; Vogeli, B. eNORA2 Exact NOE Analysis Program. J. Chem. Theory Comput. 2017, 13, 4336–4346. [Google Scholar] [CrossRef] [PubMed]
- Zwahlen, C.; Legault, P.; Vincent, S.J.F.; Greenblatt, J.; Konrat, R.; Kay, L.E. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage lambda N-peptide/boxB RNA complex. J. Am. Chem. Soc. 1997, 119, 6711–6721. [Google Scholar] [CrossRef]
- Kerfah, R.; Plevin, M.J.; Sounier, R.; Gans, P.; Boisbouvier, J. Methyl-specific isotopic labeling: A molecular tool box for solution NMR studies of large proteins. Curr. Opin. Struct. Biol. 2015, 32, 113–122. [Google Scholar] [CrossRef]
- Kerfah, R.; Plevin, M.J.; Pessey, O.; Hamelin, O.; Gans, P.; Boisbouvier, J. Scrambling free combinatorial labeling of alanine-β, isoleucine-δ1, leucine-proS and valine-proS methyl groups for the detection of long range NOEs. J. Biomol. NMR 2015, 61, 73–82. [Google Scholar] [CrossRef]
- Torres, F.; Walser, R.; Kaderli, J.; Rossi, E.; Bobby, R.; Packer, M.J.; Sarda, S.; Walker, G.; Hitchin, J.R.; Milbradt, A.G.; et al. NMR Molecular Replacement Provides New Insights into Binding Modes to Bromodomains of BRD4 and TRIM24. J. Med. Chem. 2022, 65, 5565–5574. [Google Scholar] [CrossRef]
- Mohanty, B.; Orts, J.; Wang, G.; Nebl, S.; Alwan, W.S.; Doak, B.C.; Williams, M.L.; Heras, B.; Mobli, M.; Scanlon, M.J. Methyl probes in proteins for determining ligand binding mode in weak protein-ligand complexes. Sci. Rep. 2022, 12, 11231. [Google Scholar] [CrossRef]
- Orts, J.; Riek, R. Protein—Ligand structure determination with the NMR molecular replacement tool, NMR2. J. Biomol. NMR 2020, 74, 633–642. [Google Scholar] [CrossRef]
- Guntert, P.; Mumenthaler, C.; Wuthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997, 273, 283–298. [Google Scholar] [CrossRef]
- Bax, A.; Clore, G.M.; Gronenborn, A.M. H-1-H-1 Correlation Via Isotropic Mixing of C-13 Magnetization, a New 3-Dimensional Approach for Assigning H-1 and C-13 Spectra of C-13-Enriched Proteins. J. Magn. Reson. 1990, 88, 425–431. [Google Scholar]
- Vuister, G.W.; Bax, A. Resolution Enhancement and Spectral Editing of Uniformly C-13-Enriched Proteins by Homonuclear Broad-Band C-13 Decoupling. J. Magn. Reson. 1992, 98, 428–435. [Google Scholar]
- Tanimoto, T. An Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation: Armonk, NY, USA, 1958. [Google Scholar]
- Bajusz, D.; Racz, A.; Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Orts, J.; Gossert, A.D. Structure determination of protein-ligand complexes by NMR in solution. Methods 2018, 138, 3–25. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance Restraints Tolerance | Success Rate | rmsd (Å) | TFC | Success: rl rw | Fail: rl rw |
|---|---|---|---|---|---|
| 0% | 95% | 0.88 ± 0.42 | 2.56 ± 3.00 | 1.55 ± 0.58 | 0.98 ± 0.33 |
| 0.87 ± 0.43 * | 3.68 ± 3.50 * | 0.91 ± 0.32 | 0.84 ± 0.16 | ||
| 10% | 57% | 1.05 ± 0.53 | 1.58 ± 1.99 | 1.63 ± 0.58 | 1.16 ± 0.41 |
| 0.74 ± 0.29 * | 2.16 ± 1.93 * | 0.95 ± 0.33 | 0.78 ± 0.21 | ||
| 20% | 46% | 0.96 ± 0.27 | 0.98 ± 2.85 | 1.79 ± 0.44 | 1.25 ± 0.58 |
| 0.97 ± 0.32 * | 1.50 ± 3.48 * | 1.00 ± 0.26 | 0.82 ± 0.33 | ||
| 10% (PA) | 92% | 0.85 ± 0.34 | 3.20 ± 3.80 | 1.57 ± 0.56 | 0.88 ± 0.29 |
| 0.76 ± 0.26 * | 4.51 ± 4.50 * | 0.94 ± 0.28 | 0.56 ± 0.50 | ||
| 20% (PA) | 70% | 1.09 ± 0.51 | 2.29 ± 3.73 | 1.74 ± 0.49 | 0.99 ± 0.39 |
| 0.93 ± 0.29 * | 3.63 ± 4.45 * | 0.99 ± 0.26 | 0.72 ± 0.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, F.; Stadler, G.; Kwiatkowski, W.; Orts, J. A Benchmark Study of Protein–Fragment Complex Structure Calculations with NMR2. Int. J. Mol. Sci. 2023, 24, 14329. https://doi.org/10.3390/ijms241814329
Torres F, Stadler G, Kwiatkowski W, Orts J. A Benchmark Study of Protein–Fragment Complex Structure Calculations with NMR2. International Journal of Molecular Sciences. 2023; 24(18):14329. https://doi.org/10.3390/ijms241814329
Chicago/Turabian StyleTorres, Felix, Gabriela Stadler, Witek Kwiatkowski, and Julien Orts. 2023. "A Benchmark Study of Protein–Fragment Complex Structure Calculations with NMR2" International Journal of Molecular Sciences 24, no. 18: 14329. https://doi.org/10.3390/ijms241814329
APA StyleTorres, F., Stadler, G., Kwiatkowski, W., & Orts, J. (2023). A Benchmark Study of Protein–Fragment Complex Structure Calculations with NMR2. International Journal of Molecular Sciences, 24(18), 14329. https://doi.org/10.3390/ijms241814329