
The Potential for Targeting AVIL and Other Actin-Binding Proteins in Rhabdomyosarcoma

 , , ,
, , ,
Abstract
:1. Introduction
2. Improving VAC Chemotherapy and Molecular Targeted Therapies
3. Targeting Actin and Actin-Binding Proteins
4. Targeting Nucleation Factors
5. Targeting Actin Polymerization and Depolymerization
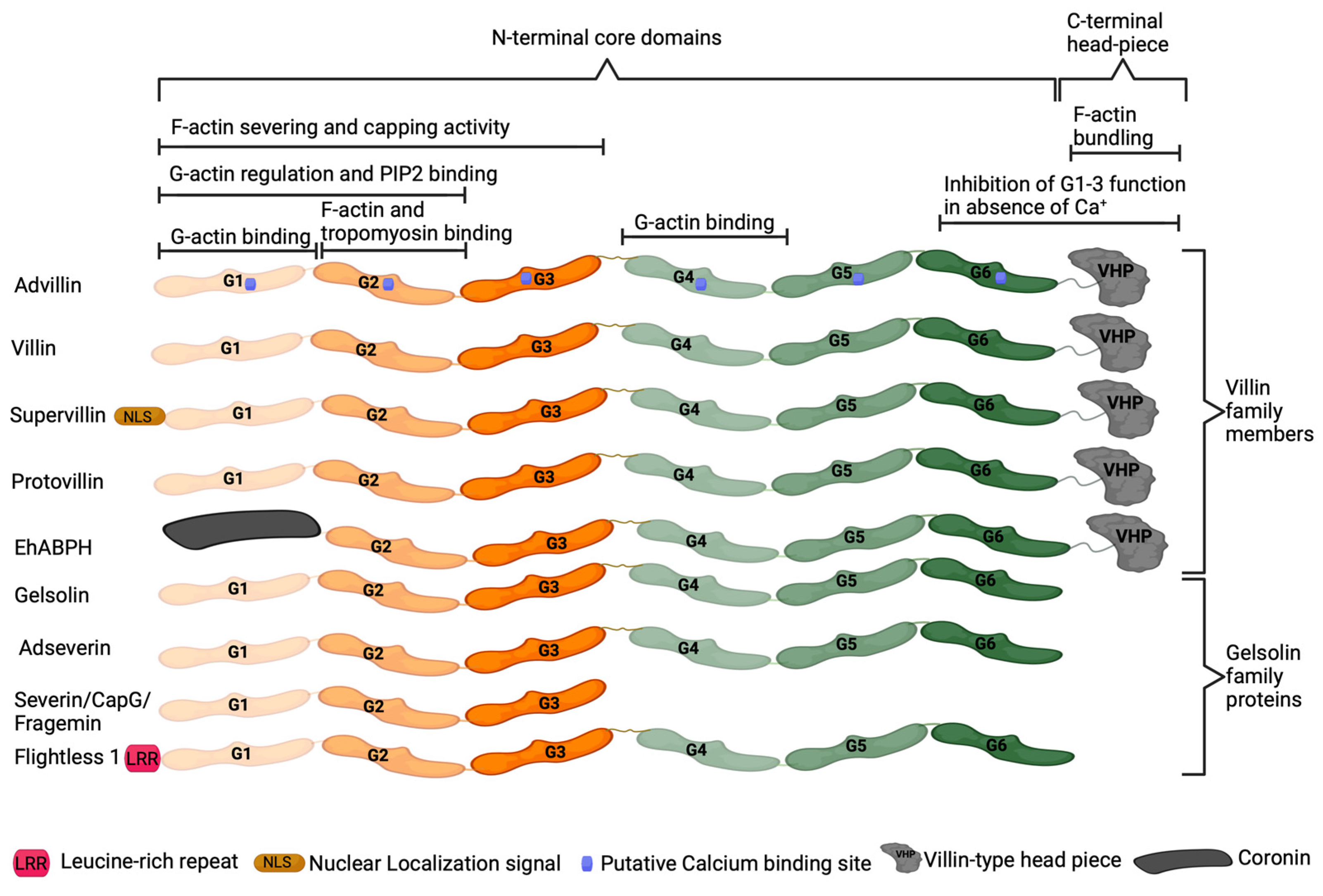
6. Advillin Background
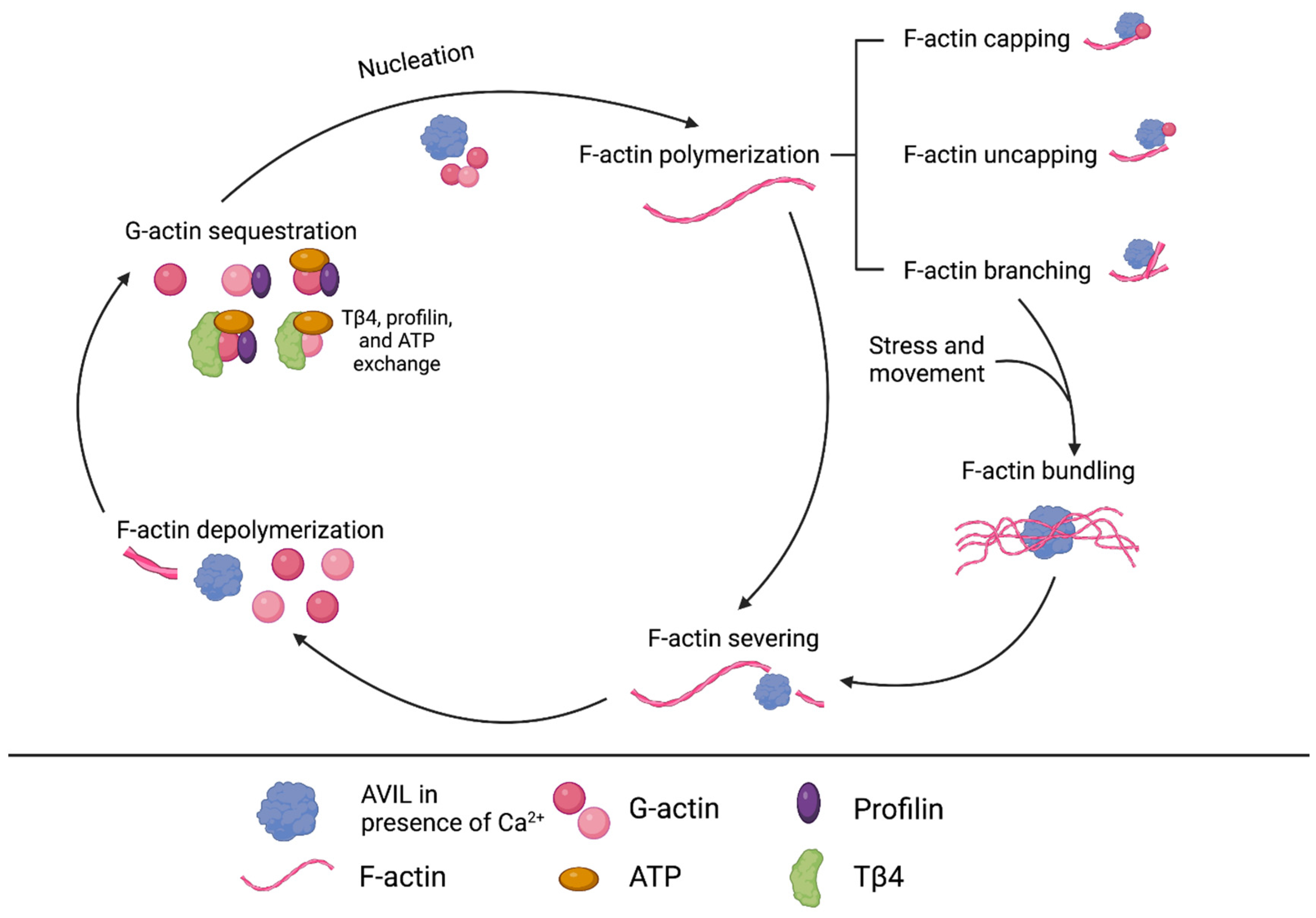
7. AVIL Functionality in ERMS and ARMS Subtypes
8. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dasgupta, R.; Fuchs, J.; Rodeberg, D. Rhabdomyosarcoma. Semin. Pediatr. Surg. 2016, 25, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, I.; Kurzawa, P.; Dopierała, M.; Larque, A.B.; Januszkiewicz-Lewandowska, D. Rhabdomyosarcoma in children—Current pathologic and molecular classification. Pol. J. Pathol. 2018, 69, 20–32. [Google Scholar] [CrossRef]
- Rudzinski, E.R.; Kelsey, A.; Vokuhl, C.; Linardic, C.M.; Shipley, J.; Hettmer, S.; Koscielniak, E.; Hawkins, D.S.; Bisogno, G. Pathology of childhood rhabdomyosarcoma: A consensus opinion document from the Children’s Oncology Group, European Paediatric Soft Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2020, 68, e28798. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Garcia, H.D.; Scheer, M.; Henssen, A.G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 2019, 9, 1458. [Google Scholar] [CrossRef]
- Agaram, N.P. Evolving classification of rhabdomyosarcoma. Histopathology 2021, 80, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef]
- Noujaim, J.; Thway, K.; Jones, R.L.; Miah, A.; Khabra, K.; Langer, R.; Kasper, B.; Judson, I.; Benson, C.; Kollàr, A. Adult Pleomorphic Rhabdomyosarcoma: A Multicentre Retrospective Study. Anticancer Res. 2015, 35, 6213. [Google Scholar]
- Xu, M.; Chen, X.; Chen, D.; Yu, B.; Huang, Z. FoxO1: A novel insight into its molecular mechanisms in the regulation of skeletal muscle differentiation and fiber type specification. Oncotarget 2017, 8, 10662–10674. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1. [Google Scholar] [CrossRef]
- Bennicelli, J.L.; Advani, S.; Schäfer, B.W.; Barr, F.G. PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene 1999, 18, 4348–4356. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Diller, L.; Sexsmith, E.; Gottlieb, A.; Li, F.P.; Malkin, D. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J. Clin. Investig. 1995, 95, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Martin-Giacalone, B.A.; Weinstein, P.A.; Plon, S.E.; Lupo, P.J. Pediatric rhabdomyosarcoma: Epidemiology and genetic susceptibility. J. Clin. Med. 2021, 10, 2028. [Google Scholar] [CrossRef]
- Halstead, N.V.; Cost, N.G.; Hecht, S.L.; Walker, J.P. Neurofibromatosis-1 and Rhabdomyosarcoma: An Unusual Recurrence. Urology 2020, 137, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Steenman, M.; Westerveld, A.; Mannens, M. Genetics of Beckwith-Wiedemann syndrome-associated tumors: Common genetic pathways. Genes Chromosomes Cancer 2000, 28, 1–13. [Google Scholar] [CrossRef]
- Doros, L.; Yang, J.; Dehner, L.; Rossi, C.T.; Skiver, K.; Jarzembowski, J.A.; Messinger, Y.; Schultz, K.A.; Williams, G.; André, N.; et al. DICER1 Mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr. Blood Cancer 2012, 59, 558–560. [Google Scholar] [CrossRef]
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric rhabdomyosarcoma. Crit. Rev. Oncog. 2015, 20, 227–243. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Melcón, S.G.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Bisogno, G.; Jenney, M.; Bergeron, C.; Melcón, S.G.; Ferrari, A.; Oberlin, O.; Carli, M.; Stevens, M.; Kelsey, A.; De Paoli, A.; et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): A multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1061–1071. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Chi, Y.-Y.; Anderson, J.R.; Tian, J.; Arndt, C.A.; Bomgaars, L.; Donaldson, S.S.; Hayes-Jordan, A.; Mascarenhas, L.; McCarville, M.B.; et al. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: A report from the children’s oncology group. J. Clin. Oncol. 2018, 36, 2770–2777. [Google Scholar] [CrossRef]
- Kim, A.; Widemann, B.C.; Krailo, M.; Jayaprakash, N.; Fox, E.; Weigel, B.; Blaney, S.M. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R.; et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kieran, M.W.; Grupp, S.; Perek, D.; Clancy, J.; Krygowski, M.; Ananthakrishnan, R.; Boni, J.P.; Berkenblit, A.; Spunt, S.L. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 2012, 48, 253–262. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Chi, Y.-Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination With Chemotherapy for First Relapse Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Recent Advances and Challenges in the Treatment of Rhabdomyosarcoma. Cancers 2020, 12, 1758. [Google Scholar] [CrossRef]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Rosenbaum, E.; Wu, N.; Dickson, M.A.; Sheikh, T.N.; D’Angelo, S.P.; Chi, P.; Keohan, M.L.; Erinjeri, J.P.; Antonescu, C.R.; et al. A Phase Ib/II Randomized Study of RO4929097, a Gamma-Secretase or Notch Inhibitor with or without Vismodegib, a Hedgehog Inhibitor, in Advanced Sarcoma. Clin. Cancer Res. 2022, 28, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Niu, J.-T.; Wu, H.-W.; Si, X.-L.; Zhang, S.-J.; Li, D.-H.; Bian, T.-T.; Li, Y.-F.; Yan, X.-K. Actin-Binding Proteins as Potential Biomarkers for Chronic Inflammation-Induced Cancer Diagnosis and Therapy. Anal. Cell. Pathol. 2021, 2021, 6692811. [Google Scholar] [CrossRef]
- Calaghan, S.C.; White, E.; Bedut, S.; Guennec, J.-Y. Cytochalasin D reduces Ca2+ sensitivity and maximum tension via interactions with myofilaments in skinned rat cardiac myocytes. J. Physiol. 2000, 529, 405–411. [Google Scholar] [CrossRef]
- Chao, J.I.; Liu, H.F. The blockage of survivin and securin expression increases the cytochalasin B-induced cell death and growth inhibition in human cancer cells. Mol. Pharmacol. 2006, 69, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Glinsukon, T.; Lekutai, S. Comparative toxicity in the rat of cytochalasins B and E. Toxicon 1979, 17, 137–144. [Google Scholar] [CrossRef]
- Van Goietsenoven, G.; Mathieu, V.; Andolfi, A.; Cimmino, A.; Lefranc, F.; Kiss, R.; Evidente, A. In vitro growth inhibitory effects of cytochalasins and derivatives in cancer cells. Planta Medica 2011, 77, 711–717. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Saito, M.; Sekita, S.; Yoshihira, K.; Natori, S. Acute toxic effects of chaetoglobosin A, a new cytochalasan compound produced by Chaetomium globosum, on mice and rats. Jpn. J. Exp. Med. 1978, 48, 105–110. [Google Scholar] [PubMed]
- Knudsen, P.B.; Hanna, B.; Ohl, S.; Sellner, L.; Zenz, T.; Döhner, H.; Stilgenbauer, S.; Larsen, T.O.; Lichter, P.; Seiffert, M. Chaetoglobosin A preferentially induces apoptosis in chronic lymphocytic leukemia cells by targeting the cytoskeleton. Leukemia 2014, 28, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Schweikart, K.; Guo, L.; Shuler, Z.; Abrams, R.; Chiao, E.T.; Kolaja, K.L.; Davis, M. The effects of jaspamide on human cardiomyocyte function and cardiac ion channel activity. Toxicol. Vitr. 2013, 27, 745–751. [Google Scholar] [CrossRef]
- Sierra-Paredes, G.; Oreiro-García, T.; Núñez-Rodriguez, A.; Vázquez-López, A.; Sierra-Marcuño, G. Seizures induced by in vivo latrunculin A and jasplakinolide microperfusion in the rat hippocampus. J. Mol. Neurosci. 2006, 28, 151–160. [Google Scholar] [CrossRef]
- Konishi, H.; Kikuchi, S.; Ochiai, T.; Ikoma, H.; Kubota, T.; Ichikawa, D.; Fujiwara, H.; Okamoto, K.; Sakakura, C.; Sonoyama, T.; et al. Latrunculin A has a strong anticancer effect in a peritoneal dissemination model of human gastric cancer in mice. Anticancer Res. 2009, 29, 2091–2097. [Google Scholar]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.J.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT 077 in chemo- resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, X.; Chen, C.; Liu, B.; Ren, B.; Wang, L.; Zhao, K.; Yu, S.; Ming, H. Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol. Rep. 2013, 30, 2127–2136. [Google Scholar] [CrossRef]
- Georgopoulou, M.P.; Tosios, K.I.; Goutas, N.; Kouloukoussa, M. Arp2/3 Complex Is Expressed in Oral Squamous Cell Carcinoma: An Immunohistochemical Study of 88 Cases. Open J. Stomatol. 2019, 09, 29–38. [Google Scholar] [CrossRef]
- Semba, S.; Iwaya, K.; Matsubayashi, J.; Serizawa, H.; Kataba, H.; Hirano, T.; Kato, H.; Matsuoka, T.; Mukai, K. Coexpression of actin-related protein 2 and Wiskott-Aldrich syndrome family verproline-homologous protein 2 in adenocarcinoma of the lung. Clin. Cancer Res. 2006, 12, 2449–2454. [Google Scholar] [CrossRef]
- Iwaya, K.; Norio, K.; Mukai, K. Coexpression of Arp2 and WAVE2 predicts poor outcome in invasive breast carcinoma. Mod. Pathol. 2007, 20, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C.; Zheng, Y.-S.; Li, X.-H.; Takahashi, H.; Hara, T.; Masuda, S.; Yang, X.-H.; Guan, Y.-F.; Takano, Y. Arp2/3 overexpression contributed to pathogenesis, growth and invasion of gastric carcinoma. Anticancer. Res. 2008, 28, 2225–2232. [Google Scholar]
- Biber, G.; Ben-Shmuel, A.; Noy, E.; Joseph, N.; Puthenveetil, A.; Reiss, N.; Levy, O.; Lazar, I.; Feiglin, A.; Ofran, Y.; et al. Targeting the actin nucleation promoting factor WASp provides a therapeutic approach for hematopoietic malignancies. Nat. Commun. 2021, 12, 5581. [Google Scholar] [CrossRef] [PubMed]
- Nolen, B.J.; Tomasevic, N.; Russell, A.; Pierce, D.W.; Jia, Z.; McCormick, C.D.; Hartman, J.; Sakowicz, R.; Pollard, T.D. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature 2009, 460, 1031–1034. [Google Scholar] [CrossRef]
- Zhu, X.L.; Liang, L.; Ding, Y.Q. Overexpression of FMNL2 is closely related to metastasis of colorectal cancer. Int. J. Color. Dis. 2008, 23, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, X.; Zeng, Y.; Wang, J.; Zhang, X.; Ding, Y.-Q.; Liang, L. FMNL2 enhances invasion of colorectal carcinoma by inducing epithelial-mesenchymal transition. Mol. Cancer Res. 2010, 8, 1579–1590. [Google Scholar] [CrossRef]
- Zhu, X.L.; Zeng, Y.-F.; Guan, J.; Li, Y.-F.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q.; Liang, L. FMNL2 is a positive regulator of cell motility and metastasis in colorectal carcinoma. J. Pathol. 2011, 224, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Wang, K.; Xu, H.; Kong, F. Silencing Formin-like 2 inhibits growth and metastasis of gastric cancer cells through suppressing internalization of integrins. Cancer Cell Int. 2018, 18, 79. [Google Scholar] [CrossRef]
- Rizvi, S.A.; Neidt, E.M.; Cui, J.; Feiger, Z.; Skau, C.T.; Gardel, M.L.; Kozmin, S.A.; Kovar, D.R. Identification and Characterization of a Small Molecule Inhibitor of Formin-Mediated Actin Assembly. Chem. Biol. 2009, 16, 1158–1168. [Google Scholar] [CrossRef]
- Takeuchi, H.; Ara, G.; Sausville, E.A.; Teicher, B. Jasplakinolide: Interaction with radiation and hyperthermia in human prostate carcinoma and Lewis lung carcinoma. Cancer Chemother. Pharmacol. 1998, 42, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Sasse, F.; Kunze, B.; Gronewold, T.M.A.; Reichenbach, H. The chondramides: Cytostatic agents from myxobacteria acting on the actin cytoskeleton. J. Natl. Cancer Inst. 1998, 90, 1559–1563. [Google Scholar] [CrossRef]
- Crews, P.; Manes, L.V.; Boehler, M. Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis Sp. Tetrahedron Lett. 1986, 27, 2797–2800. [Google Scholar] [CrossRef]
- Bubb, M.R.; Senderowicz, A.M.J.; Sausville, E.A.; Duncan, K.L.K.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Verdier-Pinard, P.; Gangwar, S.; Stessman, C.C.; McClure, K.J.; Sausville, E.A.; Pettit, G.R.; Bates, R.B.; Hamel, E. Dolastatin 11, a marine depsipeptide, arrests cells at cytokinesis and induces hyperpolymerization of purified actin. Mol. Pharmacol. 2001, 59, 462–469. [Google Scholar] [CrossRef]
- Bai, R.; Covell, D.G.; Liu, C.; Ghosh, A.K.; Hamel, E. (-)-doliculide, a new macrocyclic depsipeptide enhancer of actin assembly. J. Biol. Chem. 2002, 277, 32165–32171. [Google Scholar] [CrossRef]
- Cooper, J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987, 105, 1473–1478. [Google Scholar] [CrossRef]
- Trendowski, M.; Mitchell, J.M.; Corsette, C.M.; Acquafondata, C.; Fondy, T.P. Chemotherapy with cytochalasin congeners in vitro and in vivo against murine models. Investig. New Drugs 2015, 33, 290–299. [Google Scholar] [CrossRef]
- Trendowski, M. Using Cytochalasins to Improve Current Chemotherapeutic Approaches. Anti-Cancer Agents Med. Chem. 2015, 15, 327–335. [Google Scholar] [CrossRef]
- Ruggiero, C.; Lalli, E. Targeting the cytoskeleton against metastatic dissemination. Cancer Metastasis Rev. 2021, 40, 89–140. [Google Scholar] [CrossRef]
- Xie, Z.; Janczyk, P.L.; Shi, X.; Wang, Q.; Singh, S.; Cornelison, R.; Xu, J.; Mandell, J.W.; Barr, F.G.; Li, H. Rhabdomyosarcomas are oncogene addicted to the activation of AVIL. Proc. Natl. Acad. Sci. USA 2022, 119, e2118048119. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.W.; Arai, M.; Bandura, J.L.; Kwiatkowski, D.J. Advillin (p92): A new member of the gelsolin/villin family of actin regulatory proteins. J. Cell Sci. 1998, 111, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Silacci, P.; Mazzolai, L.; Gauci, C.; Stergiopulos, N.; Yin, H.L.; Hayoz, D. Gelsolin superfamily proteins: Key regulators of cellular functions. Cell Mol. Life Sci. 2004, 61, 2614–2623. [Google Scholar] [CrossRef]
- Nag, S.; Larsson, M.; Robinson, R.C.; Burtnick, L.D. Gelsolin: The tail of a molecular gymnast. Cytoskeleton 2013, 70, 360–384. [Google Scholar] [CrossRef]
- George, S.P.; Esmaeilniakooshkghazi, A.; Roy, S.; Khurana, S. F-actin-bundling sites are conserved in proteins with villin-type headpiece domains. Mol. Biol. Cell 2020, 31, 1857–1866. [Google Scholar] [CrossRef]
- Ravenall, S.J.; Gavazzi, I.; Wood, J.N.; Akopian, A.N. A peripheral nervous system actin-binding protein regulates neurite outgrowth. Eur. J. Neurosci. 2002, 15, 281–290. [Google Scholar] [CrossRef]
- Ruppert, A.L.; Keshavarz, M.; Winterberg, S.; Oberwinkler, J.; Kummer, W.; Schütz, B. Advillin is a tuft cell marker in the mouse alimentary tract. J. Mol. Histol. 2020, 51, 421–435. [Google Scholar] [CrossRef]
- Hunter, D.V.; Smaila, B.D.; Lopes, D.M.; Takatoh, J.; Denk, F.; Ramer, M.S. Advillin Is Expressed in All Adult Neural Crest-Derived Neurons. eNeuro 2018, 5, ENEURO.0077-18.2018. [Google Scholar] [CrossRef]
- Chuang, Y.-C.; Lee, C.-H.; Sun, W.-H.; Chen, C.-C. Involvement of advillin in somatosensory neuron subtype-specific axon regeneration and neuropathic pain. Proc. Natl. Acad. Sci. USA 2018, 115, E8557–E8566. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielińska, W.; Grzanka, D.; Gagat, M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. BioMed Res. Int. 2018, 2018, 4578373. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Janczyk, P.; Zhang, Y.; Liu, A.; Shi, X.; Singh, S.; Facemire, L.; Kubow, K.; Li, Z.; Jia, Y.; et al. A cytoskeleton regulator AVIL drives tumorigenesis in glioblastoma. Nat. Commun. 2020, 11, 3457. [Google Scholar] [CrossRef]
- Leiner, J.; Le Loarer, F. The current landscape of rhabdomyosarcomas: An update. Virchows Arch. 2020, 476, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Paulson, V.; Chandler, G.; Rakheja, D.; Galindo, R.L.; Wilson, K.; Amatruda, J.F.; Cameron, S. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer 2011, 50, 397–408. [Google Scholar] [CrossRef]
- Linardic, C.M. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. 2008, 270, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Grosveld, G.C. Alveolar rhabdomyosarcoma—The molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet. Muscle 2012, 2, 25. [Google Scholar] [CrossRef]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem.-Anti-Cancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Subtypes of RMS | Gene Alterations/Fusions | Histopathology | Common Sites |
|---|---|---|---|
| Alveolar RMS Fusion-positive | MARS-AVIL (12q14) PAX3-FOXO1 t(2;13) PAX7-FOXO1 t(1;13) PAX3-NCOA1 t(2;2) PAX3-NCOA2 t(2;8) PAX3-IN080D t(2;2) PAX3- FKHR t(2;13) PAX7-FKHR t(1;13) | Enlarged nuclei with scanty cytoplasm of rhabdomyoblast, not well-differentiated. Characterized by the expression of diffused MYOD1 and myogenin; 80–90% associated with recurrent FOXO1 fusions. | Perineal region Paraspinal region Extremities |
| Embryonal RMS Fusion-negative | Mutations in KRAS, NRAS, and HRAS Aneuploidy Activation of Hedgehog (Hh) signaling Inactivation of the master regulator of p53 and Rb pathways FGFR4 mutation PIK3KA mutation NF1 mutation FBXW7 mutation | Varying degrees of skeletal muscle differentiation with moderate cellularity. | Head Neck Genitourinary tract |
| Sclerosing/Spindle Cell RMS | VGLL2/NCOA2 gene fusions MYOD1 gene mutation | Fascicles of spindle cells. Elongated and fusiform nuclei, small nucleoli. Eosinophilic cytoplasm. | Testicular area Head Neck Trunk (MYOD1 mutation) |
| Pleomorphic RMS | Complex alterations | Pleomorphic rhabdomyoblasts. | Extremities |
| Type of Therapy | Regimen | Phase | Patient Group | Disease/Event-Free Survival (DFS/EVF) | Overall Survival (OS) | References |
|---|---|---|---|---|---|---|
| Chemotherapy | IVA + maintenance chemotherapy | III | 371 patients with non-metastatic RMS | 5-year DFS With maintenance chemotherapy: 77.6% (95% CI 70.6–83.2) Without maintenance chemotherapy: 69.8% (95% CI 62.2–76.2) | 5-year OS With maintenance chemotherapy: 86.5% (95% CI 80.2–90.9) Without maintenance chemotherapy: 73.7% (65.8–80.1) | [18] |
| Chemotherapy | IVA + Doxorubicin | III | 484 patients with non-metastatic RMS | 3-year EFS With Doxorubicin: 67.5% (95% CI 61.2–73.1) Without Doxorubicin: 63.3% (56.8–69.0) p-value: 0.33 | [19] | |
| Chemotherapy | VAC or VAC/VI | III | 488 patients with intermediate-risk RMS | 4-year EFS VAC: 63% VAC/VI: 59% p-value: 0.51 | 4-year OS VAC: 73% VAC/VI: 72% p-value: 0.80 | [20] |
| Molecular targeted drugs | Sorafenib, Inhibitors of PDGFRs, VEGFRs, and MAPK | II | 20 participants presenting both RMS and Wilms tumor | No objective response | [21] | |
| Molecular targeted drugs | Crizotinib, inhibitors of MET, ALK, ROS1, and RON | II | 13 patients with advanced and metastatic ARMS | No clinically relevant efficacy as a single agent | [22] | |
| Molecular targeted drugs | Temsirolimus | II | 16, 17, and 19 patients with RMS, high-grade glioma, and neuroblastoma, respectively | No clinically meaningful efficacy as a single agent in RMS | [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornelison, R.; Marrah, L.; Fierti, A.; Piczak, C.; Glowczyk, M.; Tajammal, A.; Lynch, S.; Li, H. The Potential for Targeting AVIL and Other Actin-Binding Proteins in Rhabdomyosarcoma. Int. J. Mol. Sci. 2023, 24, 14196. https://doi.org/10.3390/ijms241814196
Cornelison R, Marrah L, Fierti A, Piczak C, Glowczyk M, Tajammal A, Lynch S, Li H. The Potential for Targeting AVIL and Other Actin-Binding Proteins in Rhabdomyosarcoma. International Journal of Molecular Sciences. 2023; 24(18):14196. https://doi.org/10.3390/ijms241814196
Chicago/Turabian StyleCornelison, Robert, Laine Marrah, Adelaide Fierti, Claire Piczak, Martyna Glowczyk, Anam Tajammal, Sarah Lynch, and Hui Li. 2023. "The Potential for Targeting AVIL and Other Actin-Binding Proteins in Rhabdomyosarcoma" International Journal of Molecular Sciences 24, no. 18: 14196. https://doi.org/10.3390/ijms241814196
APA StyleCornelison, R., Marrah, L., Fierti, A., Piczak, C., Glowczyk, M., Tajammal, A., Lynch, S., & Li, H. (2023). The Potential for Targeting AVIL and Other Actin-Binding Proteins in Rhabdomyosarcoma. International Journal of Molecular Sciences, 24(18), 14196. https://doi.org/10.3390/ijms241814196