

Genomic and Phenotypic Characterization of Mastitis-Causing Staphylococci and Probiotic Lactic Acid Bacteria Isolated from Raw Sheep’s Milk

 ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Assembly Statistics
2.2. Lactic Acid Bacteria
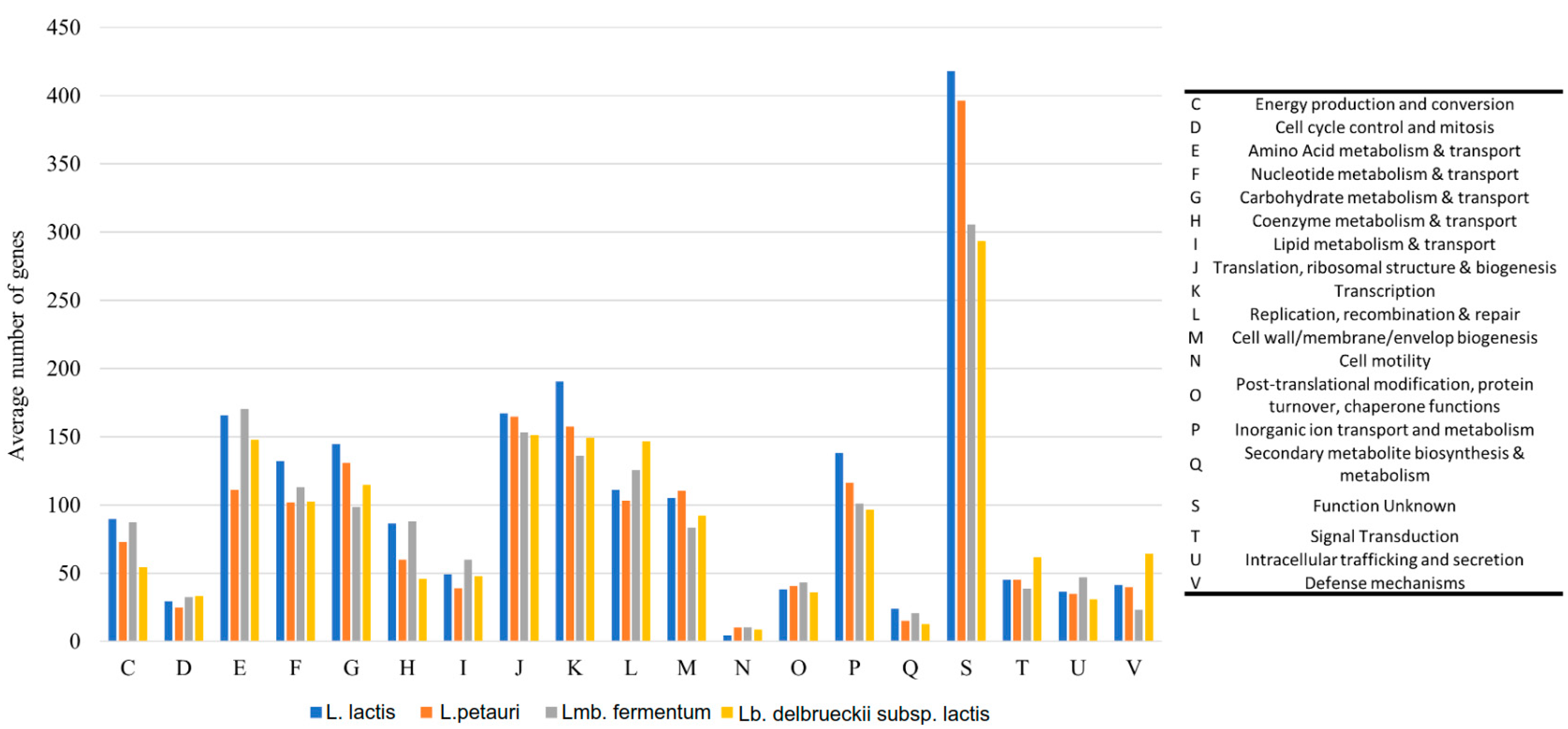
2.2.1. Phylogenetic Comparison and Functional Analysis
2.2.2. Virulence and Antimicrobial Resistance Determinants
2.2.3. Bacteriocins and Carbohydrate-Active Enzymes (CAZymes)
2.2.4. Mobile Genetic Elements and Prophages
2.2.5. Probiotic Status of the Isolated LAB Strains
2.3. Staphylococci
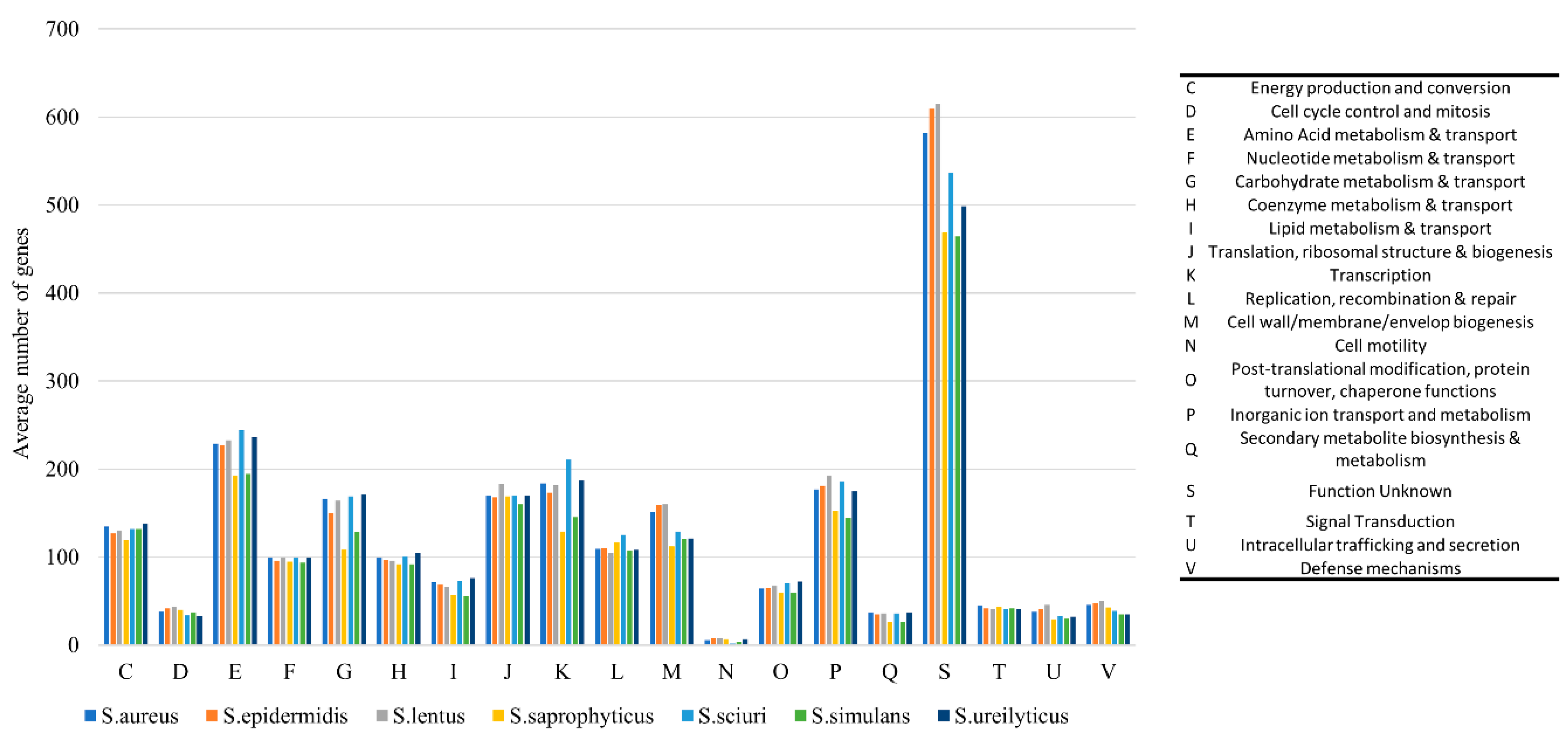
2.3.1. Phylogenetic Analysis and Comparative Genomics
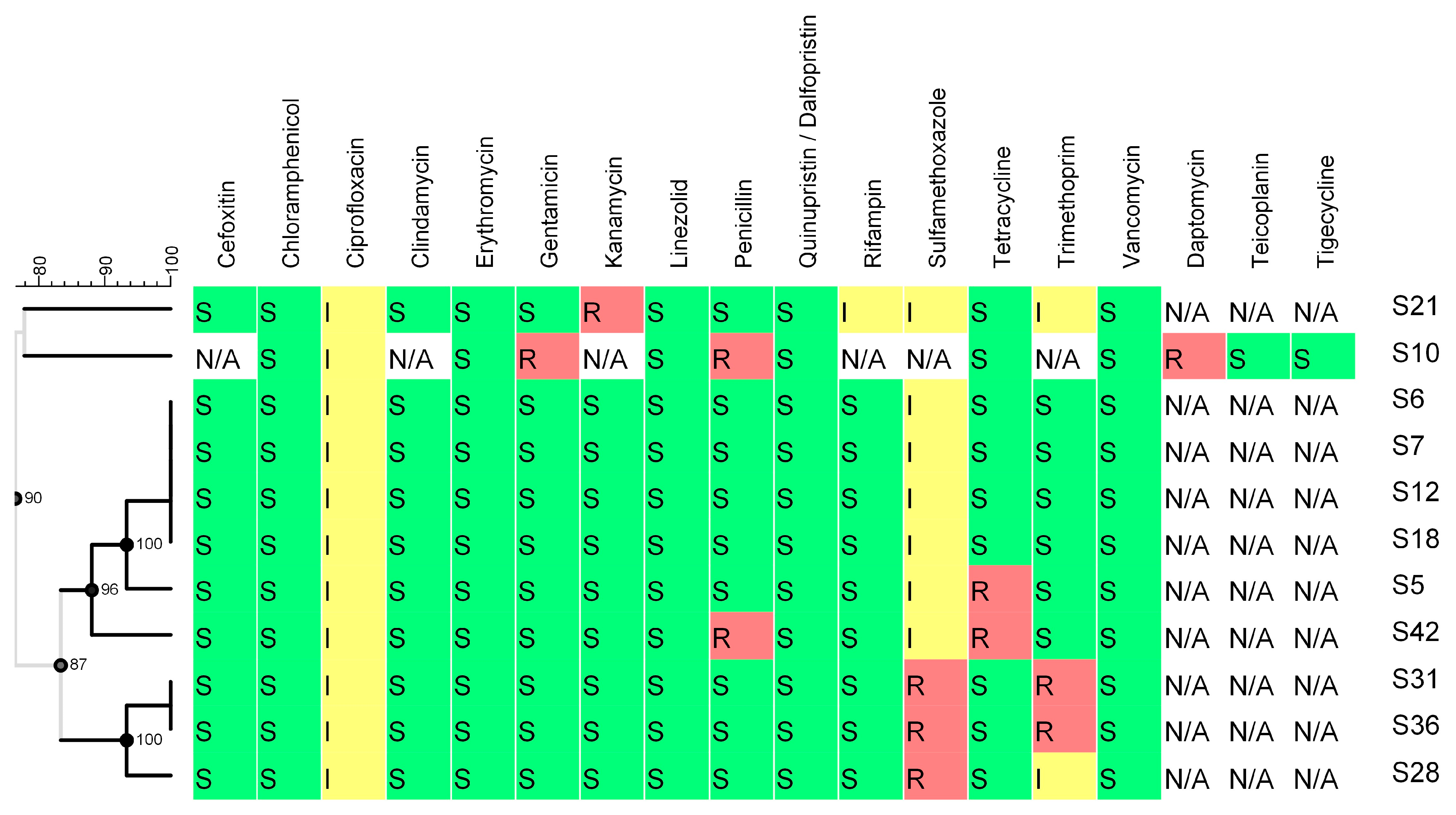
2.3.2. Virulence and Antimicrobial Resistance Determinants
2.3.3. Bacteriocins (Auto-Inducing Peptides)
2.3.4. Mobile Genetic Elements and Prophages
3. Materials and Methods
3.1. Microbial Strains and Culture Conditions
3.2. Whole Genome Sequencing, Assembly, and Quality Control
3.3. Genotyping and Comparative Genomics
3.4. Phenotype Testing
3.4.1. Antimicrobial Resistance
3.4.2. Staphylococcal Enterotoxin Production
3.4.3. Probiotic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Romero-Velarde, E.; Delgado-Franco, D.; García-Gutiérrez, M.; Gurrola-Díaz, C.; Larrosa-Haro, A.; Montijo-Barrios, E.; Muskiet, F.A.J.; Vargas-Guerrero, B.; Geurts, J. The Importance of Lactose in the Human Diet: Outcomes of a Mexican Consensus Meeting. Nutrients 2019, 11, 2737. [Google Scholar] [CrossRef] [PubMed]
- König, H.; Unden, G.; Fröhlich, J. (Eds.) Biology of Microorganisms on Grapes, in Must and in Wine; Springer International Publishing: Cham, Switzerland, 2017; ISBN 978-3-319-60020-8. [Google Scholar]
- Ayivi, R.D.; Gyawali, R.; Krastanov, A.; Aljaloud, S.O.; Worku, M.; Tahergorabi, R.; da Silva, R.C.; Ibrahim, S.A. Lactic Acid Bacteria: Food Safety and Human Health Applications. Dairy 2020, 1, 202–232. [Google Scholar] [CrossRef]
- Zapaśnik, A.; Sokołowska, B.; Bryła, M. Role of Lactic Acid Bacteria in Food Preservation and Safety. Foods 2022, 11, 1283. [Google Scholar] [CrossRef]
- Irlinger, F. Safety Assessment of Dairy Microorganisms: Coagulase-Negative Staphylococci. Int. J. Food Microbiol. 2008, 126, 302–310. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Chou, C.-C. Factors Affecting the Susceptibility of Staphylococcus aureus CCRC 12657 to Water Soluble Lactose Chitosan Derivative. Food Microbiol. 2005, 22, 29–35. [Google Scholar] [CrossRef]
- Vernozy-Rozand, C.; Mazuy, C.; Meugnier, H.; Bes, M.; Lasne, Y.; Fiedler, F.; Etienne, J.; Freney, J. Staphylococcus Fleurettii Sp. Nov., Isolated from Goat’s Milk Cheeses. Int. J. Syst. Evol. Microbiol. 2000, 50, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Apostolakos, I.; Paramithiotis, S.; Mataragas, M. Comparative Genomic Analysis Reveals the Functional Traits and Safety Status of Lactic Acid Bacteria Retrieved from Artisanal Cheeses and Raw Sheep Milk. Foods 2023, 12, 599. [Google Scholar] [CrossRef]
- Apostolakos, I.; Paramithiotis, S.; Mataragas, M. Functional and Safety Characterization of Weissella Paramesenteroides Strains Isolated from Dairy Products through Whole-Genome Sequencing and Comparative Genomics. Dairy 2022, 3, 799–813. [Google Scholar] [CrossRef]
- Apostolakos, I.; Tsigkrimani, M.; Paramithiotis, S.; Mataragas, M. Whole-Genome Sequencing and Comparative Genomic Analysis of Enterococcus Spp. Isolated from Dairy Products: Genomic Diversity, Functional Characteristics, and Pathogenic Potential. Appl. Sci. 2022, 12, 9620. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. ntroducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2019, 51, D678–D689. [Google Scholar] [CrossRef]
- Wels, M.; Siezen, R.; van Hijum, S.; Kelly, W.J.; Bachmann, H. Comparative Genome Analysis of Lactococcus Lactis Indicates Niche Adaptation and Resolves Genotype/Phenotype Disparity. Front. Microbiol. 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Lubelski, J.; de Jong, A.; van Merkerk, R.; Agustiandari, H.; Kuipers, O.P.; Kok, J.; Driessen, A.J.M. LmrCD Is a Major Multidrug Resistance Transporter in Lactococcus Lactis. Mol. Microbiol. 2006, 61, 771–781. [Google Scholar] [CrossRef]
- Whitford, M.F.; McPherson, M.A.; Forster, R.J.; Teather, R.M. Identification of Bacteriocin-like Inhibitors from Rumen Streptococcus Spp. and Isolation and Characterization of Bovicin 255. Appl. Environ. Microbiol. 2001, 67, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Pan, Y.; Li, B.; Ou, J.; Zhang, J.; Chen, Y.; Peng, X.; Chen, L. Molecular Cloning and Antimicrobial Activity of Enterolysin A and Helveticin J of Bacteriolysins from Metagenome of Chinese Traditional Fermented Foods. Food Control 2013, 31, 499–507. [Google Scholar] [CrossRef]
- Tulini, F.L.; Lohans, C.T.; Bordon, K.C.F.; Zheng, J.; Arantes, E.C.; Vederas, J.C.; De Martinis, E.C.P. Purification and Characterization of Antimicrobial Peptides from Fish Isolate Carnobacterium Maltaromaticum C2: Carnobacteriocin X and Carnolysins A1 and A2. Int. J. Food Microbiol. 2014, 173, 81–88. [Google Scholar] [CrossRef]
- Yıldırım, Z.; Yerlikaya, S.; Öncül, N.; Sakin, T. Inhibitory Effect of Lactococcin BZ Against Listeria Innocua and Indigenous Microbiota of Fresh Beef. Food Technol. Biotechnol. 2016, 54, 317–323. [Google Scholar] [CrossRef]
- Sheoran, P.; Tiwari, S.K. Anti-Staphylococcal Activity of Bacteriocins of Food Isolates Enterococcus Hirae LD3 and Lactobacillus Plantarum LD4 in Pasteurized Milk. 3 Biotech 2019, 9, 8. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef]
- Coombs, J.; Brenchley, J.E. Characterization of Two New Glycosyl Hydrolases from the Lactic Acid Bacterium Carnobacterium Piscicola Strain BA. Appl. Environ. Microbiol. 2001, 67, 5094–5099. [Google Scholar] [CrossRef]
- Sidar, A.; Albuquerque, E.D.; Voshol, G.P.; Ram, A.F.J.; Vijgenboom, E.; Punt, P.J. Carbohydrate Binding Modules: Diversity of Domain Architecture in Amylases and Cellulases From Filamentous Microorganisms. Front. Bioeng. Biotechnol. 2020, 8, 871. [Google Scholar] [CrossRef]
- Nicoloff, H.; Bringel, F. IS Lpl1 Is a Functional IS 30 -Related Insertion Element in Lactobacillus Plantarum That Is Also Found in Other Lactic Acid Bacteria. Appl. Environ. Microbiol. 2003, 69, 6032–6040. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Durmaz, E.; Higgins, D.L.; Klaenhammer, T.R. Molecular Characterization of a Second Abortive Phage Resistance Gene Present in Lactococcus Lactis Subsp. Lactis ME2. J. Bacteriol. 1992, 174, 7463–7469. [Google Scholar] [CrossRef][Green Version]
- Martinovic, A.; Cabal, A.; Nisic, A.; Sucher, J.; Stöger, A.; Allerberger, F.; Ruppitsch, W. Genome Sequences of Lactococcus Garvieae and Lactococcus Petauri Strains Isolated from Traditional Montenegrin Brine Cheeses. Microbiol. Resour. Announc. 2021, 10, e0054621. [Google Scholar] [CrossRef]
- Fayad, N.; Kallassy Awad, M.; Mahillon, J. IS982 and Kin: New Insights into an Old IS Family. Mob. DNA 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Eraclio, G.; Ricci, G.; Fortina, M.G. Insertion Sequence Elements in Lactococcus Garvieae. Gene 2015, 555, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Moineau, S. Bacteriophages of Lactic Acid Bacteria and Their Impact on Milk Fermentations. Microb. Cell Fact. 2011, 10, S20. [Google Scholar] [CrossRef]
- Aucouturier, A.; Chain, F.; Langella, P.; Bidnenko, E. Characterization of a Prophage-Free Derivative Strain of Lactococcus Lactis Ssp. Lactis IL1403 Reveals the Importance of Prophages for Phenotypic Plasticity of the Host. Front. Microbiol. 2018, 9, 2032. [Google Scholar] [CrossRef]
- Riaz Rajoka, M.S.; Mehwish, H.M.; Siddiq, M.; Haobin, Z.; Zhu, J.; Yan, L.; Shao, D.; Xu, X.; Shi, J. Identification, Characterization, and Probiotic Potential of Lactobacillus Rhamnosus Isolated from Human Milk. LWT 2017, 84, 271–280. [Google Scholar] [CrossRef]
- De Buck, J.; Ha, V.; Naushad, S.; Nobrega, D.B.; Luby, C.; Middleton, J.R.; De Vliegher, S.; Barkema, H.W. Non-Aureus Staphylococci and Bovine Udder Health: Current Understanding and Knowledge Gaps. Front. Vet. Sci. 2021, 8, 658031. [Google Scholar] [CrossRef]
- Supré, K.; Haesebrouck, F.; Zadoks, R.N.; Vaneechoutte, M.; Piepers, S.; De Vliegher, S. Some Coagulase-Negative Staphylococcus Species Affect Udder Health More than Others. J. Dairy Sci. 2011, 94, 2329–2340. [Google Scholar] [CrossRef]
- Jian, Y.; Zhao, L.; Zhao, N.; Lv, H.-Y.; Liu, Y.; He, L.; Liu, Q.; Li, M. Increasing Prevalence of Hypervirulent ST5 Methicillin Susceptible Staphylococcus aureus Subtype Poses a Serious Clinical Threat. Emerg. Microbes Infect. 2021, 10, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Sobkowiak, B.; Parreira, R.; Edgeworth, J.D.; Viveiros, M.; Clark, T.G.; Couto, I. Genetic Diversity of NorA, Coding for a Main Efflux Pump of Staphylococcus aureus. Front. Genet. 2019, 9, 710. [Google Scholar] [CrossRef]
- Kwak, Y.G.; Truong-Bolduc, Q.C.; Bin Kim, H.; Song, K.-H.; Kim, E.S.; Hooper, D.C. Association of NorB Overexpression and Fluoroquinolone Resistance in Clinical Isolates of Staphylococcus aureus from Korea. J. Antimicrob. Chemother. 2013, 68, 2766–2772. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Aras, R.; Hooper, D.C. Expression of the Multidrug Resistance Transporter NorA from Staphylococcus aureus Is Modified by a Two-Component Regulatory System. J. Bacteriol. 2000, 182, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Truong-Bolduc, Q.C.; Dunman, P.M.; Strahilevitz, J.; Projan, S.J.; Hooper, D.C. MgrA Is a Multiple Regulator of Two New Efflux Pumps in Staphylococcus aureus. J. Bacteriol. 2005, 187, 2395–2405. [Google Scholar] [CrossRef]
- Boundy, S.; Safo, M.K.; Wang, L.; Musayev, F.N.; O’Farrell, H.C.; Rife, J.P.; Archer, G.L. Characterization of the Staphylococcus aureus RRNA Methyltransferase Encoded by OrfX, the Gene Containing the Staphylococcal Chromosome Cassette Mec (SCCmec) Insertion Site. J. Biol. Chem. 2013, 288, 132–140. [Google Scholar] [CrossRef]
- Apostolakos, I.; Mughini-Gras, L.; Fasolato, L.; Piccirillo, A. Impact of Selective and Non-Selective Media on Prevalence and Genetic Makeup of ESBL/PAmpC-Producing Escherichia Coli in the Broiler Production Pyramid. Vet. Microbiol. 2020, 240, 108536. [Google Scholar] [CrossRef]
- Luong, T.T.; Lee, C.Y. Overproduction of Type 8 Capsular Polysaccharide Augments Staphylococcus aureus Virulence. Infect. Immun. 2002, 70, 3389–3395. [Google Scholar] [CrossRef]
- Schmidt, T.; Kock, M.M.; Ehlers, M.M. Molecular Characterization of Staphylococcus aureus Isolated from Bovine Mastitis and Close Human Contacts in South African Dairy Herds: Genetic Diversity and Inter-Species Host Transmission. Front. Microbiol. 2017, 8, 511. [Google Scholar] [CrossRef]
- Xiao, M.; Zhao, R.; Zhang, Q.; Fan, X.; O’Sullivan, M.V.N.; Li, D.-F.; Wang, X.-Y.; Wu, H.-L.; Kong, F.; Xu, Y.-C. Genotypic Diversity of Staphylococcus aureus α-Hemolysin Gene (Hla) and Its Association with Clonal Background: Implications for Vaccine Development. PLoS ONE 2016, 11, e0149112. [Google Scholar] [CrossRef]
- Spaan, A.N.; Vrieling, M.; Wallet, P.; Badiou, C.; Reyes-Robles, T.; Ohneck, E.A.; Benito, Y.; de Haas, C.J.C.; Day, C.J.; Jennings, M.P.; et al. The Staphylococcal Toxins γ-Haemolysin AB and CB Differentially Target Phagocytes by Employing Specific Chemokine Receptors. Nat. Commun. 2014, 5, 5438. [Google Scholar] [CrossRef] [PubMed]
- Karimzadeh, R.; Ghassab, R.K. Identification of Nuc Nuclease and Sea Enterotoxin Genes in Staphylococcus aureus Isolates from Nasal Mucosa of Burn Hospital Staff: A Cross-Sectional Study. New Microbes New Infect. 2022, 47, 100992. [Google Scholar] [CrossRef]
- Tam, K.; Torres, V.J. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Speziale, P.; Pietrocola, G.; Rindi, S.; Provenzano, M.; Provenza, G.; Di Poto, A.; Visai, L.; Arciola, C.R. Structural and Functional Role of Staphylococcus aureus Surface Components Recognizing Adhesive Matrix Molecules of the Host. Future Microbiol. 2009, 4, 1337–1352. [Google Scholar] [CrossRef]
- O’Gara, J.P. Ica and beyond: Biofilm Mechanisms and Regulation in Staphylococcus Epidermidis and Staphylococcus aureus. FEMS Microbiol. Lett. 2007, 270, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, J.K.; Blackwell, H.E. Simplified Autoinducing Peptide Mimetics with Single-Nanomolar Activity Against the Staphylococcus aureus AgrC Quorum Sensing Receptor. ACS Infect. Dis. 2019, 5, 484–492. [Google Scholar] [CrossRef]
- Kim, M.K.; Zhao, A.; Wang, A.; Brown, Z.Z.; Muir, T.W.; Stone, H.A.; Bassler, B.L. Surface-Attached Molecules Control Staphylococcus aureus Quorum Sensing and Biofilm Development. Nat. Microbiol. 2017, 2, 17080. [Google Scholar] [CrossRef] [PubMed]
- Karki, A.B.; Neyaz, L.; Fakhr, M.K. Comparative Genomics of Plasmid-Bearing Staphylococcus aureus Strains Isolated From Various Retail Meats. Front. Microbiol. 2020, 11, 574923. [Google Scholar] [CrossRef]
- Malachowa, N.; DeLeo, F.R. Mobile Genetic Elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef]
- Raue, S.; Fan, S.-H.; Rosenstein, R.; Zabel, S.; Luqman, A.; Nieselt, K.; Götz, F. The Genome of Staphylococcus Epidermidis O47. Front. Microbiol. 2020, 11, 2061. [Google Scholar] [CrossRef]
- Schouls, L.M.; Veldman, K.; Brouwer, M.S.M.; Dierikx, C.; Witteveen, S.; van Santen-Verheuvel, M.; Hendrickx, A.P.A.; Landman, F.; Hengeveld, P.; Wullings, B.; et al. Cfr and FexA Genes in Methicillin-Resistant Staphylococcus aureus from Humans and Livestock in the Netherlands. Commun. Med. 2022, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Dini, M.; Shokoohizadeh, L.; Jalilian, F.A.; Moradi, A.; Arabestani, M.R. Genotyping and Characterization of Prophage Patterns in Clinical Isolates of Staphylococcus aureus. BMC Res. Notes 2019, 12, 669. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.-S.; Seong, W.-J.; Kim, D.; Kim, E.-K.; Kim, N.-H.; Lee, C.-Y.; Kim, J.-H.; Kwon, H.-J. Molecular Prophage Typing of Staphylococcus aureus Isolates from Bovine Mastitis. J. Vet. Sci. 2018, 19, 771–781. [Google Scholar] [CrossRef]
- Syrokou, M.K.; Themeli, C.; Paramithiotis, S.; Mataragas, M.; Bosnea, L.; Argyri, A.A.; Chorianopoulos, N.G.; Skandamis, P.N.; Drosinos, E.H. Microbial Ecology of Greek Wheat Sourdoughs, Identified by a Culture-Dependent and a Culture-Independent Approach. Foods 2020, 9, 1603. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 January 2023).
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing Bacterial Genome Assemblies with Multiplex MinION Sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding Data and Analysis Capabilities. Nucleic Acids Res. 2019, 48, D606–D612. [Google Scholar] [CrossRef]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A Multi-Draft Based Scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Lu, J.; Salzberg, S.L. SkewIT: The Skew Index Test for Large-Scale GC Skew Analysis of Bacterial Genomes. PLoS Comput. Biol. 2020, 16, e1008439. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: An Improved Algorithm and Software for Calculating Average Nucleotide Identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for Finding Prophage in Bacterial Genomes. Brief. Bioinform. 2019, 20, 1560–1567. [Google Scholar] [CrossRef]
- Seemann, T. Abricate, Github 2020. Available online: https://github.com/tseemann/abricate (accessed on 27 June 2023).
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis—10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella Enterica Using a Newly Developed Web Tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. DbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Bartels, M.D.; Petersen, A.; Worning, P.; Nielsen, J.B.; Larner-Svensson, H.; Johansen, H.K.; Andersen, L.P.; Jarløv, J.O.; Boye, K.; Larsen, A.R.; et al. Comparing Whole-Genome Sequencing with Sanger Sequencing for Spa Typing of Methicillin-Resistant Staphylococcus aureus. J. Clin. Microbiol. 2014, 52, 4305–4308. [Google Scholar] [CrossRef] [PubMed]
- International Working Group on the Classification of Staphylococcal Cassette Chromosome Elements (IWG-SCC). Classification of Staphylococcal Cassette Chromosome Mec (SCCmec): Guidelines for Reporting Novel SCCmec Elements. Antimicrob. Agents Chemother. 2009, 53, 4961–4967. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, Y.; Huang, L. ImageGP: An Easy-to-use Data Visualization Web Server for Scientific Researchers. iMeta 2022, 1, e5. [Google Scholar] [CrossRef]
- Huang, L.; Goda, H.A.; Abdel-Hamid, M.; Renye, J.A., Jr.; Yang, P.; Huang, Z.; Zeng, Q.-K.; Li, L. Partial Characterization of Probiotic Lactic Acid Bacteria Isolated from Chinese Dairy Products. Int. J. Food Prop. 2021, 24, 446–456. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain ID | Species | Genome Size (Mbp) | GC Content (%) | No. of Scaffolds | N50 (Mbp) | No. of CDSs |
|---|---|---|---|---|---|---|
| S87 | Limosilactobacillus fermentum | 1.86 | 52.71 | 3 | 1.52 | 1765 |
| S94 | Limosilactobacillus fermentum | 1.83 | 52.63 | 2 | 1.57 | 1746 |
| S54 | Lactobacillus delbrueckii subsp. lactis | 1.92 | 49.69 | 3 | 0.60 | 1794 |
| S70 | Lactobacillus delbrueckii subsp. lactis | 1.91 | 49.71 | 2 | 1.91 | 1791 |
| S88 | Lactobacillus delbrueckii subsp. lactis | 1.85 | 49.94 | 2 | 1.85 | 1725 |
| S96 | Lactobacillus delbrueckii subsp. lactis | 1.91 | 49.7 | 4 | 0.66 | 1790 |
| S43 | Lactococcus lactis | 2.27 | 35.06 | 1 | 2.27 | 2052 |
| S44 | Lactococcus lactis | 2.27 | 35.06 | 1 | 2.27 | 2051 |
| S49 | Lactococcus lactis | 2.25 | 35.12 | 1 | 2.25 | 2053 |
| S58 | Lactococcus lactis | 2.25 | 35.12 | 1 | 2.25 | 2053 |
| S71 | Lactococcus lactis | 2.35 | 35 | 2 | 2.27 | 2103 |
| S73 | Lactococcus lactis | 2.45 | 34.99 | 3 | 1.28 | 2232 |
| S76 | Lactococcus lactis | 2.32 | 34.93 | 2 | 2.20 | 2098 |
| S9 | Lactococcus lactis | 2.27 | 35.06 | 1 | 2.27 | 2052 |
| S27 | Lactococcus petauri | 1.96 | 38.19 | 2 | 1.77 | 1864 |
| S46 | Lactococcus petauri | 1.95 | 38.19 | 2 | 1.76 | 1848 |
| S77 | Lactococcus petauri | 1.95 | 38.23 | 2 | 1.76 | 1839 |
| S78 | Lactococcus petauri | 1.96 | 38.2 | 2 | 1.77 | 1856 |
| S82 | Lactococcus petauri | 1.92 | 38.26 | 2 | 1.70 | 1820 |
| S84 | Lactococcus petauri | 1.96 | 38.2 | 2 | 1.77 | 1855 |
| S12 | Staphylococcus aureus | 2.75 | 32.78 | 2 | 1.50 | 2565 |
| S18 | Staphylococcus aureus | 2.75 | 32.78 | 2 | 1.39 | 2379 |
| S5 | Staphylococcus aureus | 2.80 | 32.74 | 3 | 2.76 | 2536 |
| S6 | Staphylococcus aureus | 2.75 | 32.78 | 1 | 2.75 | 2534 |
| S7 | Staphylococcus aureus | 2.75 | 32.79 | 3 | 2.64 | 2579 |
| S8 | Staphylococcus aureus | 2.87 | 32.93 | 3 | 2.42 | 2531 |
| S42 | Staphylococcus epidermidis | 2.32 | 32.05 | 1 | 2.32 | 2534 |
| S74 | Staphylococcus lentus | 2.58 | 31.79 | 48 | 0.17 | 2594 |
| S85 | Staphylococcus saprophyticus | 2.56 | 33.02 | 1 | 2.56 | 2153 |
| S10 | Staphylococcus sciuri | 2.40 | 32.41 | 2 | 2.17 | 2510 |
| S13 | Staphylococcus simulans | 2.68 | 35.89 | 7 | 2.65 | 2531 |
| S21 | Staphylococcus simulans | 2.66 | 35.87 | 11 | 0.66 | 2582 |
| S28 | Staphylococcus simulans | 2.36 | 35.96 | 1 | 2.36 | 2278 |
| S29 | Staphylococcus simulans | 2.57 | 36.03 | 10 | 0.55 | 2425 |
| S31 | Staphylococcus simulans | 2.23 | 36.12 | 1 | 2.23 | 2141 |
| S34 | Staphylococcus simulans | 2.57 | 36.05 | 2 | 1.33 | 2396 |
| S36 | Staphylococcus simulans | 2.57 | 36.05 | 1 | 2.57 | 2396 |
| S98 | Staphylococcus simulans | 2.57 | 36.04 | 1 | 2.57 | 2388 |
| S99 | Staphylococcus ureilyticus | 2.53 | 32.51 | 2 | 2.49 | 2379 |
| Function | Gene | Number of LAB Strains |
|---|---|---|
| Acid stress | atpABCDEFGH | 20/20 |
| gadB | 14/20 | |
| nhaC | 12/20 | |
| Adhesion | eno, lspA, srtA, tpiA, tuf | 20/20 |
| epsB | 6/20 | |
| mapA | 16/20 | |
| pgi | 18/20 | |
| Antioxidant | fnr | 3/20 |
| mntH, nrdH | 16/20 | |
| msrAB, trxAB | 20/20 | |
| ndh | 14/20 | |
| nox | 1/20 | |
| npr | 2/20 | |
| poxL | 8/20 | |
| tpx | 10/20 | |
| Bile tolerance | cfa | 16/20 |
| ppaC | 20/20 | |
| Cold stress | cspA | 18/20 |
| Heat stress | clpB | 14/20 |
| clpCEPX, dnaJK, grpE, hrcA, hslO, lon | 20/20 | |
| clpL, hslUV | 4/20 | |
| ctsR | 16/20 | |
| htpX | 6/20 | |
| Immunomodulation | dltABCD | 20/20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apostolakos, I.; Skarlatoudi, T.; Vatavali, K.; Giannouli, A.; Bosnea, L.; Mataragas, M. Genomic and Phenotypic Characterization of Mastitis-Causing Staphylococci and Probiotic Lactic Acid Bacteria Isolated from Raw Sheep’s Milk. Int. J. Mol. Sci. 2023, 24, 13883. https://doi.org/10.3390/ijms241813883
Apostolakos I, Skarlatoudi T, Vatavali K, Giannouli A, Bosnea L, Mataragas M. Genomic and Phenotypic Characterization of Mastitis-Causing Staphylococci and Probiotic Lactic Acid Bacteria Isolated from Raw Sheep’s Milk. International Journal of Molecular Sciences. 2023; 24(18):13883. https://doi.org/10.3390/ijms241813883
Chicago/Turabian StyleApostolakos, Ilias, Theodora Skarlatoudi, Kornilia Vatavali, Agathi Giannouli, Loulouda Bosnea, and Marios Mataragas. 2023. "Genomic and Phenotypic Characterization of Mastitis-Causing Staphylococci and Probiotic Lactic Acid Bacteria Isolated from Raw Sheep’s Milk" International Journal of Molecular Sciences 24, no. 18: 13883. https://doi.org/10.3390/ijms241813883
APA StyleApostolakos, I., Skarlatoudi, T., Vatavali, K., Giannouli, A., Bosnea, L., & Mataragas, M. (2023). Genomic and Phenotypic Characterization of Mastitis-Causing Staphylococci and Probiotic Lactic Acid Bacteria Isolated from Raw Sheep’s Milk. International Journal of Molecular Sciences, 24(18), 13883. https://doi.org/10.3390/ijms241813883