
Single-Cell RNA Sequencing Reveals Immuno-Oncology Characteristics of Tumor-Infiltrating T Lymphocytes in Photodynamic Therapy-Treated Colorectal Cancer Mouse Model

 , , ,
, , ,
Abstract
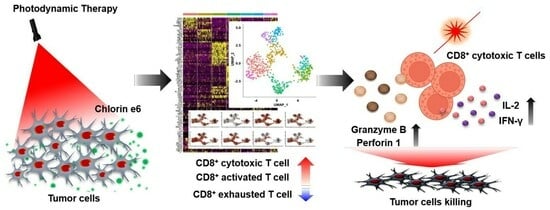
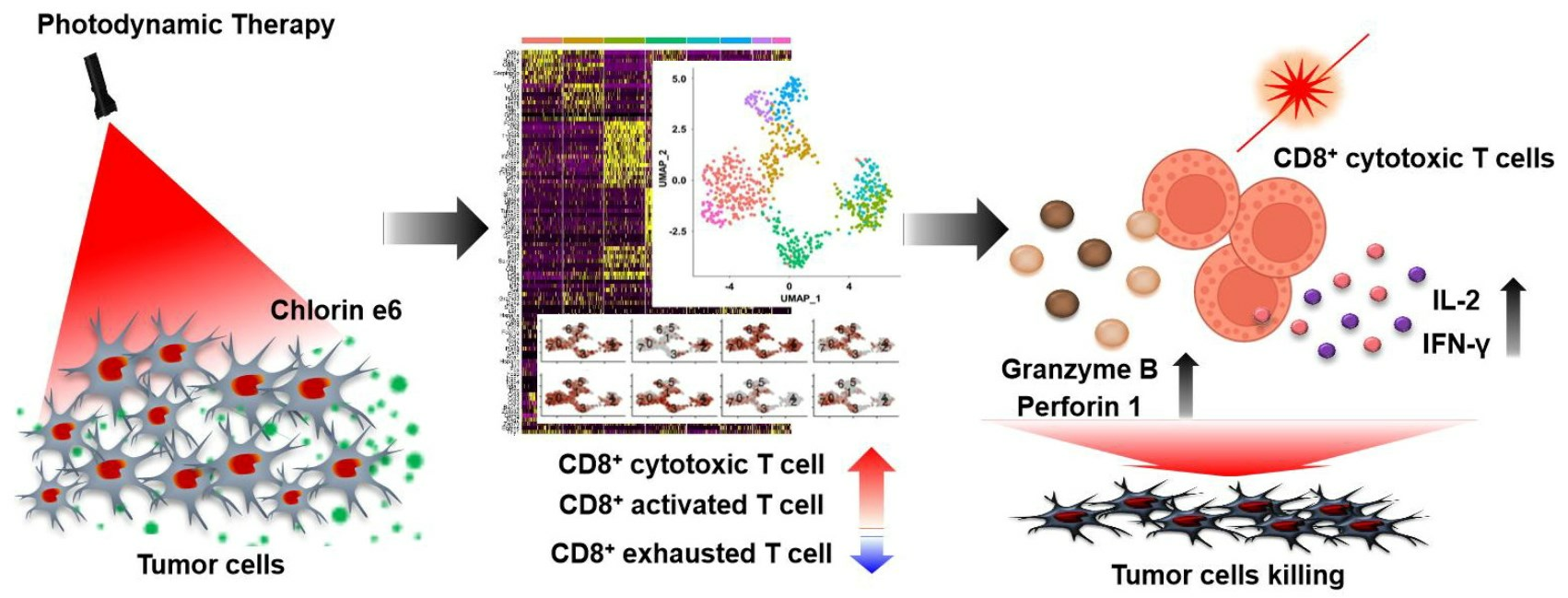{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
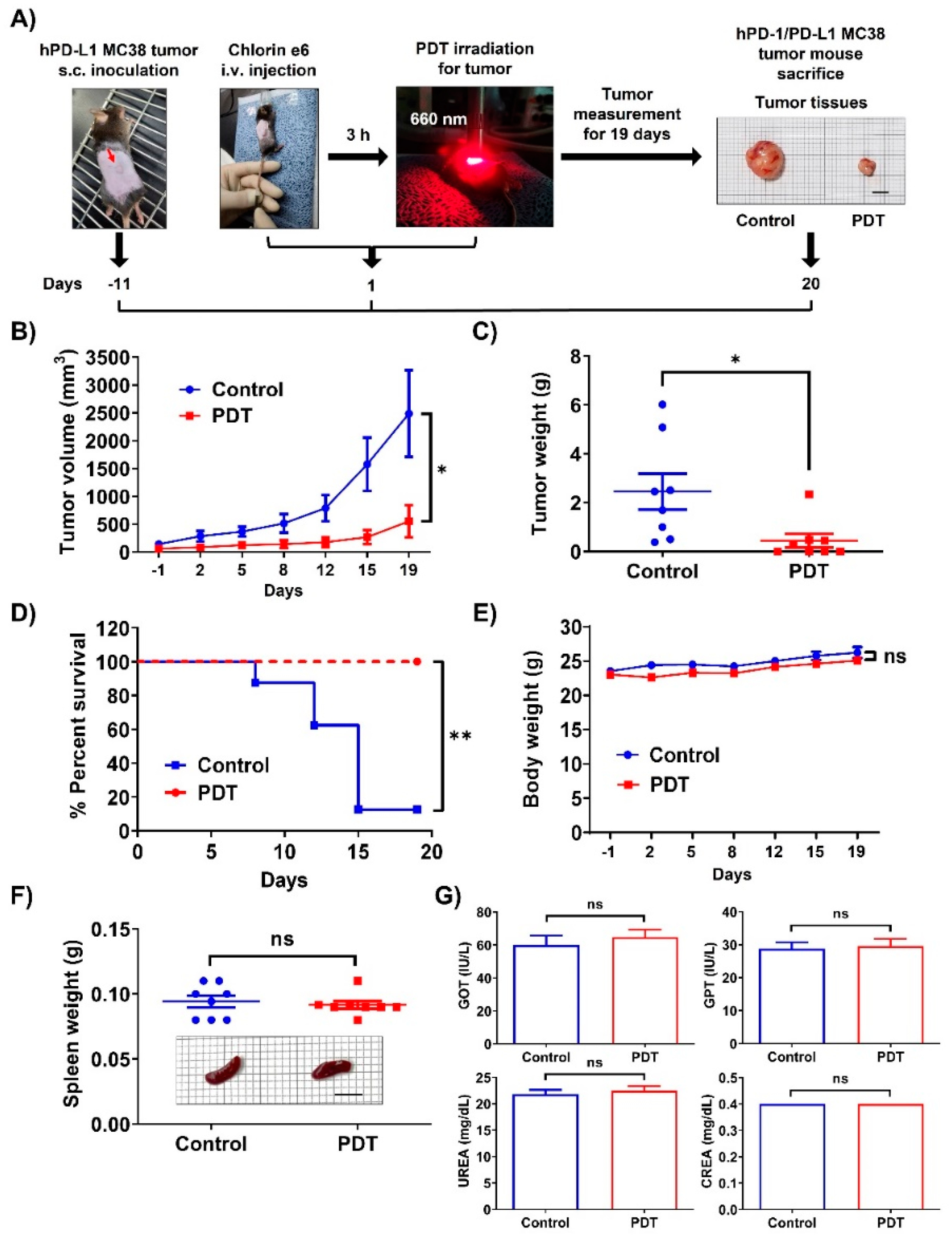
2.1. Ce6-Mediated PDT Induces Antitumor Effects in Mice Bearing Humanized PD-1/PD-L1 Knock-In MC38 Tumors
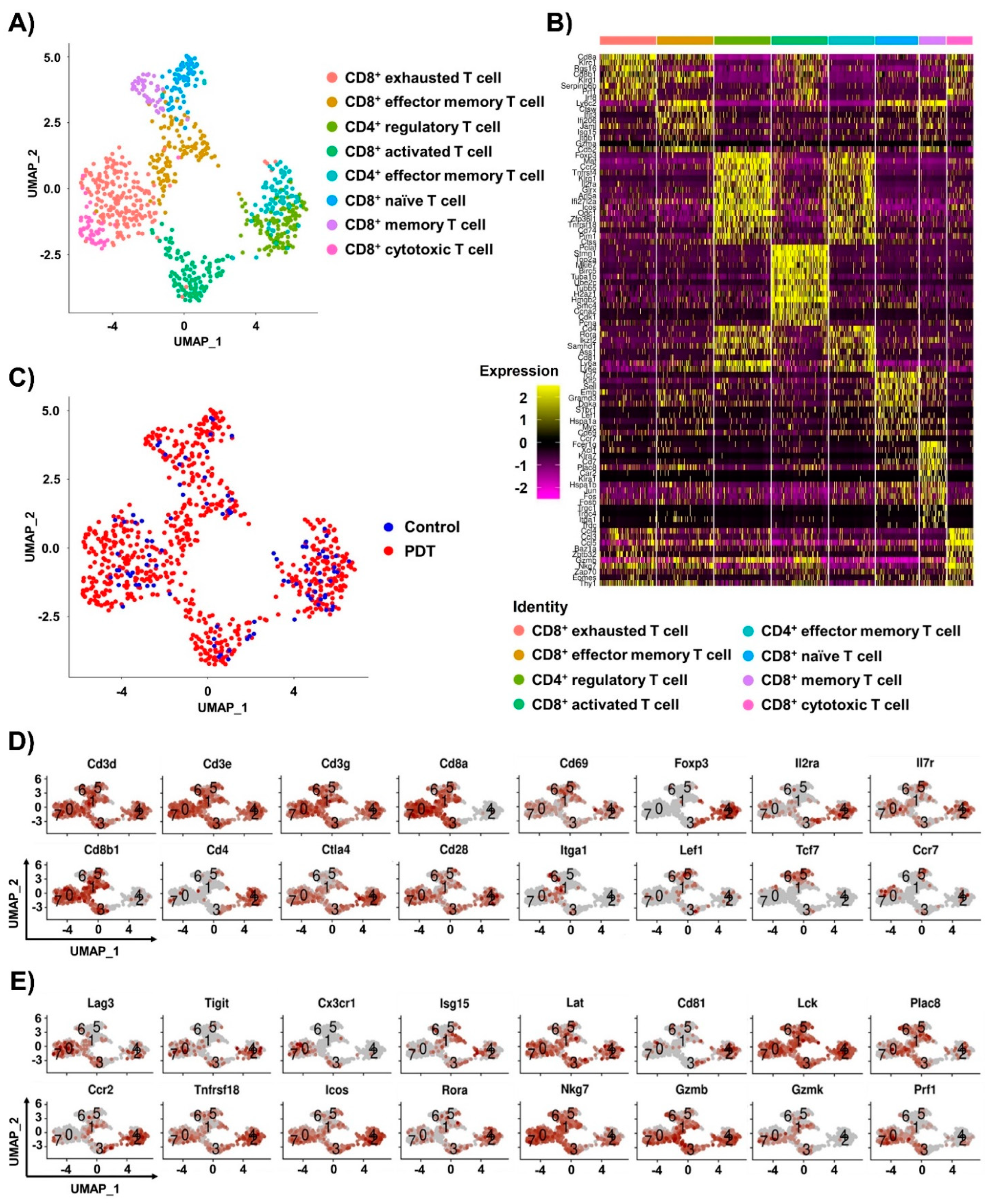
2.2. Single-Cell RNA-seq Analysis of T Cells in Tumor Tissues by Ce6-Mediated PDT Treatment
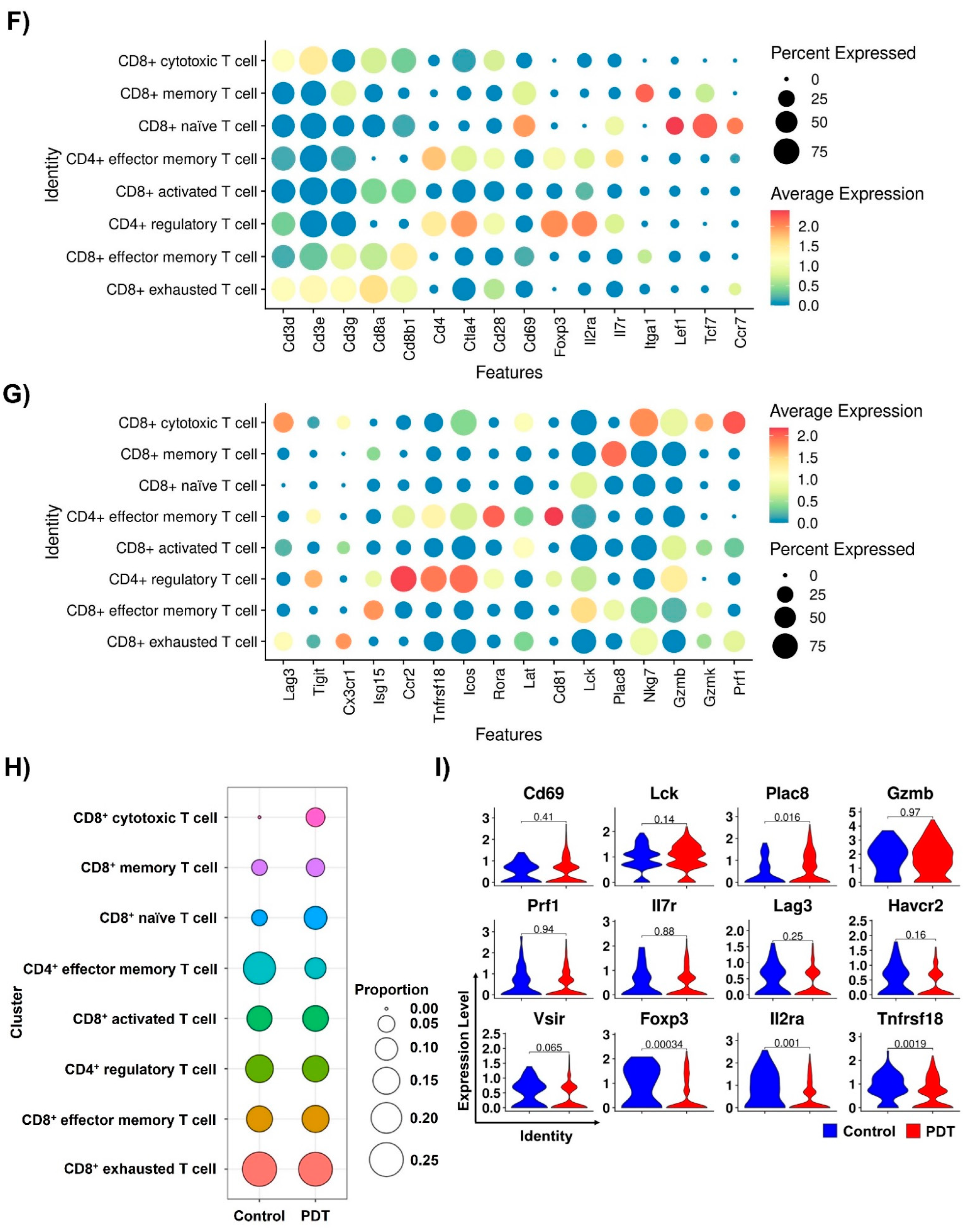
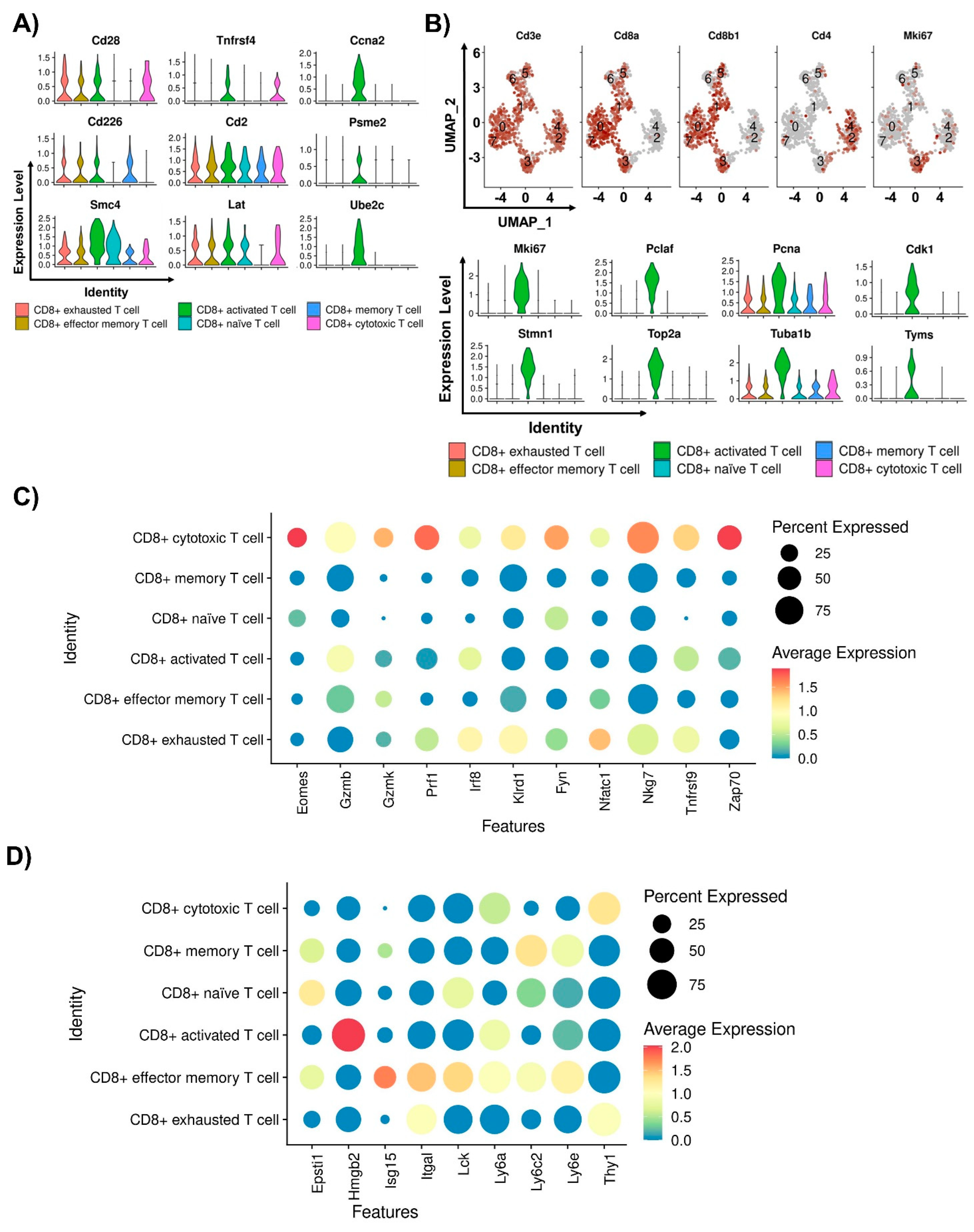
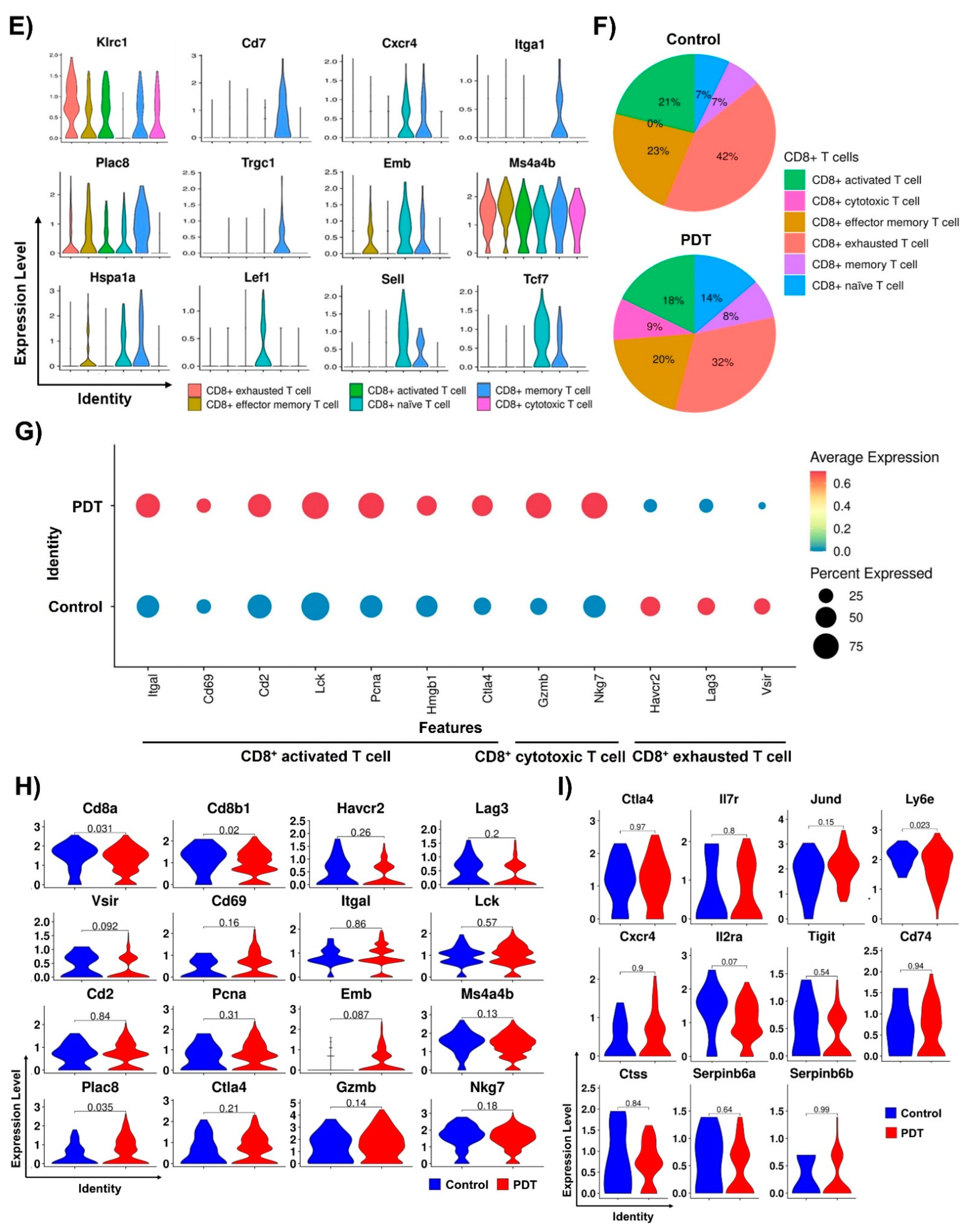
2.3. Characterizations of CD8+ T Cells in Humanized PD-L1 MC38 Tumors of PDT-Treated Mice
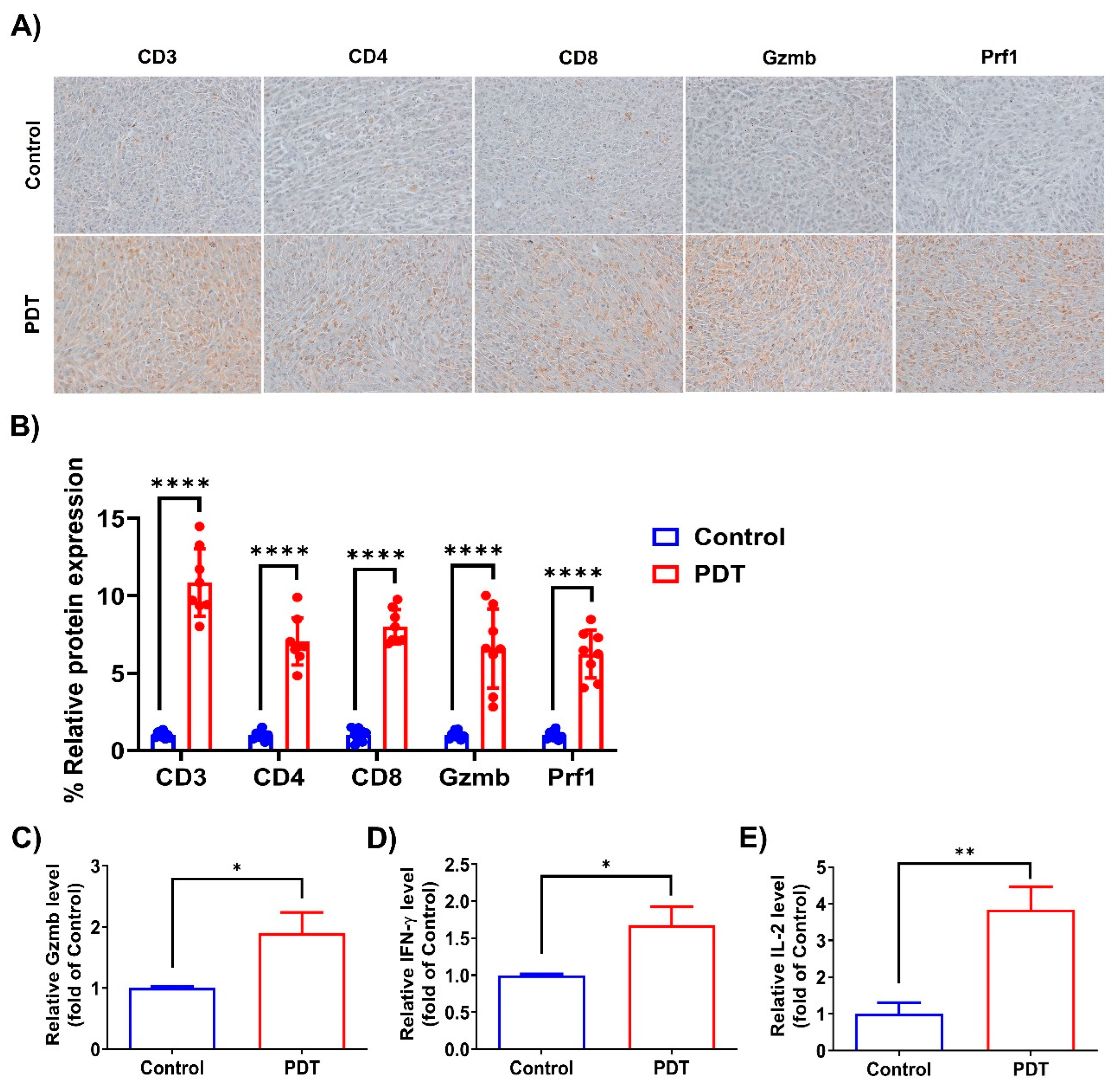
2.4. Ce6-Mediated PDT Enhances Antitumor Immunity in Humanized PD-L1 MC38 Tumors
3. Discussion
4. Materials and Methods
4.1. Preparation of Ce6
4.2. Humanized PD-1/PD-L1 Knock-In Colorectal Cancer Mouse Model
4.3. Blood Biochemistry
4.4. Isolation of Tumor-Infiltrating T Cells
4.5. Single-Cell RNA Sequencing
4.6. Single-Cell RNA Sequencing Data Processing
4.7. Immunohistochemistry
4.8. Granzyme B Measurement
4.9. IFN-γ or IL-2 Measurement
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Ganzleben, I.; Hohmann, M.; Grünberg, A.; Gonzales-Menezes, J.; Vieth, M.; Liebing, E.; Günther, C.; Thonn, V.; Beß, D.; Becker, C.; et al. Topical application of Chlorin e6-PVP (Ce6-PVP) for improved endoscopic detection of neoplastic lesions in a murine colitis-associated cancer model. Sci. Rep. 2020, 10, 13129. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, H.; Amano, T.; Qin, H.; Zheng, L.; Takahashi, A.; Zhao, S.; Tooyama, I.; Murakami, T.; Komatsu, N. Efficient delivery of chlorin e6 into ovarian cancer cells with octalysine conjugated superparamagnetic iron oxide nanoparticles for effective photodynamic therapy. J. Mater. Chem. B 2016, 4, 7741–7748. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Shen, X.; He, Z.; Lu, Y. The Role of Photodynamic Therapy in Triggering Cell Death and Facilitating Antitumor Immunology. Front. Oncol. 2022, 12, 863107. [Google Scholar] [CrossRef]
- Falk-Mahapatra, R.; Gollnick, S.O. Photodynamic Therapy and Immunity: An Update. Photochem. Photobiol. 2020, 96, 550–559. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Chen, Z.; Sternlicht, M.D.; Hidalgo, M.; Steffensen, B. Matrix metalloproteinase-2 contributes to cancer cell migration on collagen. Cancer Res. 2005, 65, 130–136. [Google Scholar] [CrossRef]
- Huang, D.Y.; Gao, H.; Boini, K.M.; Osswald, H.; Nürnberg, B.; Lang, F. In vivo stimulation of AMP-activated protein kinase enhanced tubuloglomerular feedback but reduced tubular sodium transport during high dietary NaCl intake. Pflugers Arch. 2010, 460, 187–196. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef]
- Mroz, P.; Hashmi, J.T.; Huang, Y.Y.; Lange, N.; Hamblin, M.R. Stimulation of anti-tumor immunity by photodynamic therapy. Expert Rev. Clin. Immunol. 2011, 7, 75–91. [Google Scholar] [CrossRef]
- Hua, J.; Wu, P.; Gan, L.; Zhang, Z.; He, J.; Zhong, L.; Zhao, Y.; Huang, Y. Current Strategies for Tumor Photodynamic Therapy Combined With Immunotherapy. Front. Oncol. 2021, 11, 738323. [Google Scholar] [CrossRef]
- Kleinovink, J.W.; Fransen, M.F.; Löwik, C.W.; Ossendorp, F. Photodynamic-Immune Checkpoint Therapy Eradicates Local and Distant Tumors by CD8+ T Cells. Cancer Immunol. Res. 2017, 5, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Blaudszun, A.R.; Kim, W.J.; Um, W.; Yoon, H.Y.; Shim, M.K.; Kim, K. Adoptive Transfer of Photosensitizer-Loaded Cytotoxic T Cells for Combinational Photodynamic Therapy and Cancer Immuno-Therapy. Pharmaceutics 2023, 15, 1295. [Google Scholar] [CrossRef]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Thapa Magar, T.B.; Shrestha, R.; Gurung, P.; Lim, J.; Kim, Y.-W. Improved Pilot-Plant-Scale Synthesis of Chlorin e6 and Its Efficacy as a Photosensitizer for Photodynamic Therapy and Photoacoustic Contrast Agent. Processes 2022, 10, 2215. [Google Scholar] [CrossRef]
- Shrestha, R.; Lee, H.J.; Lim, J.; Gurung, P.; Thapa Magar, T.B.; Kim, Y.T.; Lee, K.; Bae, S.; Kim, Y.W. Effect of Photodynamic Therapy with Chlorin e6 on Canine Tumors. Life 2022, 12, 2102. [Google Scholar] [CrossRef]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e1017. [Google Scholar] [CrossRef]
- Liu, W.; Sheng, S.; Zhu, C.; Li, C.; Zou, Y.; Yang, C.; Chen, Z.J.; Wang, F.; Jiao, X. Increased NKG2A+CD8+ T-cell exhaustion in patients with adenomyosis. Mucosal Immunol. 2023, 16, 121–134. [Google Scholar] [CrossRef]
- Ducoin, K.; Oger, R.; Bilonda Mutala, L.; Deleine, C.; Jouand, N.; Desfrançois, J.; Podevin, J.; Duchalais, E.; Cruard, J.; Benlalam, H.; et al. Targeting NKG2A to boost anti-tumor CD8 T-cell responses in human colorectal cancer. Oncoimmunology 2022, 11, 2046931. [Google Scholar] [CrossRef]
- Villarreal, D.O.; Wise, M.C.; Siefert, R.J.; Yan, J.; Wood, L.M.; Weiner, D.B. Ubiquitin-like Molecule ISG15 Acts as an Immune Adjuvant to Enhance Antigen-specific CD8 T-cell Tumor Immunity. Mol. Ther. 2015, 23, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, B.P.; Guislain, A.; van Alphen, F.P.J.; Gomez-Eerland, R.; Schumacher, T.N.M.; van den Biggelaar, M.; Wolkers, M.C. CD29 identifies IFN-γ-producing human CD8+ T cells with an increased cytotoxic potential. Proc. Natl. Acad. Sci. USA 2020, 117, 6686–6696. [Google Scholar] [CrossRef]
- Johnson, B.J.; Costelloe, E.O.; Fitzpatrick, D.R.; Haanen, J.B.; Schumacher, T.N.; Brown, L.E.; Kelso, A. Single-cell perforin and granzyme expression reveals the anatomical localization of effector CD8+ T cells in influenza virus-infected mice. Proc. Natl. Acad. Sci. USA 2003, 100, 2657–2662. [Google Scholar] [CrossRef]
- Miyagawa, F.; Zhang, H.; Terunuma, A.; Ozato, K.; Tagaya, Y.; Katz, S.I. Interferon regulatory factor 8 integrates T-cell receptor and cytokine-signaling pathways and drives effector differentiation of CD8 T cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12123–12128. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, H.S.; Baek, S.Y.; Lee, G.R. Transcription factor IRF8 controls Th1-like regulatory T-cell function. Cell. Mol. Immunol. 2016, 13, 785–794. [Google Scholar] [CrossRef]
- Di Rosa, F.; Cossarizza, A.; Hayday, A.C. To Ki or Not to Ki: Re-Evaluating the Use and Potentials of Ki-67 for T Cell Analysis. Front. Immunol. 2021, 12, 653974. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S. Roles of SMC Complexes During T Lymphocyte Development and Function. Adv. Protein Chem. Struct. Biol. 2017, 106, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Bjorkdahl, O.; Barber, K.A.; Brett, S.J.; Daly, M.G.; Plumpton, C.; Elshourbagy, N.A.; Tite, J.P.; Thomsen, L.L. Characterization of CC-chemokine receptor 7 expression on murine T cells in lymphoid tissues. Immunology 2003, 110, 170–179. [Google Scholar] [CrossRef]
- Chen, Y.; Zander, R.; Khatun, A.; Schauder, D.M.; Cui, W. Transcriptional and Epigenetic Regulation of Effector and Memory CD8 T Cell Differentiation. Front. Immunol. 2018, 9, 2826. [Google Scholar] [CrossRef]
- Hänninen, A.; Maksimow, M.; Alam, C.; Morgan, D.J.; Jalkanen, S. Ly6C supports preferential homing of central memory CD8+ T cells into lymph nodes. Eur. J. Immunol. 2011, 41, 634–644. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Lins, D.C.; Mescher, M.F. CD8+ memory T cells (CD44high, Ly-6C+) are more sensitive than naive cells to (CD44low, Ly-6C-) to TCR/CD8 signaling in response to antigen. J. Immunol. 1998, 160, 3236–3243. [Google Scholar] [CrossRef] [PubMed]
- Aandahl, E.M.; Sandberg, J.K.; Beckerman, K.P.; Taskén, K.; Moretto, W.J.; Nixon, D.F. CD7 is a differentiation marker that identifies multiple CD8 T cell effector subsets. J. Immunol. 2003, 170, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Slade, C.D.; Reagin, K.L.; Lakshmanan, H.G.; Klonowski, K.D.; Watford, W.T. Placenta-specific 8 limits IFNγ production by CD4 T cells in vitro and promotes establishment of influenza-specific CD8 T cells in vivo. PLoS ONE 2020, 15, e0235706. [Google Scholar] [CrossRef] [PubMed]
- Reilly, E.C.; Sportiello, M.; Emo, K.L.; Amitrano, A.M.; Jha, R.; Kumar, A.B.R.; Laniewski, N.G.; Yang, H.; Kim, M.; Topham, D.J. CD49a Identifies Polyfunctional Memory CD8 T Cell Subsets that Persist in the Lungs After Influenza Infection. Front. Immunol. 2021, 12, 728669. [Google Scholar] [CrossRef]
- Yamashita, Y.; Yatabe, Y.; Tsuzuki, T.; Nakayama, A.; Hasegawa, Y.; Kojima, H.; Nagasawa, T.; Mori, N. Perforin and granzyme expression in cytotoxic T-cell lymphomas. Mod. Pathol. 1998, 11, 313–323. [Google Scholar]
- Waterhouse, N.J.; Sutton, V.R.; Sedelies, K.A.; Ciccone, A.; Jenkins, M.; Turner, S.J.; Bird, P.I.; Trapani, J.A. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J. Cell Biol. 2006, 173, 133–144. [Google Scholar] [CrossRef]
- Wen, T.; Barham, W.; Li, Y.; Zhang, H.; Gicobi, J.K.; Hirdler, J.B.; Liu, X.; Ham, H.; Peterson Martinez, K.E.; Lucien, F.; et al. NKG7 Is a T-cell-Intrinsic Therapeutic Target for Improving Antitumor Cytotoxicity and Cancer Immunotherapy. Cancer Immunol. Res. 2022, 10, 162–181. [Google Scholar] [CrossRef] [PubMed]
- Llaó-Cid, L.; Roessner, P.M.; Chapaprieta, V.; Öztürk, S.; Roider, T.; Bordas, M.; Izcue, A.; Colomer, D.; Dietrich, S.; Stilgenbauer, S.; et al. EOMES is essential for antitumor activity of CD8+ T cells in chronic lymphocytic leukemia. Leukemia 2021, 35, 3152–3162. [Google Scholar] [CrossRef]
- Valencia, X.; Lipsky, P.E. CD4+CD25+FoxP3+ regulatory T cells in autoimmune diseases. Nat. Clin. Pract. Rheumatol. 2007, 3, 619–626. [Google Scholar] [CrossRef]
- Wheaton, J.D.; Yeh, C.H.; Ciofani, M. Cutting Edge: C-Maf Is Required for Regulatory T Cells To Adopt RORγt(+) and Follicular Phenotypes. J. Immunol. 2017, 199, 3931–3936. [Google Scholar] [CrossRef]
- Zhan, Y.; Wang, N.; Vasanthakumar, A.; Zhang, Y.; Chopin, M.; Nutt, S.L.; Kallies, A.; Lew, A.M. CCR2 enhances CD25 expression by FoxP3+ regulatory T cells and regulates their abundance independently of chemotaxis and CCR2+ myeloid cells. Cell. Mol. Immunol. 2020, 17, 123–132. [Google Scholar] [CrossRef]
- Beyersdorf, N.; Ding, X.; Tietze, J.K.; Hanke, T. Characterization of mouse CD4 T cell subsets defined by expression of KLRG1. Eur. J. Immunol. 2007, 37, 3445–3454. [Google Scholar] [CrossRef]
- Moore, T.V.; Clay, B.S.; Ferreira, C.M.; Williams, J.W.; Rogozinska, M.; Cannon, J.L.; Shilling, R.A.; Marzo, A.L.; Sperling, A.I. Protective effector memory CD4 T cells depend on ICOS for survival. PLoS ONE 2011, 6, e16529. [Google Scholar] [CrossRef] [PubMed]
- Haim-Vilmovsky, L.; Henriksson, J.; Walker, J.A.; Miao, Z.; Natan, E.; Kar, G.; Clare, S.; Barlow, J.L.; Charidemou, E.; Mamanova, L.; et al. Mapping Rora expression in resting and activated CD4+ T cells. PLoS ONE 2021, 16, e0251233. [Google Scholar] [CrossRef]
- Maecker, H.T. Human CD81 directly enhances Th1 and Th2 cell activation, but preferentially induces proliferation of Th2 cells upon long-term stimulation. BMC Immunol. 2003, 4, 1. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parsa, R.; London, M.; Rezende de Castro, T.B.; Reis, B.; Buissant des Amorie, J.; Smith, J.G.; Mucida, D. Newly recruited intraepithelial Ly6A+CCR9+CD4+ T cells protect against enteric viral infection. Immunity 2022, 55, 1234–1249. [Google Scholar] [CrossRef]
- Cray, C.; Keane, R.W.; Malek, T.R.; Levy, R.B. Regulation and selective expression of Ly-6A/E, a lymphocyte activation molecule, in the central nervous system. Brain Res. Mol. Brain Res. 1990, 8, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.Z.; Eshel, R.; Sagi-Assif, O.; Witz, I.P. An association between high Ly-6A/E expression on tumor cells and a highly malignant phenotype. Int. J. Cancer 1994, 59, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.C.; Kamdar, M.M.; Bamezai, A. Ly-6A.2 expression regulates antigen-specific CD4+ T cell proliferation and cytokine production. J. Immunol. 2002, 168, 118–126. [Google Scholar] [CrossRef]
- Engelhardt, J.J.; Sullivan, T.J.; Allison, J.P. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J. Immunol. 2006, 177, 1052–1061. [Google Scholar] [CrossRef]
- Berg, M.; Zavazava, N. Regulation of CD28 expression on CD8+ T cells by CTLA-4. J. Leukoc. Biol. 2008, 83, 853–863. [Google Scholar] [CrossRef]
- Colpitts, S.L.; Dalton, N.M.; Scott, P. IL-7 receptor expression provides the potential for long-term survival of both CD62Lhigh central memory T cells and Th1 effector cells during Leishmania major infection. J. Immunol. 2009, 182, 5702–5711. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Hoki, T.; Oba, T.; Saito, H.; Attwood, K.; Sabel, M.S.; Chang, A.E.; Odunsi, K.; Ito, F. CX3CR1-CD8+ T cells are critical in antitumor efficacy but functionally suppressed in the tumor microenvironment. JCI Insight. 2020, 5, e133920. [Google Scholar] [CrossRef]
- Dolina, J.S.; Van Braeckel-Budimir, N.; Thomas, G.D.; Salek-Ardakani, S. CD8+ T Cell Exhaustion in Cancer. Front. Immunol. 2021, 12, 715234. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Ricci, E.; Petrillo, M.G.; Cari, L.; Migliorati, G.; Nocentini, G.; Riccardi, C. Glucocorticoid-induced tumour necrosis factor receptor-related protein: A key marker of functional regulatory T cells. J. Immunol. Res. 2015, 2015, 171520. [Google Scholar] [CrossRef] [PubMed]
- Wange, R.L. LAT, the linker for activation of T cells: A bridge between T cell-specific and general signaling pathways. Sci. STKE 2000, 2000, re1. [Google Scholar] [CrossRef]
- Grimm, E.A. Human lymphokine-activated killer cells (LAK cells) as a potential immunotherapeutic modality. Biochim. Biophys. Acta 1986, 865, 267–279. [Google Scholar] [CrossRef]
- Wuerdemann, N.; Pütz, K.; Eckel, H.; Jain, R.; Wittekindt, C.; Huebbers, C.U.; Sharma, S.J.; Langer, C.; Gattenlöhner, S.; Büttner, R.; et al. LAG-3, TIM-3 and VISTA Expression on Tumor-Infiltrating Lymphocytes in Oropharyngeal Squamous Cell Carcinoma-Potential Biomarkers for Targeted Therapy Concepts. Int. J. Mol. Sci. 2020, 22, 379. [Google Scholar] [CrossRef]
- Zhu, W.; Li, Y.; Han, M.; Jiang, J. Regulatory Mechanisms and Reversal of CD8+T Cell Exhaustion: A Literature Review. Biology 2023, 12, 541. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Philip, M. CD226 Throttles up CD8+ T Cell Antitumor Activity. Immunity 2020, 53, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.; Cvetkovski, F.; Sellberg, F.; Berg, S.; Paternina Visbal, H.; Sachs, D.H.; Berglund, E.; Berglund, D. CD2 Immunobiology. Front. Immunol. 2020, 11, 1090. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, K.; Suzuki, T.; Kojima, M.; Shimomura, M.; Sakuyama, N.; Tsukada, Y.; Sasaki, T.; Nishizawa, Y.; Taketomi, A.; Ito, M.; et al. Ki67 expression and localization of T cells after neoadjuvant therapies as reliable predictive markers in rectal cancer. Cancer Sci. 2020, 111, 23–35. [Google Scholar] [CrossRef]
- Borcherding, N.; Vishwakarma, A.; Voigt, A.P.; Bellizzi, A.; Kaplan, J.; Nepple, K.; Salem, A.K.; Jenkins, R.W.; Zakharia, Y.; Zhang, W. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun. Biol. 2021, 4, 122. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Chen, R.; Peng, P.C.; Wen, B.; Li, F.Y.; Xie, S.; Chen, G.; Lu, J.; Peng, Z.; Tang, S.B.; Liang, Y.M.; et al. Anti-Programmed Cell Death (PD)-1 Immunotherapy for Malignant Tumor: A Systematic Review and Meta-Analysis. Transl. Oncol. 2016, 9, 32–40. [Google Scholar] [CrossRef]
- Li, X.Y.; Corvino, D.; Nowlan, B.; Aguilera, A.R.; Ng, S.S.; Braun, M.; Cillo, A.R.; Bald, T.; Smyth, M.J.; Engwerda, C.R. NKG7 Is Required for Optimal Antitumor T-cell Immunity. Cancer Immunol. Res. 2022, 10, 154–161. [Google Scholar] [CrossRef]
- Dai, Q.; Han, P.; Qi, X.; Li, F.; Li, M.; Fan, L.; Zhang, H.; Zhang, X.; Yang, X. 4-1BB Signaling Boosts the Anti-Tumor Activity of CD28-Incorporated 2(nd) Generation Chimeric Antigen Receptor-Modified T Cells. Front. Immunol. 2020, 11, 539654. [Google Scholar] [CrossRef]
- Figueiredo, C.; Wittmann, M.; Wang, D.; Dressel, R.; Seltsam, A.; Blasczyk, R.; Eiz-Vesper, B. Heat shock protein 70 (HSP70) induces cytotoxicity of T-helper cells. Blood 2009, 113, 3008–3016. [Google Scholar] [CrossRef]
- Buang, N.; Tapeng, L.; Gray, V.; Sardini, A.; Whilding, C.; Lightstone, L.; Cairns, T.D.; Pickering, M.C.; Behmoaras, J.; Ling, G.S.; et al. Type I interferons affect the metabolic fitness of CD8(+) T cells from patients with systemic lupus erythematosus. Nat. Commun. 2021, 12, 1980. [Google Scholar] [CrossRef]
- Meixner, A.; Karreth, F.; Kenner, L.; Wagner, E.F. JunD regulates lymphocyte proliferation and T helper cell cytokine expression. Embo J. 2004, 23, 1325–1335. [Google Scholar] [CrossRef]
- Wang, H.X.; Zhang, Q.; Zhang, J.; Luan, R.; Liang, Z.; Tan, L.; Xu, Y.; Zhang, P.; Zheng, L.; Zhao, Y.; et al. CD74 regulates cellularity and maturation of medullary thymic epithelial cells partially by activating the canonical NF-κB signaling pathway. FASEB J. 2021, 35, e21535. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wu, C.; Chen, T.; Santos, M.M.; Liu, C.L.; Yang, C.; Zhang, L.; Ren, J.; Liao, S.; Guo, H.; et al. Cathepsin S inhibition changes regulatory T-cell activity in regulating bladder cancer and immune cell proliferation and apoptosis. Mol. Immunol. 2017, 82, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Furner, B.B.; Stevens, C.S. Diffuse, ulcerating plaques and nodules. Necrobiotic xanthogranuloma (NXG) with paraproteinemia. Arch. Dermatol. 1989, 125, 287–288, 290. [Google Scholar] [CrossRef]
- Pigula, M.; Huang, H.C.; Mallidi, S.; Anbil, S.; Liu, J.; Mai, Z.; Hasan, T. Size-dependent Tumor Response to Photodynamic Therapy and Irinotecan Monotherapies Revealed by Longitudinal Ultrasound Monitoring in an Orthotopic Pancreatic Cancer Model. Photochem. Photobiol. 2019, 95, 378–386. [Google Scholar] [CrossRef]
- Hao, Y.; Gu, Z.; Yu, Z.; Schomann, T.; Sayedipour, S.; Aguilar, J.C.; Ten Dijke, P.; Cruz, L.J. Photodynamic Therapy in Combination with the Hepatitis B Core Virus-like Particles (HBc VLPs) to Prime Anticancer Immunity for Colorectal Cancer Treatment. Cancers 2022, 14, 2724. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chung, C.K.; Gu, Z.; Schomann, T.; Dong, X.; Veld, R.; Camps, M.G.M.; Ten Dijke, P.; Ossendorp, F.A.; Cruz, L.J. Combinatorial therapeutic approaches of photodynamic therapy and immune checkpoint blockade for colon cancer treatment. Mol. Biomed. 2022, 3, 26. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Liu, H.; Tian, H.; Wang, Y.; Zhang, G.; Lei, Y.; Xue, L.; Zheng, B.; Fan, T.; et al. The therapeutic significance of the novel photodynamic material TPE-IQ-2O in tumors. Aging 2020, 13, 1383–1409. [Google Scholar] [CrossRef]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. CD8(+) T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’h, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T Cells Use Mechanical Force to Potentiate Target Cell Killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef]
- R Core Team, R. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. R-Project Org. 2013. Available online: https://www.R-project.org/ (accessed on 3 September 2023).
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.-J.; Choi, J.-G.; Han, J.H.; Kim, Y.-W.; Lim, J.; Chung, H.-S. Single-Cell RNA Sequencing Reveals Immuno-Oncology Characteristics of Tumor-Infiltrating T Lymphocytes in Photodynamic Therapy-Treated Colorectal Cancer Mouse Model. Int. J. Mol. Sci. 2023, 24, 13913. https://doi.org/10.3390/ijms241813913
Lee E-J, Choi J-G, Han JH, Kim Y-W, Lim J, Chung H-S. Single-Cell RNA Sequencing Reveals Immuno-Oncology Characteristics of Tumor-Infiltrating T Lymphocytes in Photodynamic Therapy-Treated Colorectal Cancer Mouse Model. International Journal of Molecular Sciences. 2023; 24(18):13913. https://doi.org/10.3390/ijms241813913
Chicago/Turabian StyleLee, Eun-Ji, Jang-Gi Choi, Jung Ho Han, Yong-Wan Kim, Junmo Lim, and Hwan-Suck Chung. 2023. "Single-Cell RNA Sequencing Reveals Immuno-Oncology Characteristics of Tumor-Infiltrating T Lymphocytes in Photodynamic Therapy-Treated Colorectal Cancer Mouse Model" International Journal of Molecular Sciences 24, no. 18: 13913. https://doi.org/10.3390/ijms241813913
APA StyleLee, E.-J., Choi, J.-G., Han, J. H., Kim, Y.-W., Lim, J., & Chung, H.-S. (2023). Single-Cell RNA Sequencing Reveals Immuno-Oncology Characteristics of Tumor-Infiltrating T Lymphocytes in Photodynamic Therapy-Treated Colorectal Cancer Mouse Model. International Journal of Molecular Sciences, 24(18), 13913. https://doi.org/10.3390/ijms241813913