
Uridine as a Regulator of Functional and Ultrastructural Changes in the Brain of Rats in a Model of 6-OHDA-Induced Parkinson’s Disease

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
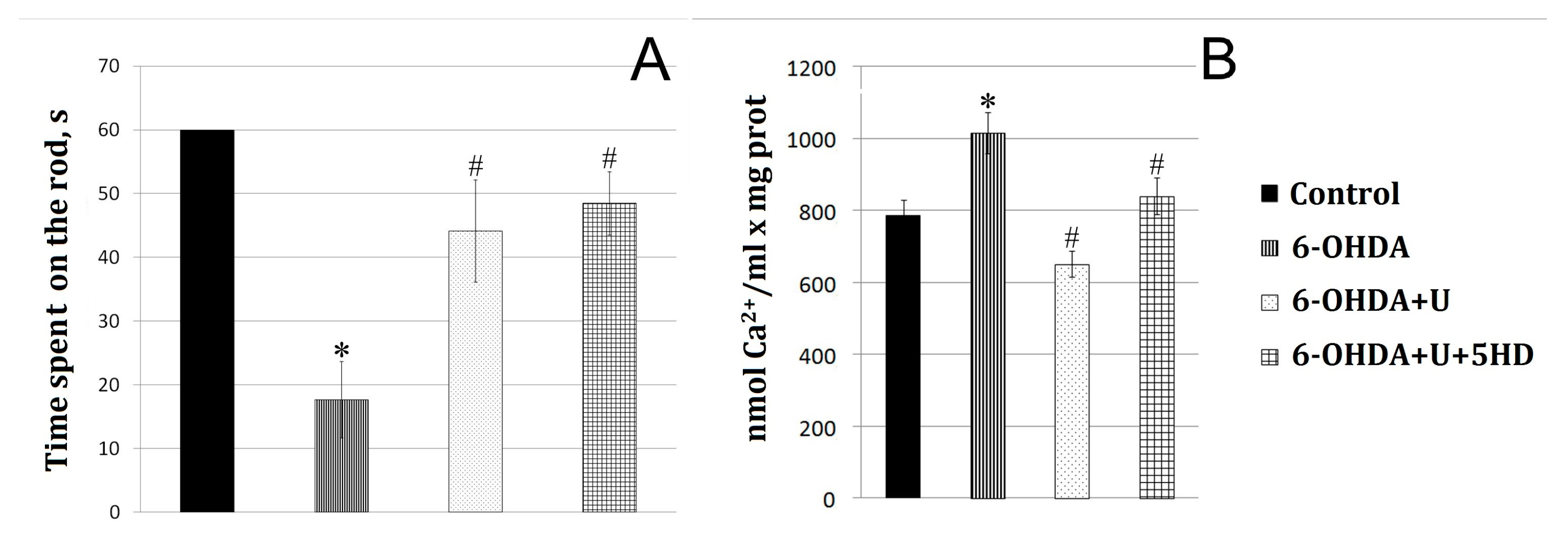
2.1. Effect of Uridine on the Behavior of Animals in a Model of PD Created by the Bilateral Injection of 6-OHDA into the Substantia Nigra
2.2. Effect of Uridine on the Calcium Exchange in Rat Brain Mitochondria after the Bilateral Administration of 6-OHDA into the Substantia Nigra
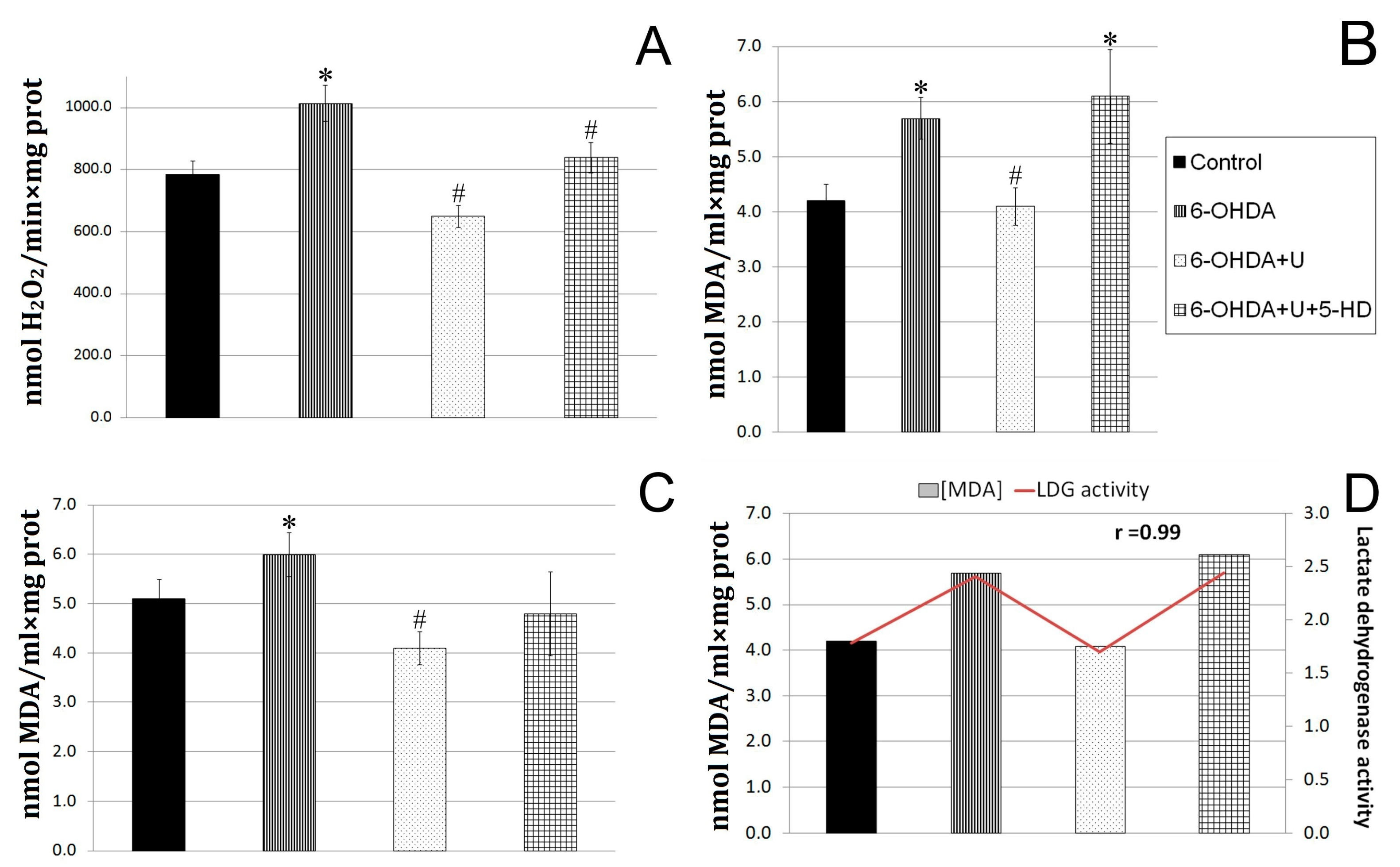
2.3. Effect of Uridine on the Oxidative Exchange in Rat Brain Mitochondria after the Bilateral Injection of 6-OHDA into the Substantia Nigra
2.4. Effect of Uridine on Succinate Dehydrogenase and Lactate Dehydrogenase Activities in Blood Lymphocytes in a PD Model
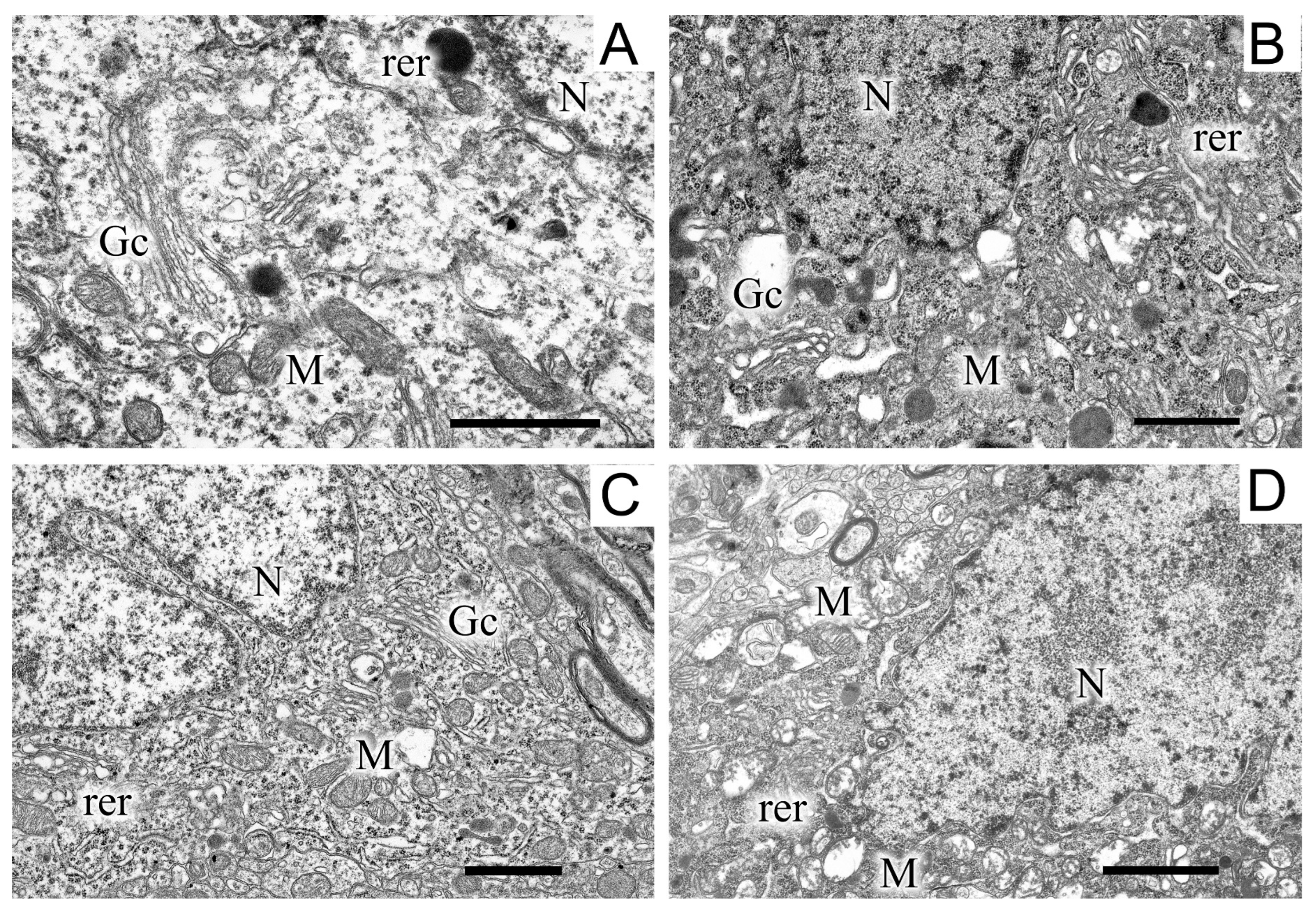
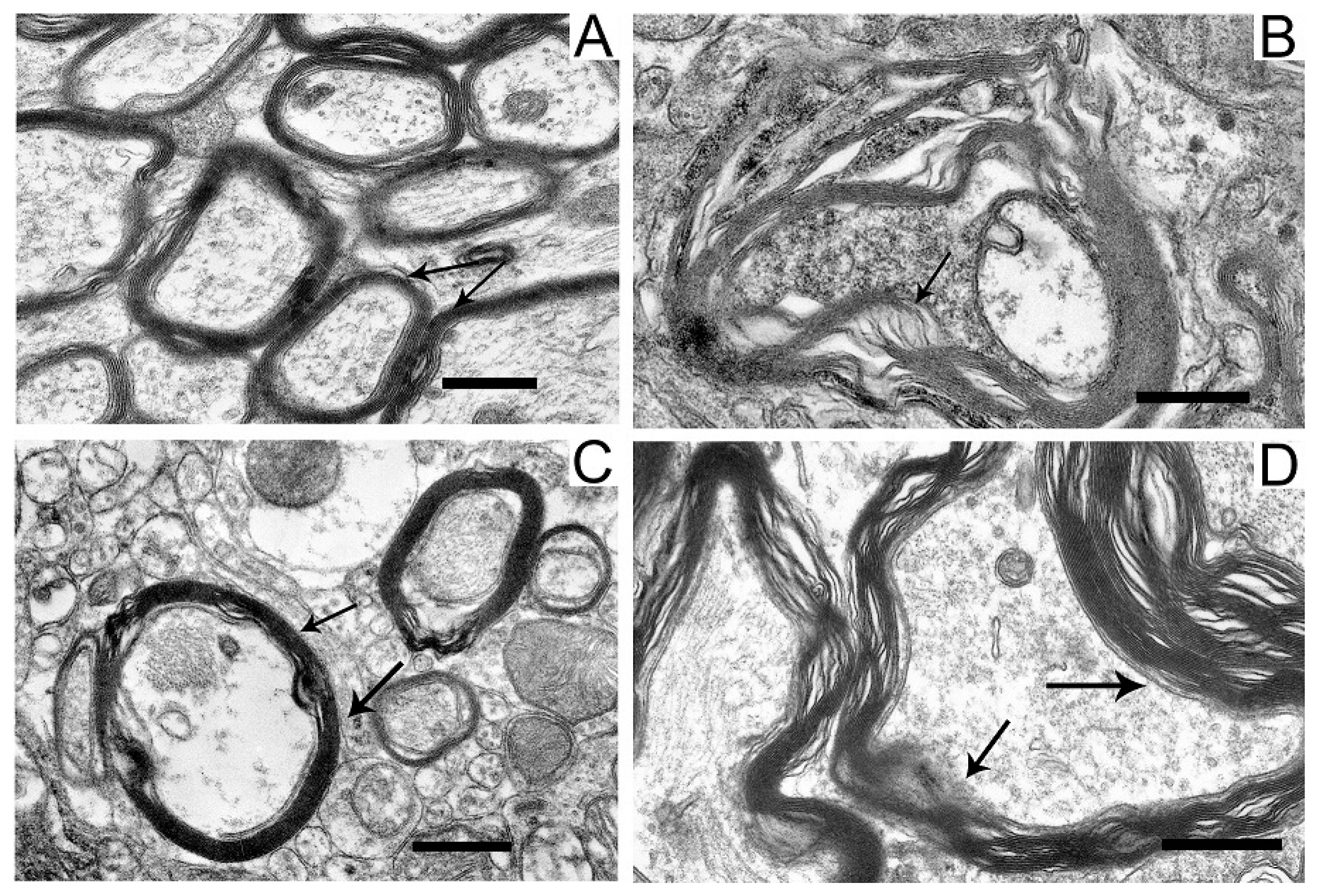
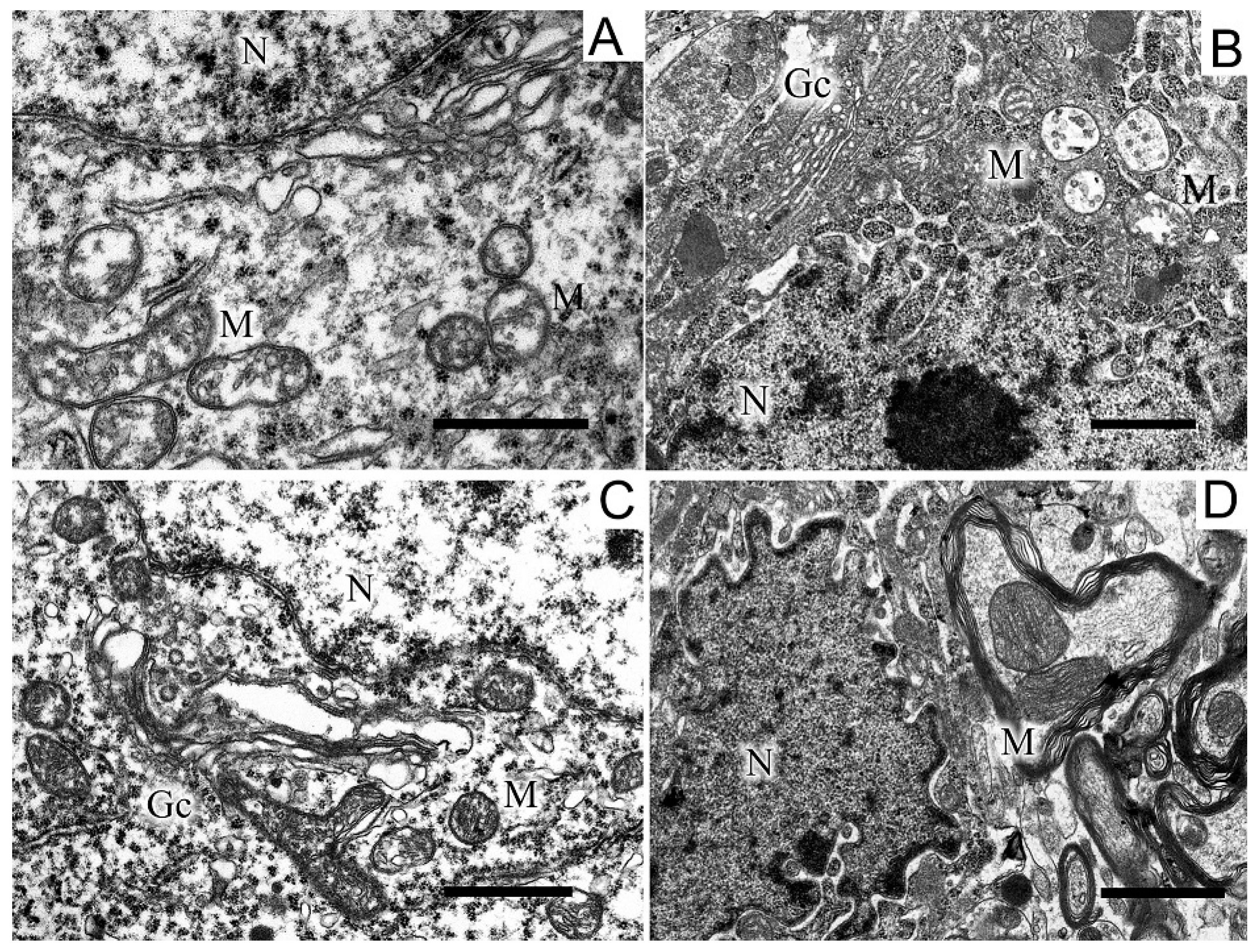
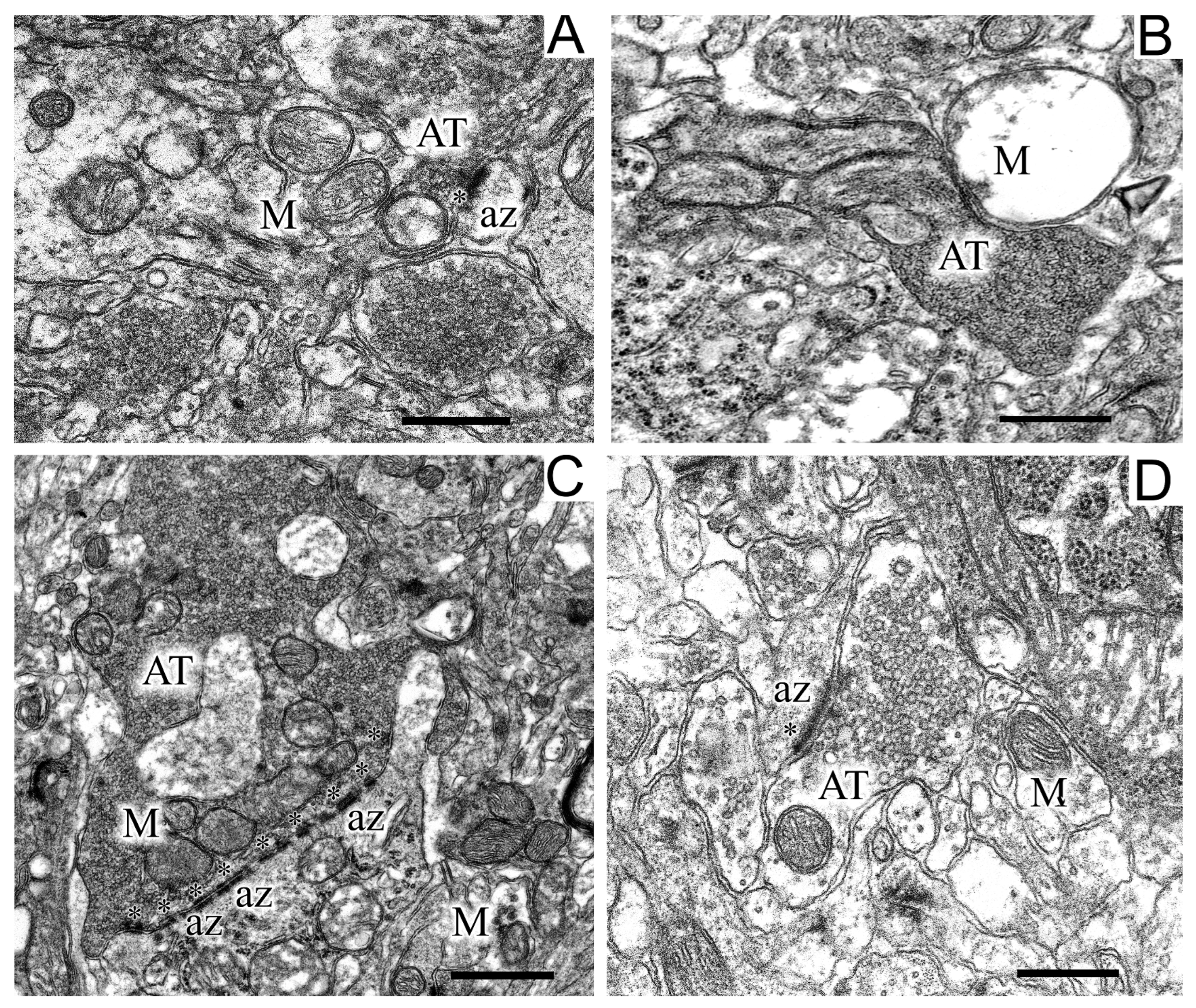
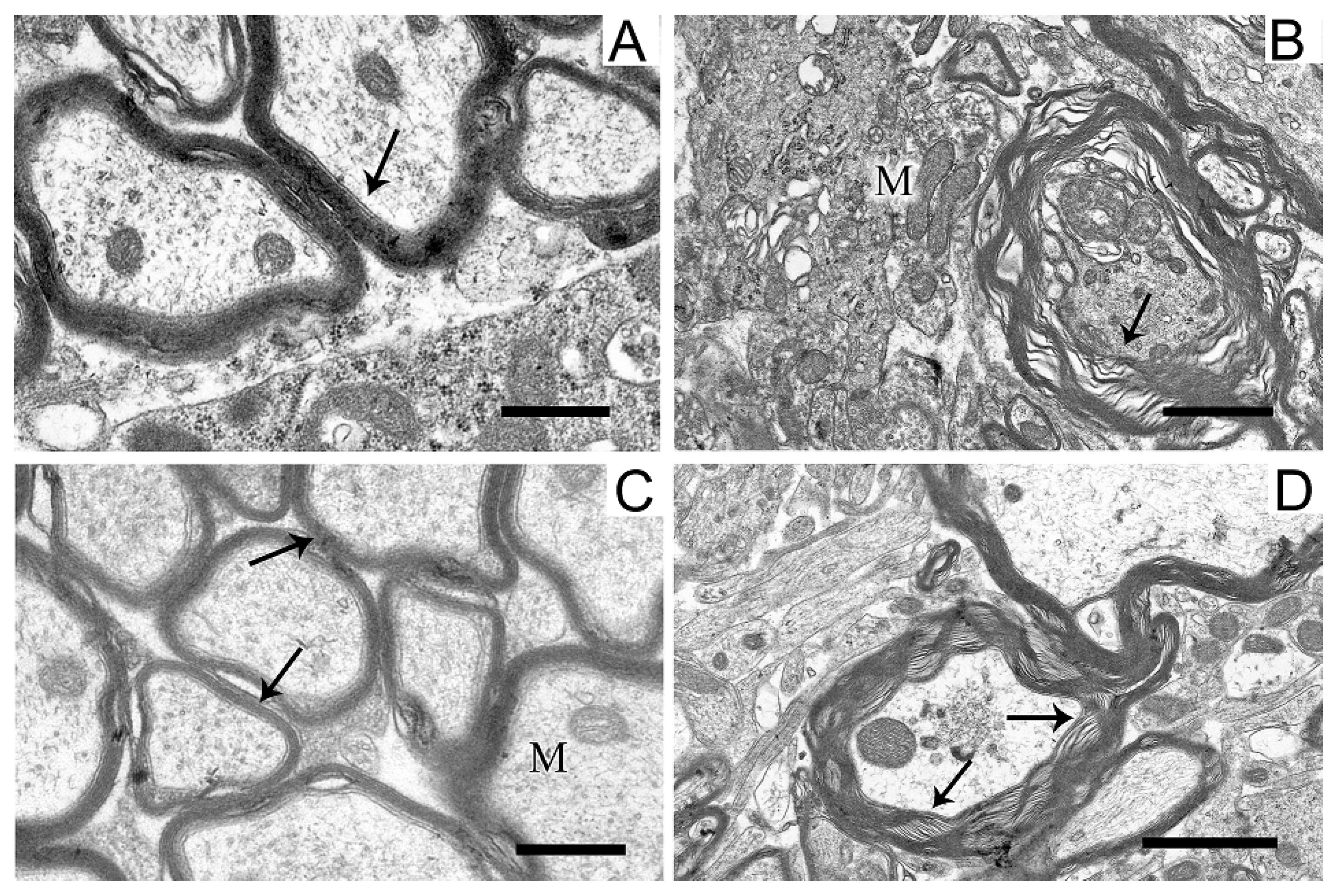
2.5. Ultrastructures of Substantia Nigra and Striatum Neurons after the Bilateral Administration of 6-OHDA and the Protective Effect of Uridine on These Structures
3. Discussion
4. Materials and Methods
4.1. Stereotaxic Intervention and Groups of Animals
4.2. Behavioral Testing
4.3. Isolation of Mitochondria
4.4. Calcium Retention Capacity
4.5. Rate of H2O2 Formation in Brain Mitochondria
4.6. Concentration of Lipid Peroxidation Products in Brain Mitochondria and Blood Serum
4.7. Succinate Dehydrogenase and Lactate Dehydrogenase Activities in Lymphocytes
4.8. Transmission Electron Microscopy
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Yankee, E.L. A review on Parkinson’s disease treatment. Neuroimmunol. Neuroinflamm. 2021, 8, 222–244. [Google Scholar] [CrossRef]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Anik, M.I.; Mahmud, N.; Masud, A.A.; Khan, M.I.; Islam, M.N.; Uddin, S.; Hossain, M.K. Role of reactive oxygen species in aging and age-related diseases: A review. ACS Appl. Bio Mater. 2022, 5, 4028–4054. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Yang, T.; Gu, Y.; Sun, X.H. Mitochondrial dysfunction in Parkinson’s disease: From mechanistic insights to therapy. Front. Aging Neurosci. 2022, 14, 885500. [Google Scholar] [CrossRef]
- Barata-Antunes, S.; Teixeira, F.G.; Mendes-Pinheiro, B.; Domingues, A.V.; Vilaça-Faria, H.; Marote, A.; Silva, D.; Sousa, R.A.; Salgado, A.J. Impact of aging on the 6-OHDA-induced rat model of Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 3459. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef]
- Krylova, I.B.; Selina, E.N.; Bulion, V.V.; Rodionova, O.M.; Evdokimova, N.R.; Belosludtseva, N.V.; Shigaeva, M.I.; Mironova, G.D. Uridine treatment prevents myocardial injury in rat models of acute ischemia and ischemia/reperfusion by activating the mitochondrial ATP-dependent potassium channel. Sci. Rep. 2021, 11, 16999. [Google Scholar] [CrossRef]
- Mironova, G.D.; Khrenov, M.O.; Talanov, E.Y.; Glushkova, O.V.; Parfenyuk, S.B.; Novoselova, T.V.; Lunin, S.M.; Belosludtseva, N.V.; Novoselova, E.G.; Lemasters, J.J. The role of mitochondrial KATP channel in anti-inflammatory effects of uridine in endotoxemic mice. Arch. Biochem. Biophys. 2018, 654, 70–76. [Google Scholar] [CrossRef]
- Mosentsov, A.A.; Rozova, E.V.; Belosludtseva, N.V.; Mankovskaya, I.N.; Putiy, Y.V.; Karaban, I.N.; Mikheeva, I.B.; Mironova, G.D. Does the operation of mitochondrial ATP-dependent potassium channels affect the structural component of mitochondrial and endothelial dysfunctions in experimental parkinsonism? Bull. Exp. Biol. Med. 2021, 170, 431–435. [Google Scholar] [CrossRef]
- Krylova, I.B.; Safonova, A.F.; Evdokimova, N.R. Correction of hypoxic state by metabolic precursors of endogenous activator of mitochondrial ATP-dependent K+ channels. Rev. Clin. Pharmacol. Drug Ther. 2018, 16, 25–31. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochem. (Mosc.) 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Abramov, A.Y. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 2018, 663, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Doenst, T.; Schwarzer, M. Metabolic Pathways and Cycles. In The Scientist’s Guide to Cardiac Metabolism, 1st ed.; Schwarzer, M., Doenst, T., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2015; pp. 39–55. ISBN 978-012-802-394-5. [Google Scholar]
- Zhu, D.; Wei, Y.; Yin, J.; Liu, D.; Ang, E.L.; Zhao, H.; Zhang, Y. A pathway for degradation of uracil to acetyl coenzyme A in Bacillus megaterium. Appl. Environ. Microbiol. 2020, 86, e02837-19. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.P.; Poli, A.; Hara, D.B.; Takahashi, R.N. Time course study of microglial and behavioral alterations induced by 6-hydroxydopamine in rats. Neurosci. Lett. 2016, 622, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, X.; Zhao, L.; Yang, C.; Pan, L.; Li, C.; Liu, K.; Bai, G.; Gao, H.; Yan, Z. Metabolic disturbances in the striatum and substantia nigra in the onset and progression of MPTP-induced parkinsonism model. Front. Neurosci. 2018, 12, 90. [Google Scholar] [CrossRef]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef]
- Todorova, V.; Blokland, A. Mitochondria and synaptic plasticity in the mature and aging mervous system. Curr. Neuropharmacol. 2017, 15, 166–173. [Google Scholar] [CrossRef]
- Soukup, S.F.; Vanhauwaert, R.; Verstreken, P. Parkinson’s disease: Convergence on synaptic homeostasis. EMBO J. 2018, 37, e98960. [Google Scholar] [CrossRef] [PubMed]
- Merino-Galán, L.; Jimenez-Urbieta, H.; Zamarbide, M.; Rodríguez-Chinchilla, T.; Belloso-Iguerategui, A.; Santamaria, E.; Fernández-Irigoyen, J.; Aiastui, A.; Doudnikoff, E.; Bézard, E.; et al. Striatal synaptic bioenergetic and autophagic decline in premotor experimental parkinsonism. Brain 2022, 145, 2092–2107. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, T.N.; Sergeev, V.G.; Vezheeva, O.A. Electron-microscopy studies of rat brain neurodegenerative changes in the model of autoimmune synucleopathie. Bull. Udmurt. Univ. Ser. Biology. Earth Sci. 2011, 4, 113–118. (In Russian) [Google Scholar]
- Papuć, E.; Rejdak, K. The role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 2020, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Mironova, G.D.; Negoda, A.E.; Marinov, B.S.; Paucek, P.; Costa, A.D.; Grigoriev, S.M.; Skarga, Y.Y.; Garlid, K.D. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). J. Biol. Chem. 2004, 279, 32562–32568. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Gerke, S.; Bohnensack, R.; Wojtczak, L. Stimulation of potassium cycling in mitochondria by long-chain fatty acids. Biochim. Biophys. Acta (BBA) Bioenerg. 2003, 1604, 125–133. [Google Scholar] [CrossRef]
- Garlid, K.D.; Paucek, P. Mitochondrial potassium transport: The K+ cycle. Biochim. Biophys. Acta (BBA) Bioenerg. 2003, 1606, 23–41. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef]
- Shigaeva, M.; Gritsenko, E.; Murzaeva, S.; Gorbacheva, O.; Talanov, E.; Mironova, G. Age-related changes in the functioning of the mitochondrial potassium-transporting system. Biofizika 2010, 55, 1030–1037. [Google Scholar] [CrossRef]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships between ion channels, mitochondrial functions and inflammation in human aging. Front. Physiol. 2019, 10, 158. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, M.N.; Dubinin, M.V.; Mironova, G.D.; Belosludtsev, K.N. Effect of chronic treatment with uridine on cardiac mitochondrial dysfunction in the C57BL/6 mouse model of high-fat diet streptozotocin-induced diabetes. Int. J. Mol. Sci. 2022, 23, 10633. [Google Scholar] [CrossRef]
- Skinner, O.S.; Blanco-Fernández, J.; Goodman, R.P.; Kawakami, A.; Shen, H.; Kemény, L.V.; Joesch-Cohen, L.; Rees, M.G.; Roth, J.A.; Fisher, D.E.; et al. Salvage of ribose from uridine or RNA supports glycolysis in nutrient-limited conditions. Nat. Metab. 2023, 5, 765–776. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Dunnett, S. 6-OHDA lesion models of Parkinson’s disease in the rat. In Animal Models of Movement Disorders; Part of the Neuromethods Book Series; Humana Press: Totowa, NJ, USA, 2011; Volume 61, pp. 267–279. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: Burlington, MA, USA, 2007; pp. 166–191. ISBN 978-008-047-515-8. [Google Scholar]
- Ferranti, R.; da Silva, M.M.; Kowaltowski, A.J. Mitochondrial ATP-sensitive K+ channel opening decreases reactive oxygen species generation. FEBS Lett. 2003, 536, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, M.; Zakharchenko, M.; Khunderyakova, N. Preservation of the in vivo state of mitochondrial network for ex vivo physiological study of mitochondria. Int. J. Biochem. Cell Biol. 2009, 41, 2036–2050. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | SDH Activity, Relative Units | LDH Activity, Relative Units |
|---|---|---|
| Control | 1.14 | 1.78 |
| 6-OHDA | 1.37 | 2.41 * |
| 6-OHDA+U | 1.82 * | 1.70 |
| 6-OHDA+U + 5-HD | 1.00 | 2.44 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uspalenko, N.I.; Mosentsov, A.A.; Khmil, N.V.; Pavlik, L.L.; Belosludtseva, N.V.; Khunderyakova, N.V.; Shigaeva, M.I.; Medvedeva, V.P.; Malkov, A.E.; Kitchigina, V.F.; et al. Uridine as a Regulator of Functional and Ultrastructural Changes in the Brain of Rats in a Model of 6-OHDA-Induced Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 14304. https://doi.org/10.3390/ijms241814304
Uspalenko NI, Mosentsov AA, Khmil NV, Pavlik LL, Belosludtseva NV, Khunderyakova NV, Shigaeva MI, Medvedeva VP, Malkov AE, Kitchigina VF, et al. Uridine as a Regulator of Functional and Ultrastructural Changes in the Brain of Rats in a Model of 6-OHDA-Induced Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(18):14304. https://doi.org/10.3390/ijms241814304
Chicago/Turabian StyleUspalenko, Nina I., Alexei A. Mosentsov, Natalia V. Khmil, Lyubov L. Pavlik, Natalia V. Belosludtseva, Natalia V. Khunderyakova, Maria I. Shigaeva, Vasilisa P. Medvedeva, Anton E. Malkov, Valentina F. Kitchigina, and et al. 2023. "Uridine as a Regulator of Functional and Ultrastructural Changes in the Brain of Rats in a Model of 6-OHDA-Induced Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 18: 14304. https://doi.org/10.3390/ijms241814304
APA StyleUspalenko, N. I., Mosentsov, A. A., Khmil, N. V., Pavlik, L. L., Belosludtseva, N. V., Khunderyakova, N. V., Shigaeva, M. I., Medvedeva, V. P., Malkov, A. E., Kitchigina, V. F., & Mironova, G. D. (2023). Uridine as a Regulator of Functional and Ultrastructural Changes in the Brain of Rats in a Model of 6-OHDA-Induced Parkinson’s Disease. International Journal of Molecular Sciences, 24(18), 14304. https://doi.org/10.3390/ijms241814304