


Dual-Target Mycobacterium tuberculosis Inhibition: Insights into the Molecular Mechanism of Antifolate Drugs



Abstract
:1. Introduction
2. Results
2.1. Predicting the Pharmacokinetic Properties of the Dual-Inhibitor INCAs
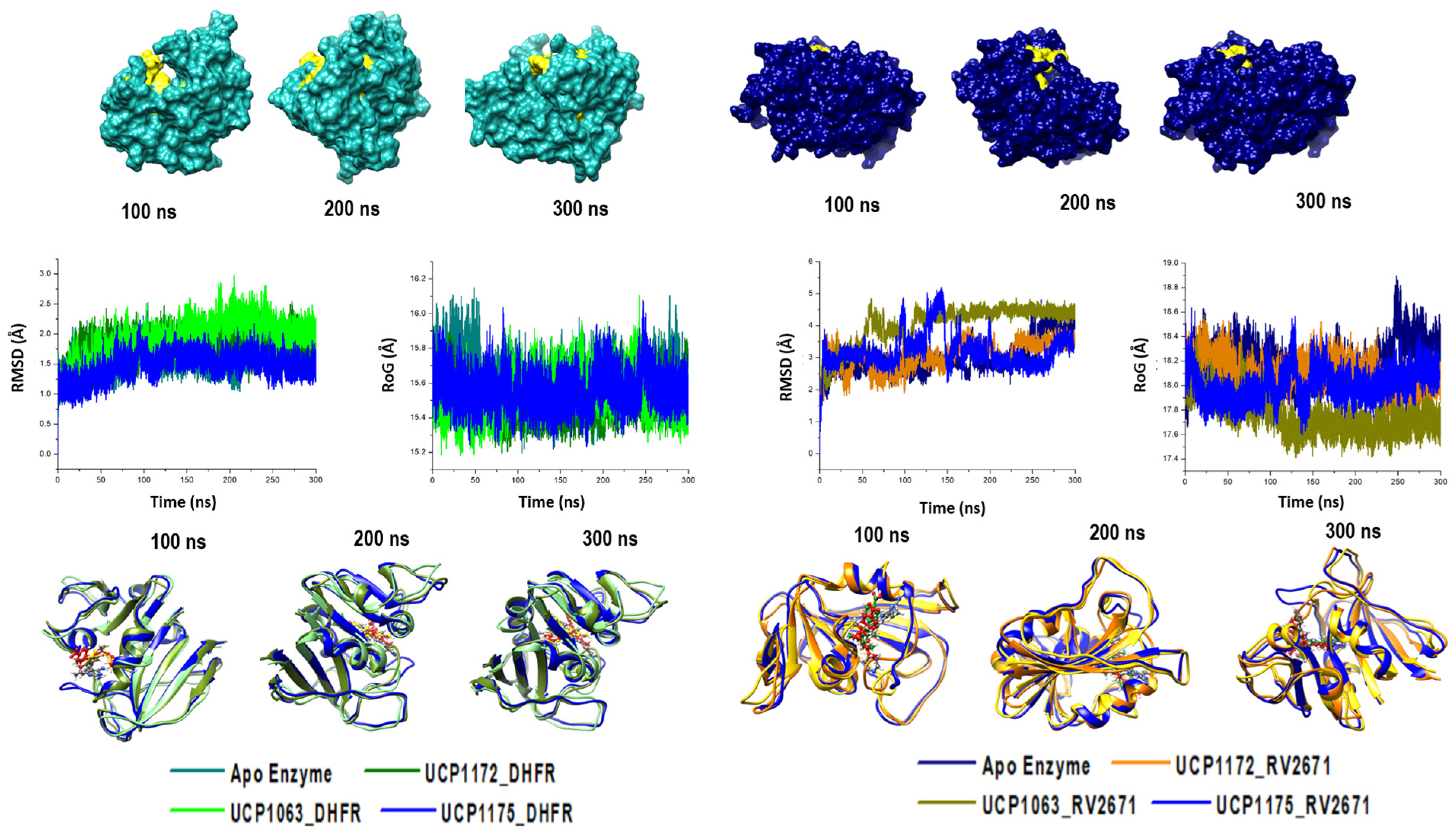
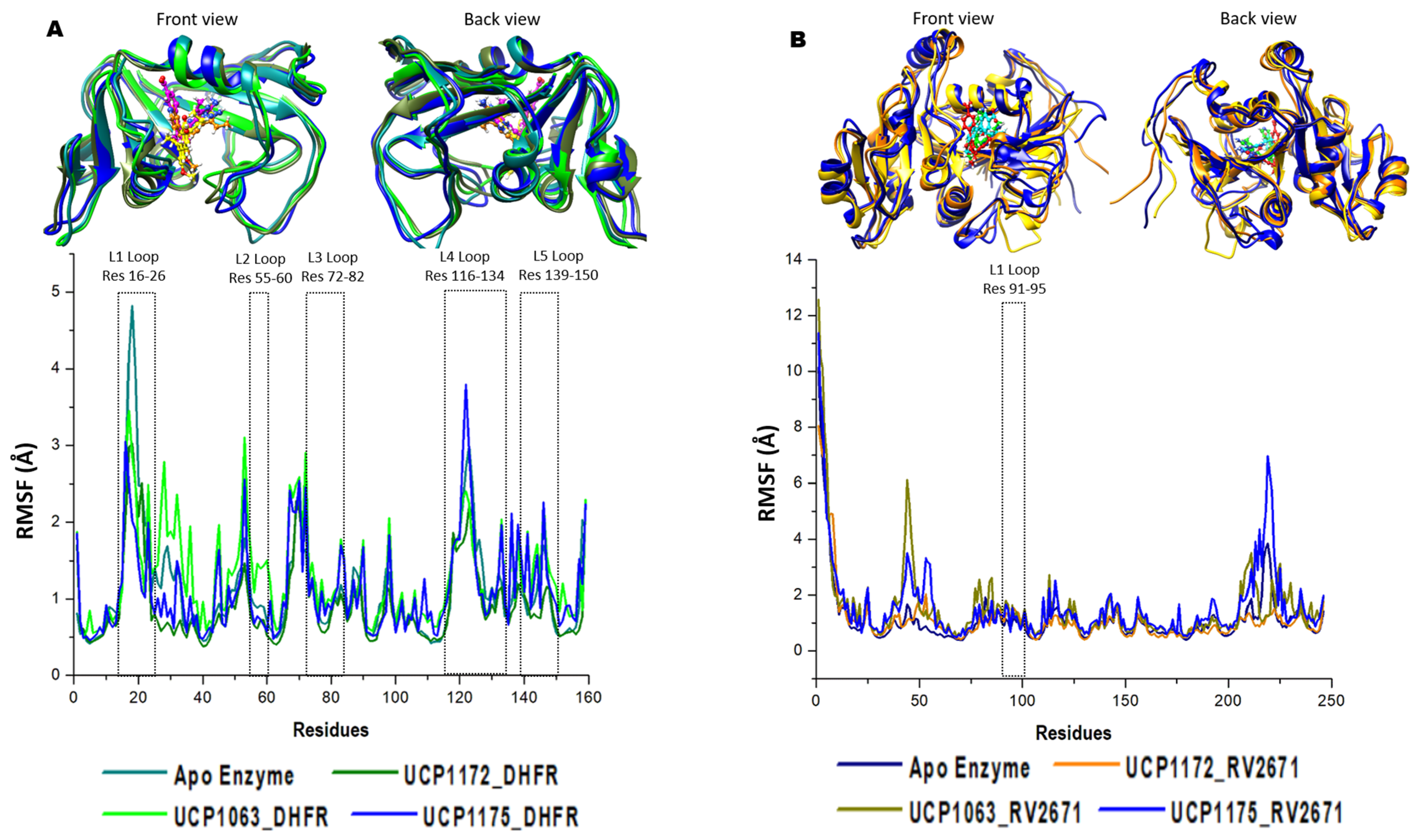
2.2. Defining the Structural Integrity of the INCAs’ Antibacterial Potency
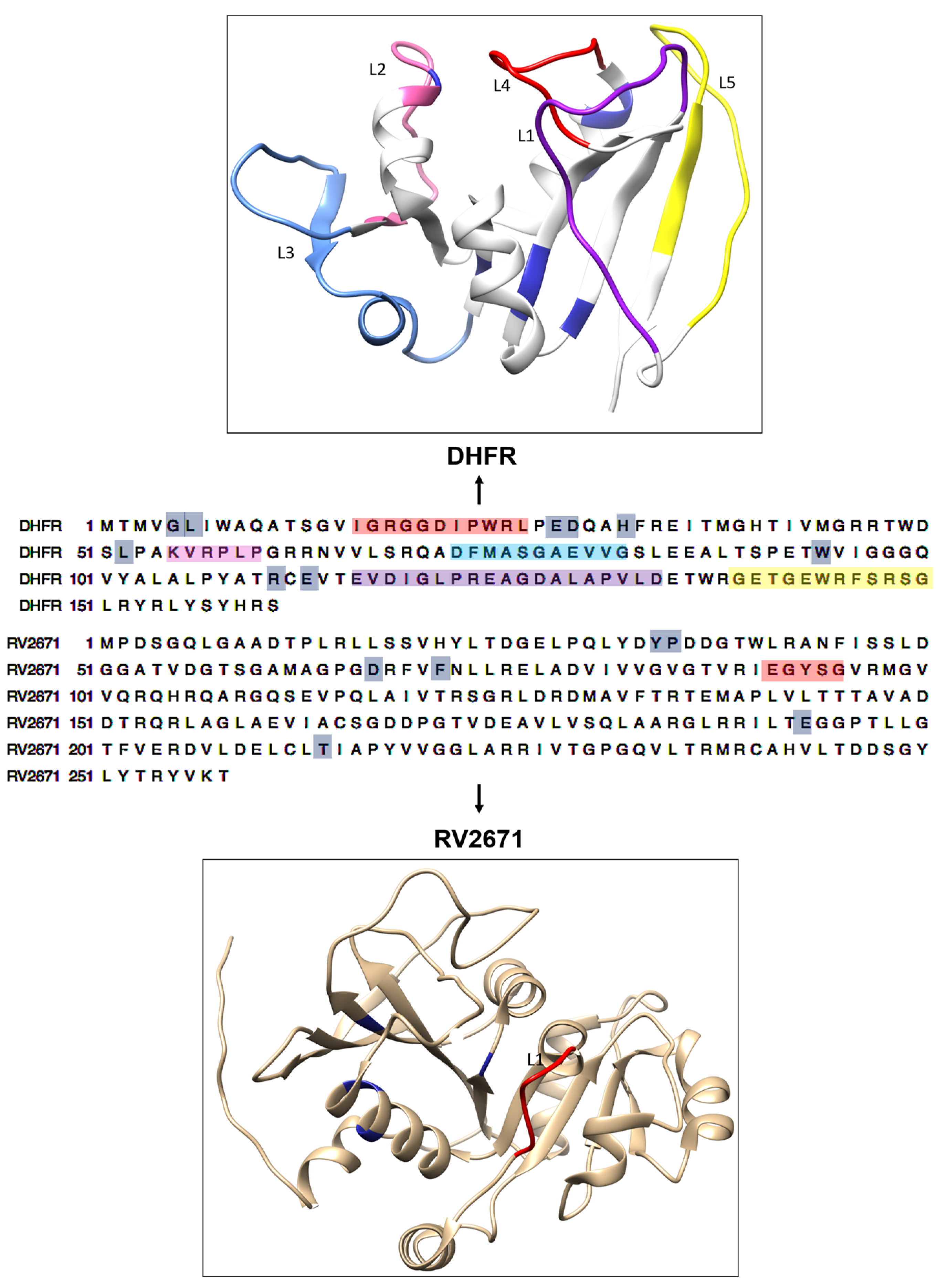
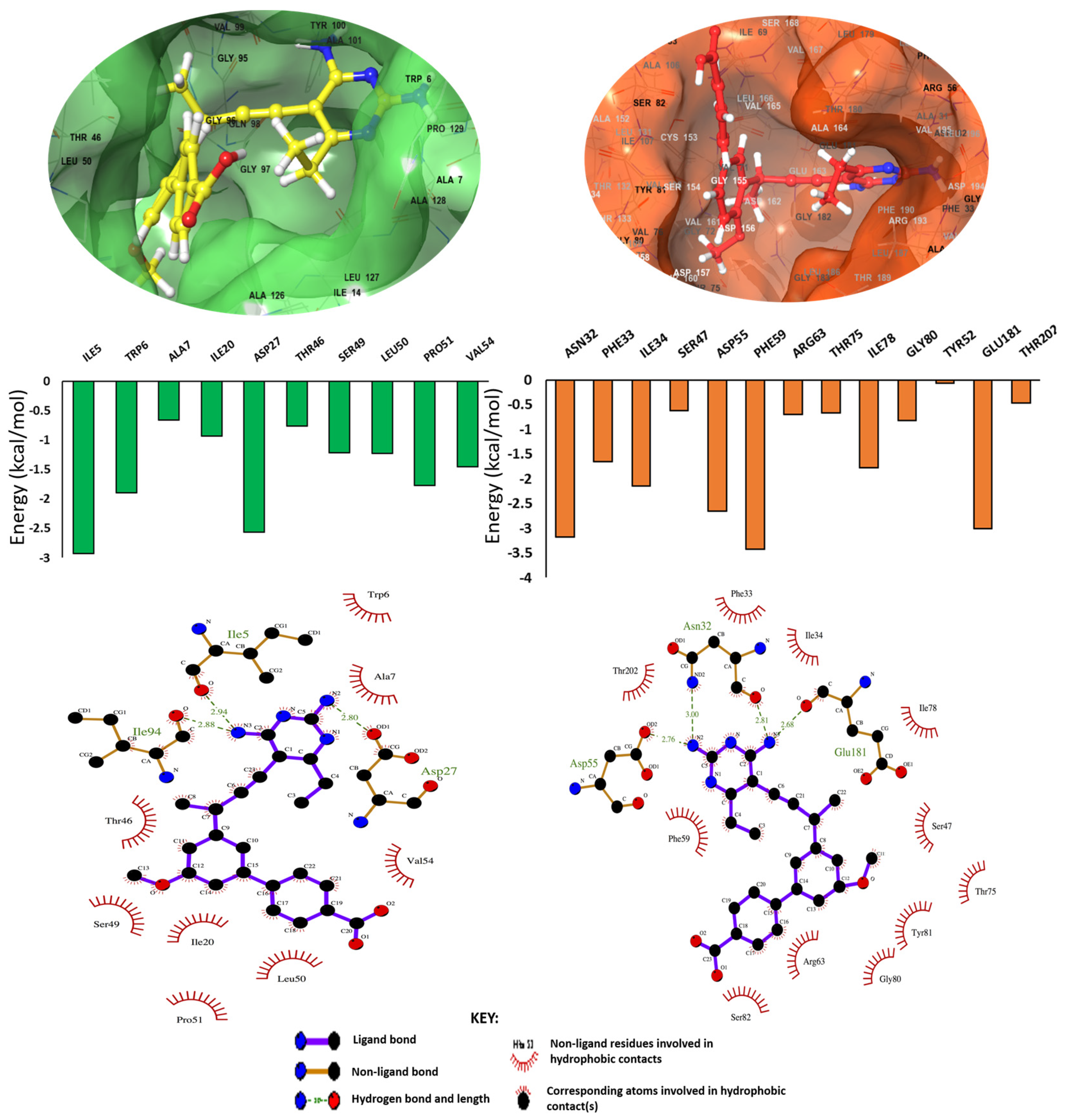
2.3. Deciphering the Commonality between the Active Site of DHFR and RV2671
3. Discussion
4. Material and Methods
4.1. Measurement of Pharmacokinetic Properties and Drug Likeliness of INCAs
4.2. DHFR System Preparation
4.3. RV2671 System Preparation
4.4. Molecular Dynamic Simulations
4.5. Post-Dynamic Simulation and Data Analysis
Binding Free Energy Analysis via MM/GBSA Method
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Furin, J.; Cox, H.; Pai, M. Tuberculosis. Lancet 2019, 393, 1642–1656. [Google Scholar] [CrossRef] [PubMed]
- Seung, K.J.; Keshavjee, S.; Rich, M.L. Drug-Resistant Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 2015, 1–20. [Google Scholar]
- Conradie, F.; Bagdasaryan, T.R.; Borisov, S.; Howell, P.; Mikiashvili, L.; Ngubane, N.; Samoilova, A.; Skornykova, S.; Tudor, E.; Variava, E.; et al. Bedaquiline–Pretomanid–Linezolid Regimens for Drug-Resistant Tuberculosis. N. Engl. J. Med. 2022, 387, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Multi-Target Pharmacology: Possibilities and Limitations of the “Skeleton Key Approach” from a Medicinal Chemist Perspective. Front. Pharmacol. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Hajian, B.; Scocchera, E.; Shoen, C.; Krucinska, J.; Viswanathan, K.; G-Dayanandan, N.; Erlandsen, H.; Estrada, A.; Mikušová, K.; Korduláková, J.; et al. Drugging the Folate Pathway in Mycobacterium tuberculosis: The Role of Multi-Targeting Agents. Cell Chem. Biol. 2019, 26, 781–791.e6. [Google Scholar] [CrossRef]
- Koźmiński, P.; Halik, P.K.; Chesori, R.; Gniazdowska, E. Overview of Dual-Acting Drug Methotrexate in Different Neurological Diseases, Autoimmune Pathologies and Cancers. Int. J. Mol. Sci. 2020, 21, 3483. [Google Scholar] [CrossRef]
- Rollins, K.D.; Lindley, C. Pemetrexed: A Multitargeted Antifolate. Clin. Ther. 2005, 27, 1343–1382. [Google Scholar] [CrossRef]
- Phan, J.; Koli, S.; Minor, W.; Dunlap, R.B.; Berger, S.H.; Lebioda, L. Human Thymidylate Synthase Is in the Closed Conformation When Complexed with DUMP and Raltitrexed, an Antifolate Drug. Biochemistry 2001, 40, 1897–1902. [Google Scholar] [CrossRef]
- Cunningham, D.; Zalcberg, J.; Smith, I.; Gore, M.; Pazdur, R.; Burris, H.; Meropol, N.J.; Kennealey, G.; Seymour, L. “Tomudex” (ZD1694): A Novel Thymidylate Synthase Inhibitor with Clinical Antitumour Activity in a Range of Solid Tumours. Ann. Oncol. 1996, 7, 179–182. [Google Scholar] [CrossRef]
- Allegra, C.J.; Kovacs, J.A.; Drake, J.C.; Swan, J.C.; Chabner, B.A.; Masur, H. Potent in Vitro and in Vivo Antitoxoplasma Activity of the Lipid-Soluble Antifolate Trimetrexate. J. Clin. Investig. 1987, 79, 478–482. [Google Scholar] [CrossRef]
- Teixeira, B.V.F.; Teles, A.L.B.; Silva, S.G.; Brito, C.C.B.; Freitas, H.F.D.; Pires, A.B.L.; Froes, T.Q.; Castilho, M.S. Dual and Selective Inhibitors of Pteridine Reductase 1 (PTR1) and Dihydrofolate Reductase-Thymidylate Synthase (DHFR-TS) from Leishmania Chagasi. J. Enzym. Inhib. Med. Chem. 2019, 34, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Cossins, E. The Fascinating World of Folate and One-Carbon Metabolism. Can. J. Bot. 2000, 78, 691–708. [Google Scholar] [CrossRef]
- Visentin, M.; Zhao, R.; Goldman, I. The Antifolates. Hematol. Oncol. Clin. N. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef]
- Cheng, Y.-S.; Sachettini, J.C. Structural Insights into Mycobacterium tuberculosis Rv2671 Protein as a Dihydrofolate Reductase Functional Analogue Contributing to Para-Aminosalicylic Acid Resistance. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Kraut, J. Loop and Subdomain Movements in the Mechanism of Escherichia Coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Bystroff, C.; Kraut, J. Crystal Structure of Unliganded Escherichia Coli Dihydrofolate Reductase. Ligand-Induced Conformational Changes and Cooperativity in Binding. Biochemistry 1991, 30, 2227–2239. [Google Scholar] [CrossRef]
- Dias, M.V.; Tyrakis, P.; Domingues, R.R.; Franco, A.; Leme, A.F. Article Mycobacterium tuberculosis Dihydrofolate Reductase Reveals Two Conformational States and a Possible Low Affinity Mechanism to Antifolate Drugs. Struct. Fold. Des. 2014, 22, 94–103. [Google Scholar] [CrossRef]
- Webborn, P. The Role of Pharmacokinetic Studies in Drug Discovery: Where Are We Now, How Did We Get Here and Where Are We Going? Future Med. Chem. 2014, 6, 1233–1235. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Kruijtzer, C.M.F. Improvement of Oral Drug Treatment by Temporary Inhibition of Drug Transporters and/or Cytochrome P450 in the Gastrointestinal Tract and Liver: An Overview. Oncologist 2002, 7, 516–530. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Ryde, U. The Normal-Mode Entropy in the MM/GBSA Method: E Ff Ect of System Truncation, Bu Ff Er Region, and Dielectric Constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef]
- Bhal, S. Log P—Making Sense of the Value; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2007; pp. 1–4. [Google Scholar]
- Hajian, B.; Scocchera, E.; Keshipeddy, S.; G-Dayanandan, N.; Shoen, C.; Krucinska, J.; Reeve, S.; Cynamon, M.; Anderson, A.C.; Wright, D.L. Propargyl-Linked Antifolates Are Potent Inhibitors of Drug-Sensitive and Drug-Resistant Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0161740. [Google Scholar] [CrossRef]
- Pan, A.C.; Borhani, D.W.; Dror, R.O.; Shaw, D.E. Molecular Determinants of Drug-Receptor Binding Kinetics. Drug Discov. Today 2013, 18, 667–673. [Google Scholar] [CrossRef]
- Sharma, K.; Tanwar, O.; Sharma, S.; Ali, S.; Alam, M.M.; Zaman, M.S.; Akhter, M. Structural Comparison of Mtb-DHFR and h-DHFR for Design, Synthesis and Evaluation of Selective Non-Pteridine Analogues as Antitubercular Agents. Bioorg. Chem. 2018, 80, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Sittikornpaiboon, P.; Toochinda, P.; Lawtrakul, L. Structural and Dynamics Perspectives on the Binding of Substrate and Inhibitors in Mycobacterium tuberculosis DHFR. Sci. Pharm. 2017, 85, 31. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina. J. Comput. Chem. 2010, 31, 445–461. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Ramharack, P.; Soliman, M.E.S. Zika Virus NS5 Protein Potential Inhibitors: An Enhanced in Silico Approach in Drug Discovery. J. Biomol. Struct. Dyn. 2018, 36, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Agoni, C.; Ramharack, P.; Soliman, M.E.S. Co-Inhibition as a Strategic Therapeutic Approach to Overcome Rifampin Resistance in Tuberculosis Therapy: Atomistic Insights. Future Med. Chem. 2018, 10, 1665–1675. [Google Scholar] [CrossRef]
- Munsamy, G.; Ramharack, P.; Soliman, M.E.S. Egress and Invasion Machinary of Malaria: An in Depth Look into the Structural and Functional Features of the Fl Ap Dynamics of Plasmepsin IX and X. RSC Adv. 2018, 8, 21829–21840. [Google Scholar] [CrossRef] [PubMed]
- Seifert, E. OriginPro 9.1: Scientific Data Analysis and Graphing Software—Software Review. J. Chem. Inf. Model. 2014, 54, 1552. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined Molecular Mechanical and Continuum Solvent Approach (MM-PBSA/GBSA) to Predict Ligand Binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Donini, O.; Cieplak, P.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; Mcgee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An E Ffi Cient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADME Profile | UCP1172 | UCP1175 | UCP1063 |
|---|---|---|---|
| Drug likeness (Lipinski) | Yes | Yes | Yes |
| Molecular weight (g/mol) | 416.47 | 402.45 | 373.45 |
| Lipophilicity (iLOGP) | 3.18 | 3.06 | 3.10 |
| Water soluble | Soluble | Moderately Soluble | Moderately Soluble |
| GIT absorption | High | High | High |
| BBB permeability | No | No | No |
| CYP P450 3A4 inhibitor | Yes | Yes | Yes |
| P-gp substrate | No | No | Yes |
| Synthetic accessibility | 3.96 | 3.40 | 3.76 |
| PAINS | No | No | No |
| System | Energy Components (kcal/mol) | ||||
|---|---|---|---|---|---|
| UCP1172-DHFR | −51.43 ± 0.37 | −21.91 ± 0.68 | −73.34 ± 0.64 | 31.71 ± 0.51 | −41.63 ± 0.46 |
| UCP1172-RV2671 | −50.93 ± 0.61 | −35.94 ± 1.09 | −86.86 ± 1.27 | 38.81 ± 0.94 | −48.04 ± 0.78 |
| UCP1063-DHFR | −41.06 ± 0.37 | −36.27 ± 1.08 | −77.33 ± 0.93 | 42.13 ± 0.61 | −35.20 ± 0.44 |
| UCP1063-RV2671 | −53.45 ± 0.30 | −30.93 ± 0.33 | −84.37 ± 0.45 | 40.56 ± 0.20 | −43.81 ± 0.37 |
| UCP1175-DHFR | −20.02 ± 0.14 | −338.37 ± 1.25 | −217.89 ± 0.93 | 191.50 ± 0.98 | −26.39 ± 0.33 |
| UCP1175-RV2671 | −37.53 ± 0.27 | −89.71 ± 2.04 | −127.24 ± 2.07 | 103.60 ± 1.89 | −23.64 ± 0.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramharack, P.; Salifu, E.Y.; Agoni, C. Dual-Target Mycobacterium tuberculosis Inhibition: Insights into the Molecular Mechanism of Antifolate Drugs. Int. J. Mol. Sci. 2023, 24, 14021. https://doi.org/10.3390/ijms241814021
Ramharack P, Salifu EY, Agoni C. Dual-Target Mycobacterium tuberculosis Inhibition: Insights into the Molecular Mechanism of Antifolate Drugs. International Journal of Molecular Sciences. 2023; 24(18):14021. https://doi.org/10.3390/ijms241814021
Chicago/Turabian StyleRamharack, Pritika, Elliasu Y. Salifu, and Clement Agoni. 2023. "Dual-Target Mycobacterium tuberculosis Inhibition: Insights into the Molecular Mechanism of Antifolate Drugs" International Journal of Molecular Sciences 24, no. 18: 14021. https://doi.org/10.3390/ijms241814021
APA StyleRamharack, P., Salifu, E. Y., & Agoni, C. (2023). Dual-Target Mycobacterium tuberculosis Inhibition: Insights into the Molecular Mechanism of Antifolate Drugs. International Journal of Molecular Sciences, 24(18), 14021. https://doi.org/10.3390/ijms241814021