
Simufilam Reverses Aberrant Receptor Interactions of Filamin A in Alzheimer’s Disease

 , and
, and 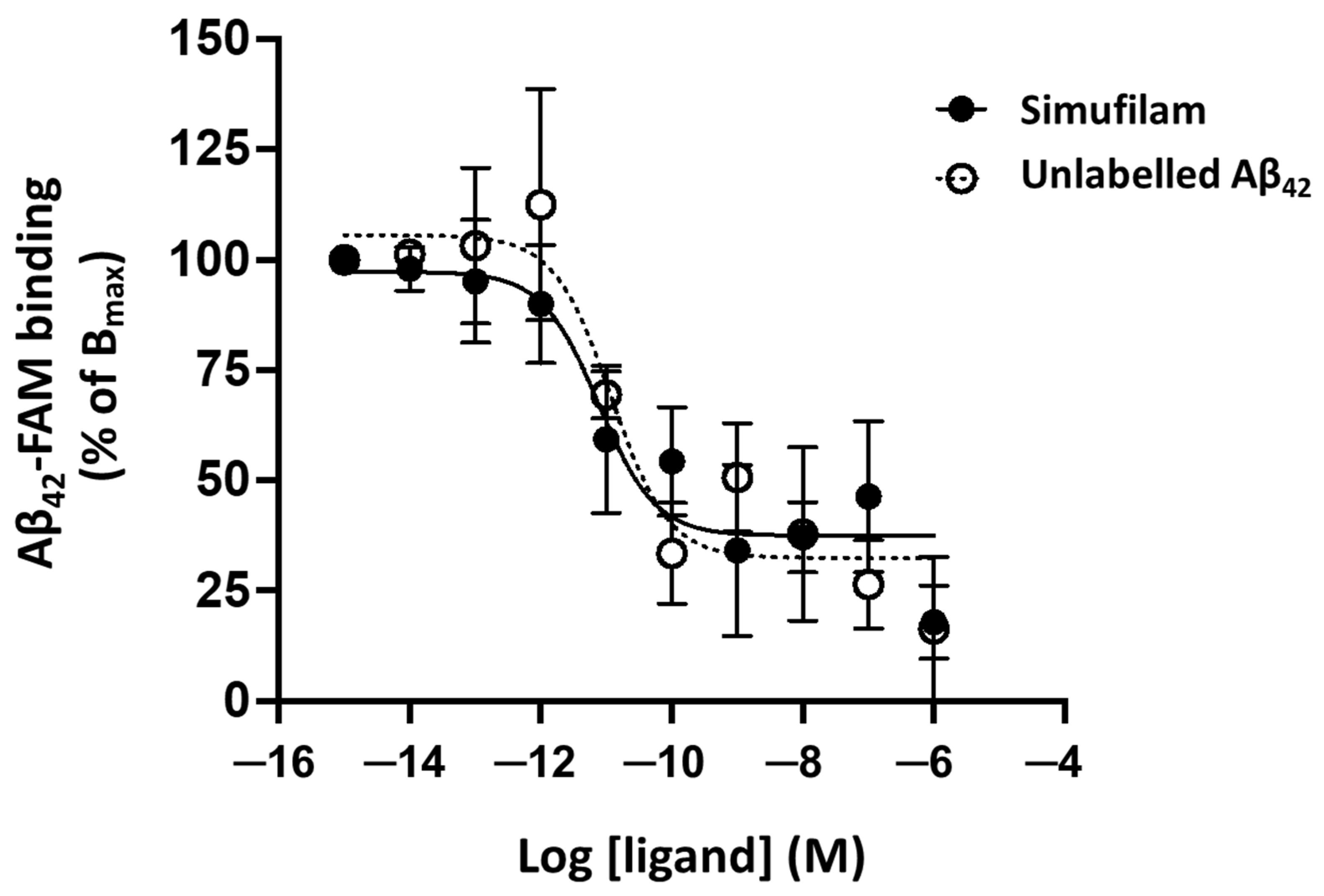{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
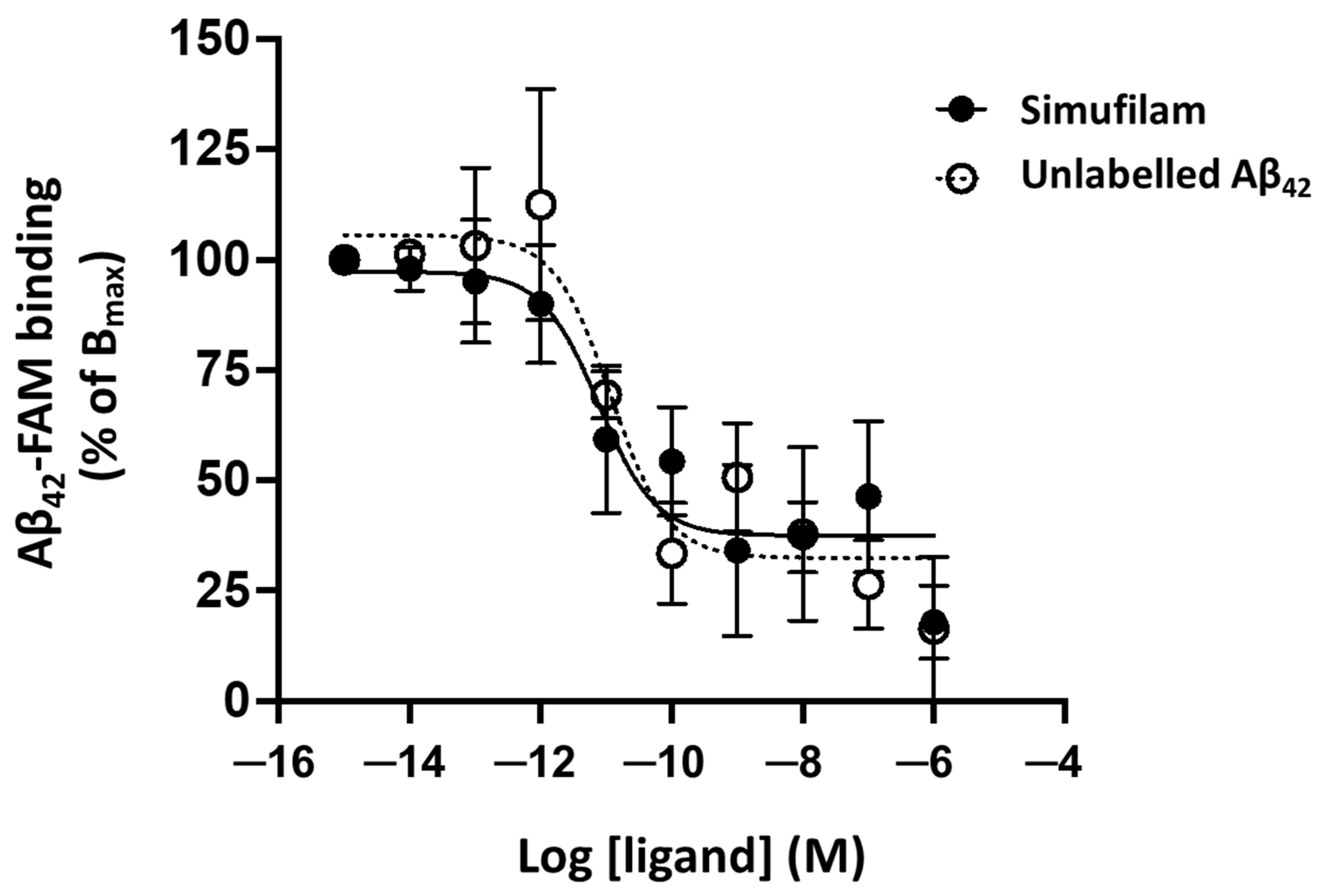
2.1. Simufilam Reduced Aβ42 Binding to α7nAChR
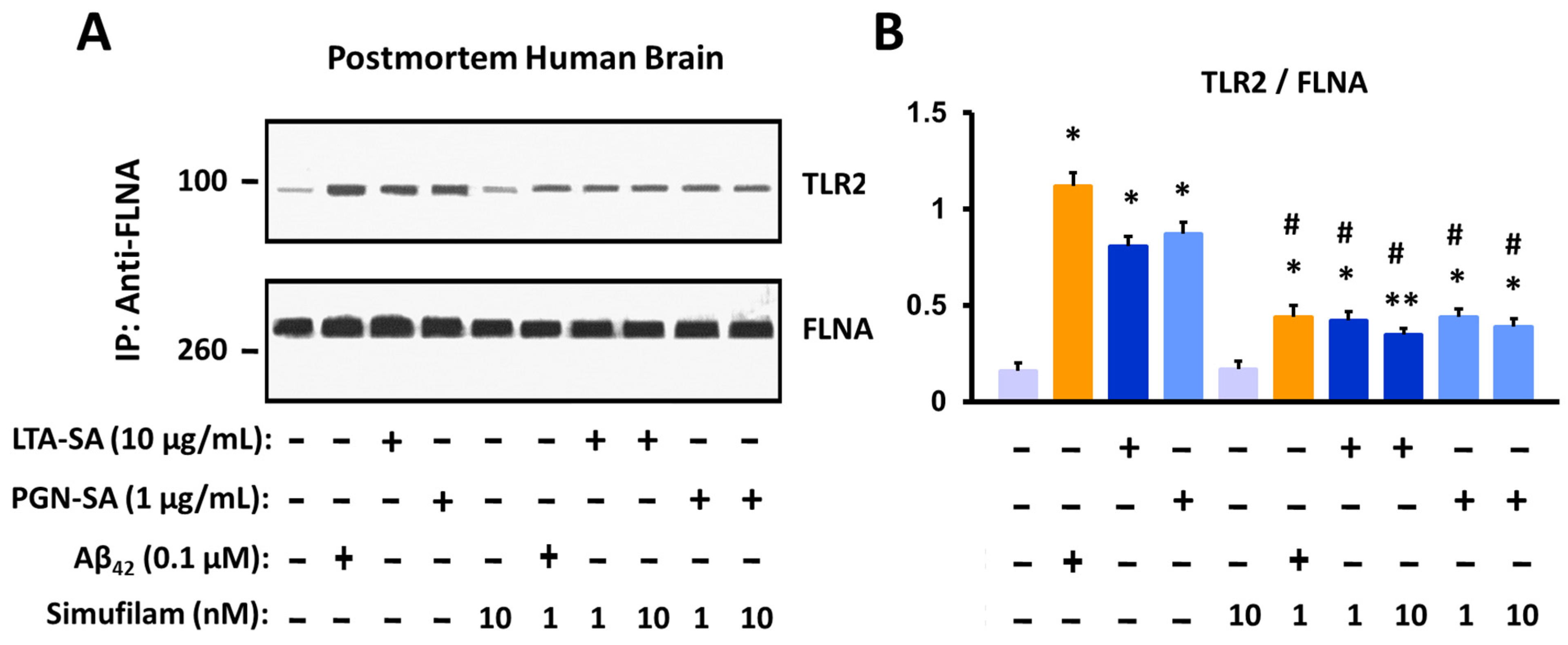
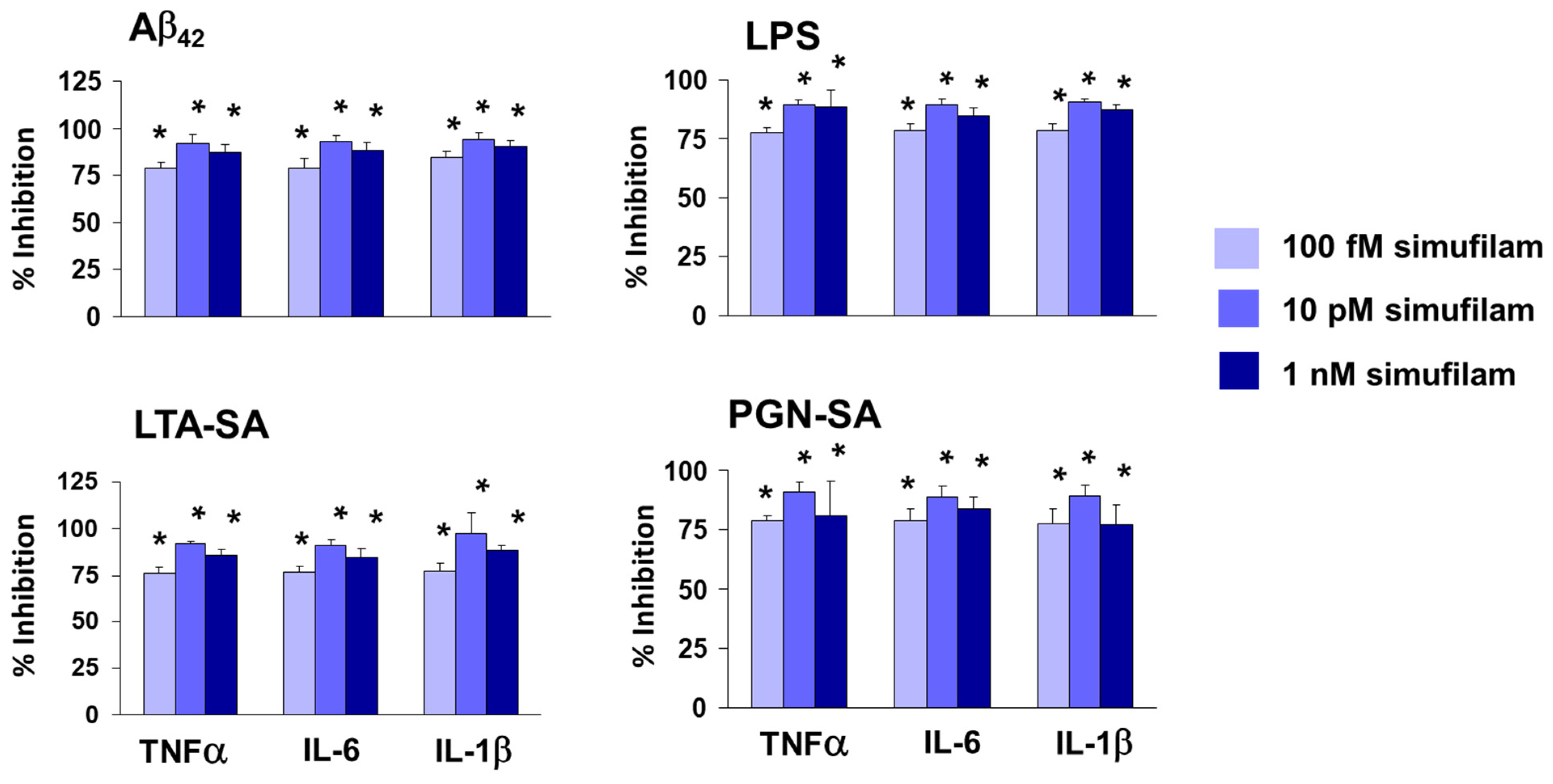
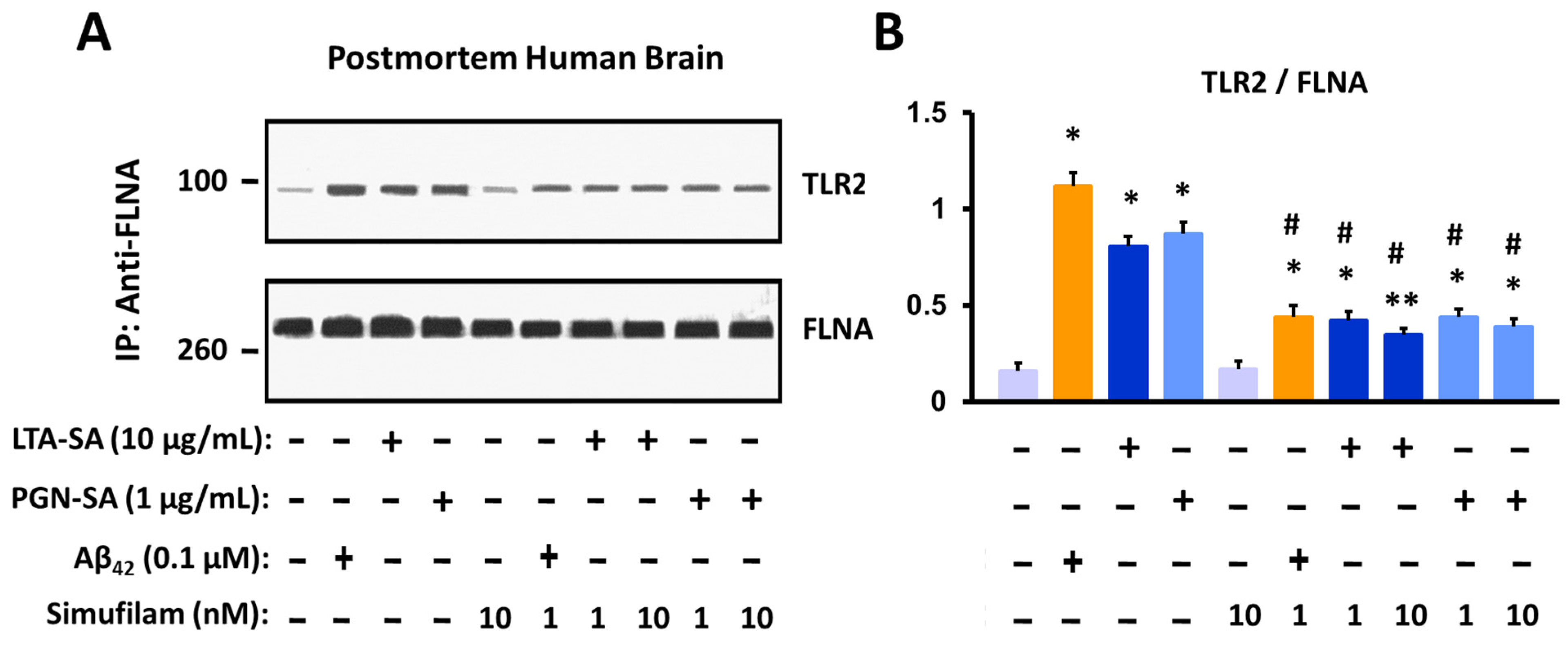
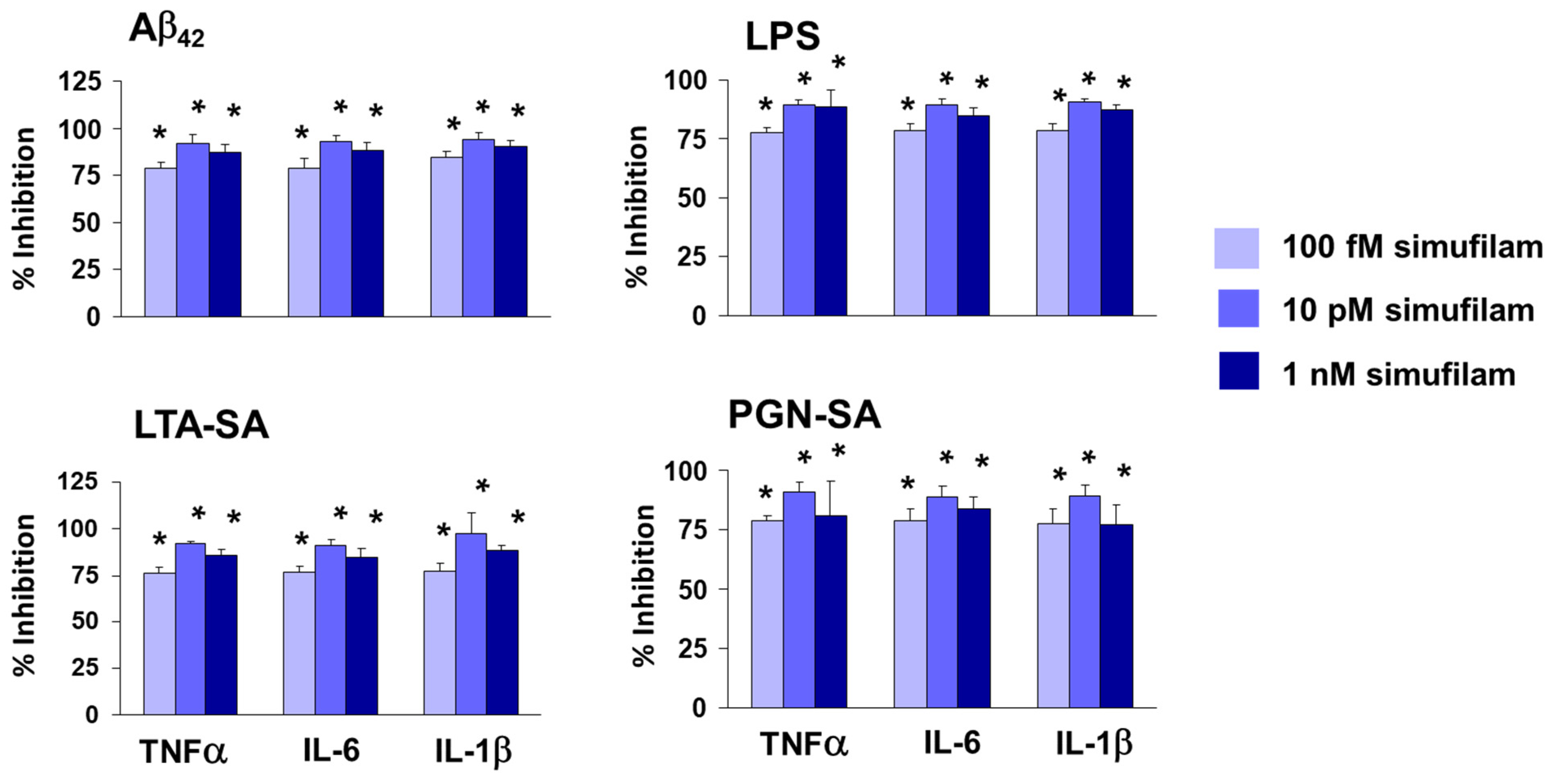
2.2. Simufilam Reduced FLNA–TLR2 Linkage and Cytokine Release Stimulated by Aβ42 and TLR2 Agonists
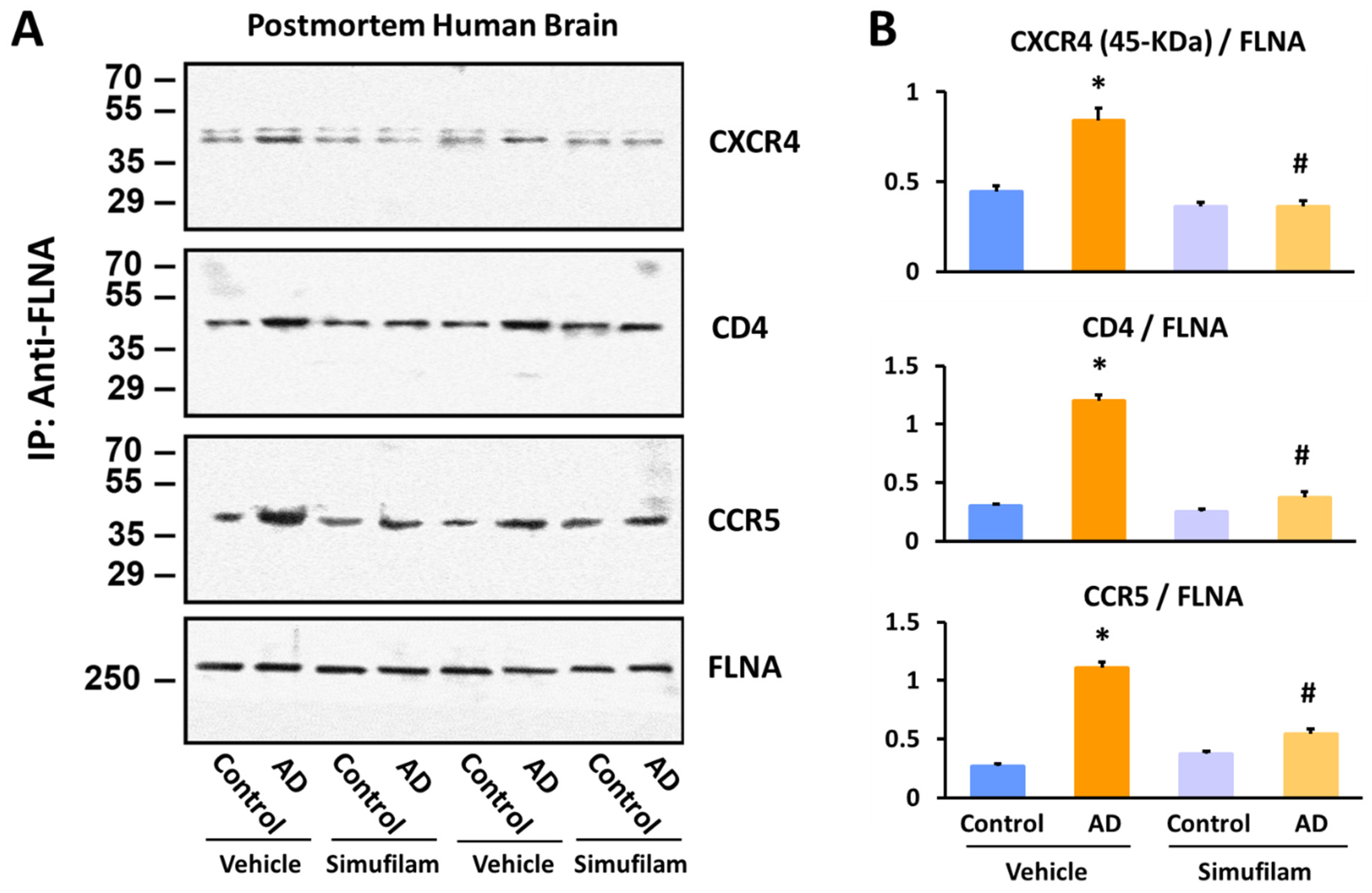
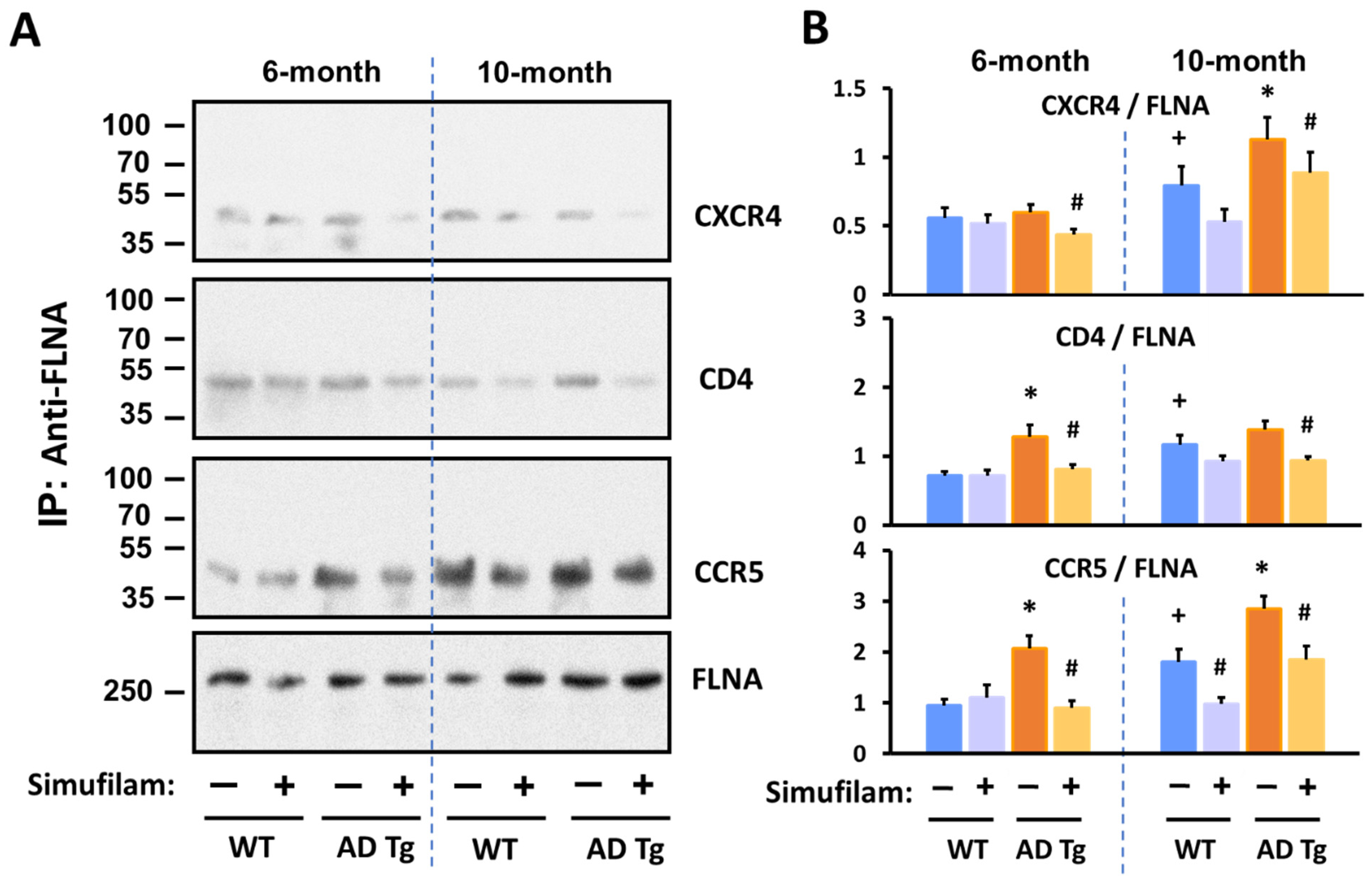
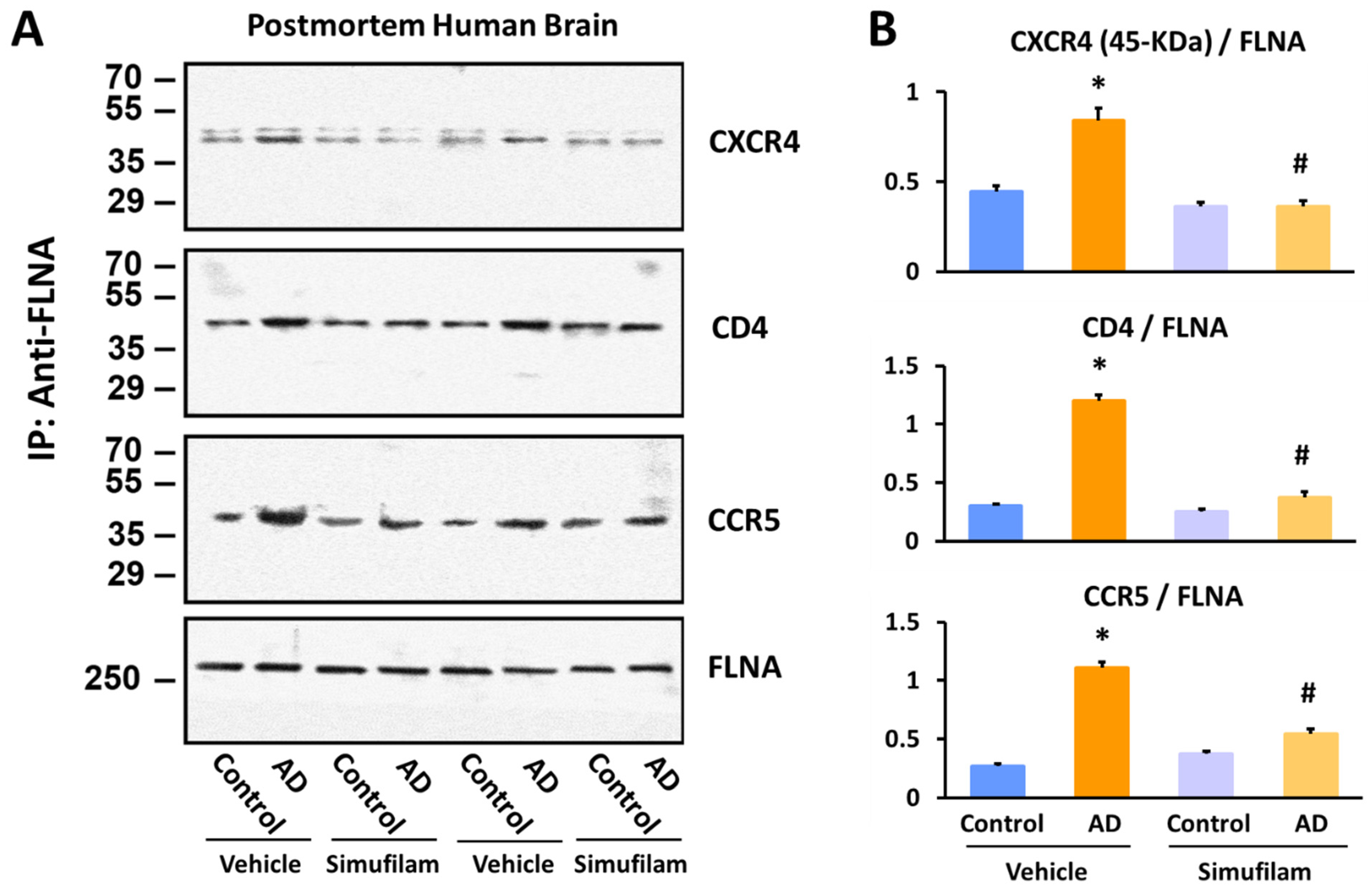
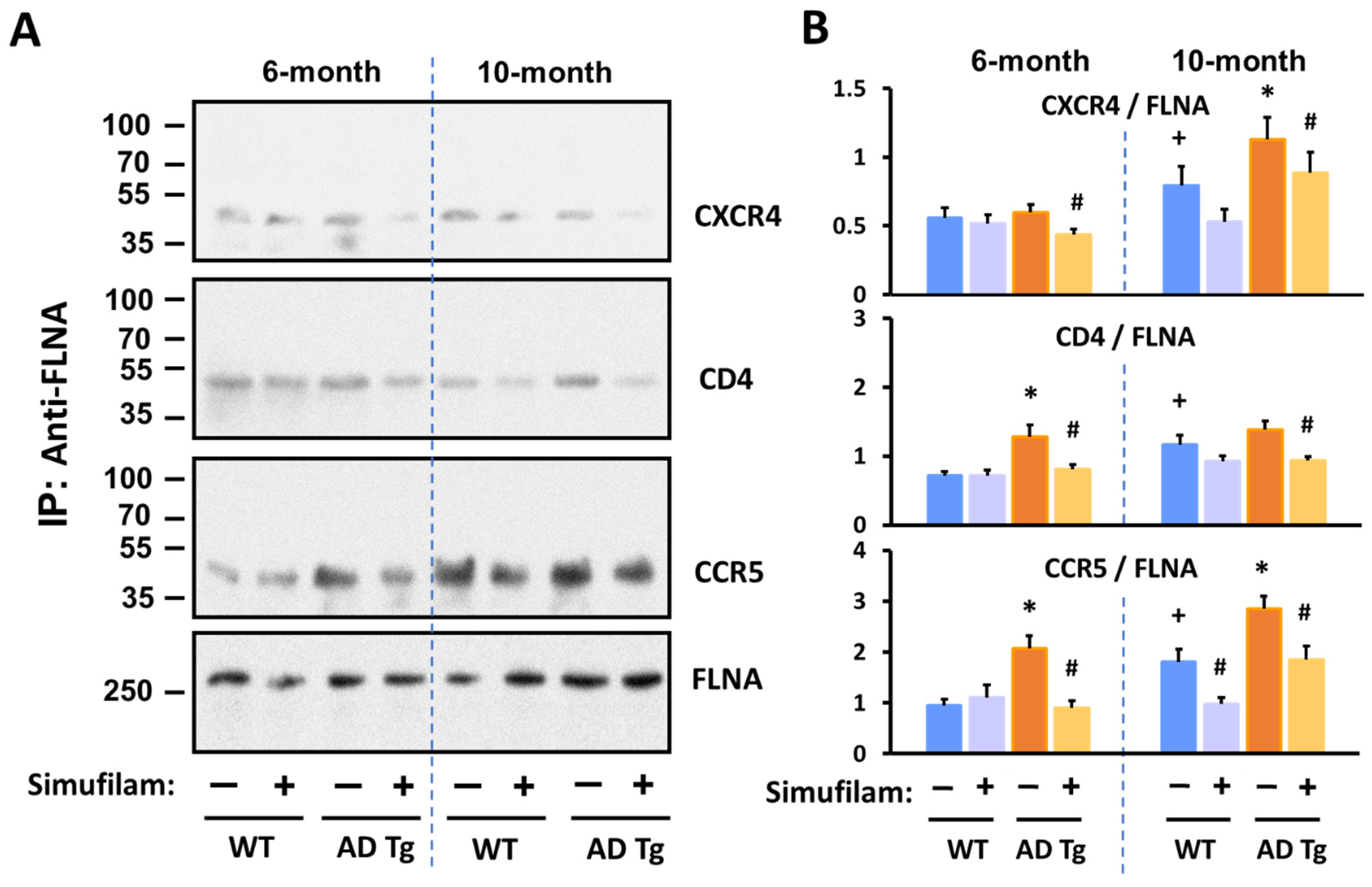
2.3. Simufilam Reduced FLNA–CXCR4/CD4/CCR5 Linkages
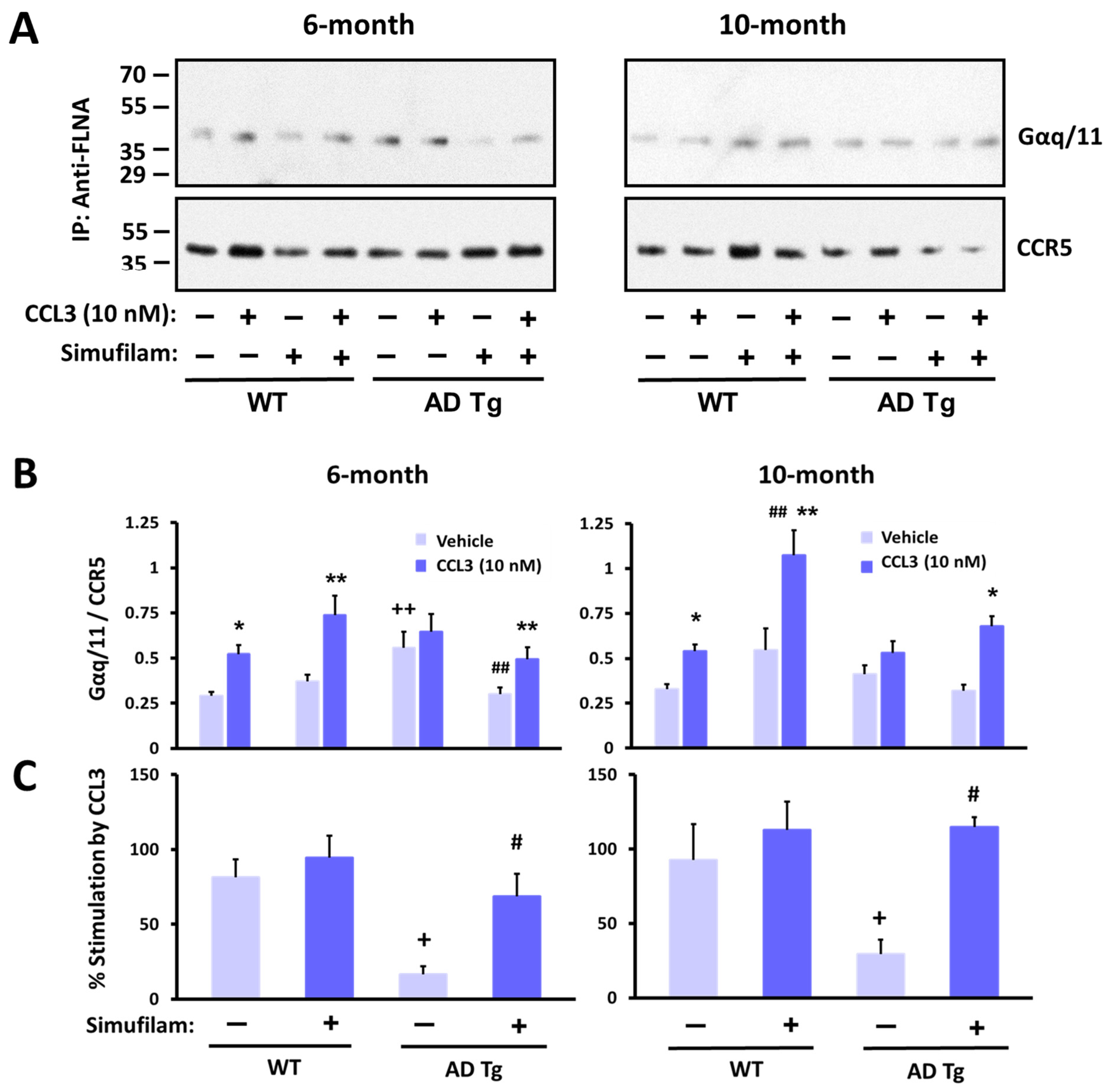
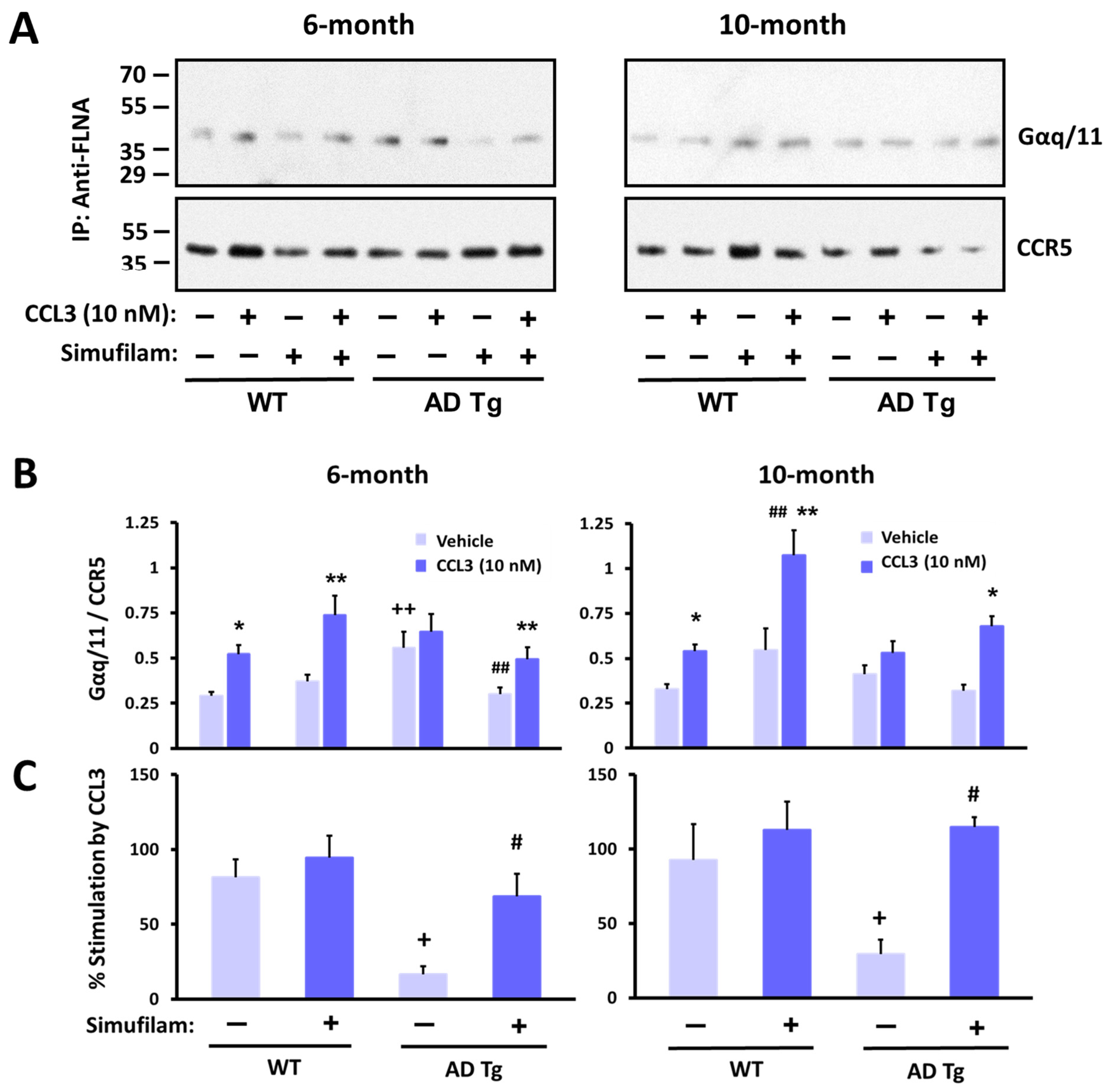
2.4. Simufilam Reduced Chronic CCR5 Activation in AD Transgenic Mice
3. Discussion
4. Materials and Methods
4.1. Materials and Chemicals
4.2. TR-FRET Binding Assay
4.3. Postmortem Human Brain Tissue
4.4. In Vivo Oral Administration of Simufilam
4.5. Assessment of Cytokine Levels in Primary Human Astrocytes
4.6. Assessment of FLNA–TLR2 Interaction in Postmortem Human Brain Tissue
4.7. Assessment of FLNA–CCR5/CD4/CXCR4 Interaction in Postmortem Human Brain and Transgenic AD Mouse Brain
4.8. CCL3-Stimulated Gq/11 Recruitment to CCR5 in Synaptic Membranes
4.9. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Status Report on the Public Health Response to Dementia; World Health Organization: Geneva, Switzerland, 2021; p. 137. [Google Scholar]
- 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Brockmann, R.; Nixon, J.; Love, B.L.; Yunusa, I. Impacts of FDA approval and Medicare restriction on antiamyloid therapies for Alzheimer’s disease: Patient outcomes, healthcare costs, and drug development. Lancet Reg. Health Am. 2023, 20, 100467. [Google Scholar] [CrossRef]
- Barkhof, F.; Knopman, D.S. Brain shrinkage in anti-β-amyloid Alzheimer trials: Neurodegeneration or pseudoatrophy? Neurology 2023, 100, 941–942. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in early symptomatic Alzheimer disease: The TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2023. Alzheimers Dement. 2023, 9, e12385. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Pei, Z.; Lee, K.-C.; Lopez-Brignoni, E.; Nikolov, B.; Crowley, C.; Marsman, M.; Barbier, R.; Friedmann, N.; Burns, L. PTI-125 reduces biomarkers of Alzheimer’s disease in patients. J. Prev. Alzheimer’s Dis. 2020, 7, 256–264. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Bakshi, K.; Frankfurt, M.; Stucky, A.; Goberdhan, M.; Shah, S.; Burns, L. Reducing amyloid-related Alzheimer’s disease pathogenesis by a small molecule targeting filamin A. J. Neurosci. 2012, 32, 9773–9784. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Lee, K.-C.; Pei, Z.; Khan, A.; Bakshi, K.; Burns, L. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol. Aging 2017, 55, 99–114. [Google Scholar] [CrossRef]
- Nakamura, F.; Stossel, T.; Hartwig, J. The filamins: Organizers of cell structure and function. Cell Adh. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef]
- Nakamura, F.; Osborn, T.; Hartemink, C.; Hartwig, J.; Stossel, T. Structural basis of filamin A functions. J. Cell Biol. 2007, 179, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Ruskamo, S.; Gilbert, R.; Hofmann, G.; Jiang, P.; Campbell, I.D.; Ylänne, J.; Pentikäinen, U. The C-terminal rod 2 fragment of filamin A forms a compact structure that can be extended. Biochem. J. 2012, 446, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kang, X.; An, H.; Lv, Y.; Liu, X. The function and pathogenic mechanism of filamin A. Gene 2021, 784, 145575. [Google Scholar] [CrossRef]
- Stossel, T.; Condeelis, J.; Cooley, L.; Hartwig, J.; Noegel, A.; Schleicher, M.; Shapiro, S. Filamins as integrators of cell mechanics and signalling. Nature 2001, 2, 138–145. [Google Scholar] [CrossRef]
- Aumont, E.; Tremblay, C.; Levert, S.; Bennett, D.A.; Calon, F.; Leclerc, N. Evidence of Filamin A loss of solubility at the prodromal stage of neuropathologically-defined Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1038343. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, X.; Cong, P.; Sheetz, M.P.; Nakamura, F.; Yan, J. Differential mechanical stability of filamin A rod segments. Biophys. J. 2011, 101, 1231–1237. [Google Scholar] [CrossRef]
- Kesner, B.A.; Ding, F.; Temple, B.R.; Dokholyan, N.V. N-terminal strands of filamin Ig domains act as a conformational switch under biological forces. Proteins 2010, 78, 12–24. [Google Scholar] [CrossRef]
- Strang, C.J.; Wales, M.E.; Brown, D.M.; Wild, J.R. Site-directed alterations to the geometry of the aspartate transcarbamoylase zinc domain: Selective alteration to regulation by heterotropic ligands, isoelectric point, and stability in urea. Biochemistry 1993, 32, 4156–4167. [Google Scholar] [CrossRef]
- Burns, L.H.; Pei, Z.; Wang, H.Y. Targeting α7 nicotinic acetylcholine receptors and their protein interactions in Alzheimer’s disease drug development. Drug Dev. Res. 2023; online ahead of print. [Google Scholar]
- Wang, H.-Y.; Lee, D.; D’Andrea, M.; Peterson, P.; Shank, R.; Reitz, A. b-Amyloid1-42 binds to a7 nicotinic acetylcholine receptor with high affinity: Implication for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Lee, D.; Davie, C.; Shank, R. Amyloid peptide Aβ1-42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J. Neurochem. 2000, 75, 1155–1161. [Google Scholar] [CrossRef]
- Dineley, K.; Bell, K.; Bui, D.; Sweatt, J. b-Amyloid peptide activates a7 nicotinic acetylcholine receptors expressed in xenopus oocytes. J. Biol. Chem. 2002, 227, 25056–25061. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Li, W.; Benedetti, N.; Lee, D. α7 nicotinic acetylcholine receptors mediate β-amyloid peptide-induced tau protein phosphorylation. J. Biol. Chem. 2003, 278, 31547–31553. [Google Scholar] [CrossRef] [PubMed]
- El Kouhen, R.; Hu, M.; Anderson, D.J.; Li, J.; Gopalakrishnan, M. Pharmacology of alpha7 nicotinic acetylcholine receptor mediated extracellular signal-regulated kinase signalling in PC12 cells. Br. J. Pharmacol. 2009, 156, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Waring, J.; Gopalakrishnan, M.; Li, J. Role of GSK-3beta activation and alpha7 nAChRs in Abeta(1-42)-induced tau phosphorylation in PC12 cells. J. Neurochem. 2008, 106, 1371–1377. [Google Scholar] [CrossRef]
- Alonso, A.; Grundke-Iqbal, I.; Barra, H.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. PNAS 1997, 94, 298–303. [Google Scholar] [CrossRef]
- Alonso, A.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. PNAS 1994, 91, 5562–5566. [Google Scholar] [CrossRef]
- Pîrşcoveanu, D.F.V.; Pirici, I.; Tudorică, V.; Bălşeanu, T.A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; Pîrşcoveanu, M. Tau protein in neurodegenerative diseases—A review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar]
- D’Andrea, M.; Nagele, R.; Wang, H.-Y.; Peterson, P.; Lee, D. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 2001, 38, 120–134. [Google Scholar] [CrossRef]
- Nagele, R.; D’Andrea, M.; Anderson, W.; Wang, H.-Y. Accumulation of beta-amyloid1-42 in neurons is facilitated by the alpha7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110, 199–211. [Google Scholar] [CrossRef]
- Povala, G.; Bellaver, B.; De Bastiani, M.A.; Brum, W.S.; Ferreira, P.C.L.; Bieger, A.; Pascoal, T.A.; Benedet, A.L.; Souza, D.O.; Araujo, R.M.; et al. Soluble amyloid-beta isoforms predict downstream Alzheimer’s disease pathology. Cell Biosci. 2021, 11, 204. [Google Scholar] [CrossRef]
- Dziewczapolski, G.; Glogowski, C.; Masliah, E.; Heinemann, S. Deletion of the α7 Nicotinic Acetylcholine Receptor Gene Improves Cognitive Deficits and Synaptic Pathology in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2009, 29, 8805–8815. [Google Scholar] [CrossRef]
- Cecon, E.; Dam, J.; Luka, M.; Gautier, C.; Chollet, A.M.; Delagrange, P.; Danober, L.; Jockers, R. Quantitative assessment of oligomeric amyloid β peptide binding to α7 nicotinic receptor. Br. J. Pharmacol. 2019, 176, 3475–3488. [Google Scholar] [CrossRef]
- Gambuzza, M.; Sofo, V.; Salmeri, F.; Soraci, L.; Marino, S.; Bramanti, P. Toll-like receptors in Alzheimer’s disease: A therapeutic perspective. CNS Neurol. Disord. Drug Targets 2014, 13, 1542–1558. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef]
- Re, F.; Strominger, J.L. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J. Biol. Chem. 2001, 276, 37692–37699. [Google Scholar] [CrossRef]
- Xia, M.Q.; Qin, S.X.; Wu, L.J.; Mackay, C.R.; Hyman, B.T. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am. J. Pathol. 1998, 153, 31–37. [Google Scholar] [CrossRef]
- Singer, I.I.; Scott, S.; Kawka, D.W.; Chin, J.; Daugherty, B.L.; DeMartino, J.A.; DiSalvo, J.; Gould, S.L.; Lineberger, J.E.; Malkowitz, L.; et al. CCR5, CXCR4, and CD4 are clustered and closely apposed on microvilli of human macrophages and T cells. J. Virol. 2001, 75, 3779–3790. [Google Scholar] [CrossRef]
- Whittaker, V.P. Thirty years of synaptosome research. J. Neurocytol. 1993, 22, 735–742. [Google Scholar] [CrossRef]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am. J. Pathol. 2008, 172, 1683–1692. [Google Scholar] [CrossRef]
- Gylys, K.H.; Fein, J.A.; Wiley, D.J.; Cole, G.M. Rapid annexin-V labeling in synaptosomes. Neurochem. Int. 2004, 44, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Tyagi, N. Method and validation of synaptosomal preparation for isolation of synaptic membrane proteins from rat brain. MethodsX 2014, 1, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Sokolow, S.; Henkins, K.M.; Williams, I.A.; Vinters, H.V.; Schmid, I.; Cole, G.M.; Gylys, K.H. Isolation of synaptic terminals from Alzheimer’s disease cortex. Cytom. A 2012, 81, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ergin, E.; Dogan, A.; Parmaksiz, M.; Elçin, A.E.; Elçin, Y.M. Time-resolved fluorescence resonance energy transfer [TR-FRET] assays for biochemical processes. Curr. Pharm. Biotechnol. 2016, 17, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Marra, G.; Treppiedi, D.; Di Muro, G.; Mangili, F.; Catalano, R.; Esposito, E.; Nozza, E.; Locatelli, M.; Lania, A.; Sala, E.; et al. A Novel Filamin A-Binding Molecule May Significantly Enhance Somatostatin Receptor Type 2 Antitumoral Actions in Growth Hormone-Secreting PitNET Cells; European Congress of Endocrinology: Istanbul, Turkey, 2023. [Google Scholar]
- Zhang, L.; Huang, T.; Teaw, S.; Nguyen, L.H.; Hsieh, L.S.; Gong, X.; Burns, L.H.; Bordey, A. Filamin A inhibition reduces seizure activity in a mouse model of focal cortical malformations. Sci. Transl. Med. 2020, 12, eaay0289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gannon, M.; Chen, Y.; Yan, S.; Zhang, S.; Feng, W.; Tao, J.; Sha, B.; Liu, Z.; Saito, T.; et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Transl. Med. 2020, 12, eaay6931. [Google Scholar] [CrossRef]
- Cecon, E.; Lhomme, T.; Maurice, T.; Luka, M.; Chen, M.; Silva, A.; Wauman, J.; Zabeau, L.; Tavernier, J.; Prévot, V.; et al. Amyloid beta peptide is an endogenous negative allosteric modulator of leptin receptor. Neuroendocrinology 2021, 111, 370–387. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef]
- Steinman, L. Inflammatory cytokines at the summits of pathological signal cascades in brain diseases. Sci. Signal. 2013, 6, pe3. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural. Regen. Res. 2022, 17, 705–716. [Google Scholar] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Clarke, J.R.; Bomfim, T.R.; De Felice, F.G. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. 2014, 10 (Suppl. 1), S76–S83. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Feinstein, R.; Kanety, H.; Papa, M.Z.; Lunenfeld, B.; Karasik, A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J. Biol. Chem. 1993, 268, 26055–26058. [Google Scholar] [CrossRef]
- Ly, M.; Yu, G.Z.; Mian, A.; Cramer, A.; Meysami, S.; Merrill, D.A.; Samara, A.; Eisenstein, S.A.; Hershey, T.; Babulal, G.M.; et al. Neuroinflammation: A modifiable pathway linking obesity, Alzheimer’s disease, and depression. Am. J. Geriatr. Psychiatry 2023, 31, 853–866. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Pei, Z.; Lee, K.-C.; Nikolov, B.; Doehner, T.; Puente, J.; Friedmann, N.; Burns, L. Simufilam suppresses overactive mTOR and restores its sensitivity to insulin in Alzheimer’s disease patient lymphocytes. Front. Aging 2023, 4, 1175601. [Google Scholar] [CrossRef]
- Gu, S.; Matta, J.A.; Lord, B.; Harrington, A.W.; Sutton, S.W.; Davini, W.B.; Bredt, D.S. Brain α7 Nicotinic Acetylcholine Receptor Assembly Requires NACHO. Neuron 2016, 89, 948–955. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Hirano, A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer’s neurofibrillary tangles. Neuropathol. Appl. Neurobiol. 1986, 12, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.; Trojanowski, J. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 1095–1097. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Friedman, E. Effects of lithium on receptor-mediated activation of G proteins in rat brain cortical membranes. Neuropharmacology 1999, 38, 403–414. [Google Scholar] [CrossRef]
- Wang, L.; Gintzler, A. Bimodal opioid regulation of cyclic AMP formation: Implications for positive and negative coupling of opiate receptors to adenylyl cyclase. J. Neurochem. 1994, 63, 1726–1730. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.-Y.; Cecon, E.; Dam, J.; Pei, Z.; Jockers, R.; Burns, L.H. Simufilam Reverses Aberrant Receptor Interactions of Filamin A in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 13927. https://doi.org/10.3390/ijms241813927
Wang H-Y, Cecon E, Dam J, Pei Z, Jockers R, Burns LH. Simufilam Reverses Aberrant Receptor Interactions of Filamin A in Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(18):13927. https://doi.org/10.3390/ijms241813927
Chicago/Turabian StyleWang, Hoau-Yan, Erika Cecon, Julie Dam, Zhe Pei, Ralf Jockers, and Lindsay H. Burns. 2023. "Simufilam Reverses Aberrant Receptor Interactions of Filamin A in Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 18: 13927. https://doi.org/10.3390/ijms241813927
APA StyleWang, H.-Y., Cecon, E., Dam, J., Pei, Z., Jockers, R., & Burns, L. H. (2023). Simufilam Reverses Aberrant Receptor Interactions of Filamin A in Alzheimer’s Disease. International Journal of Molecular Sciences, 24(18), 13927. https://doi.org/10.3390/ijms241813927