
Development of a Macrophage-Related Risk Model for Metastatic Melanoma

 ,
,
Abstract
1. Introduction
2. Results
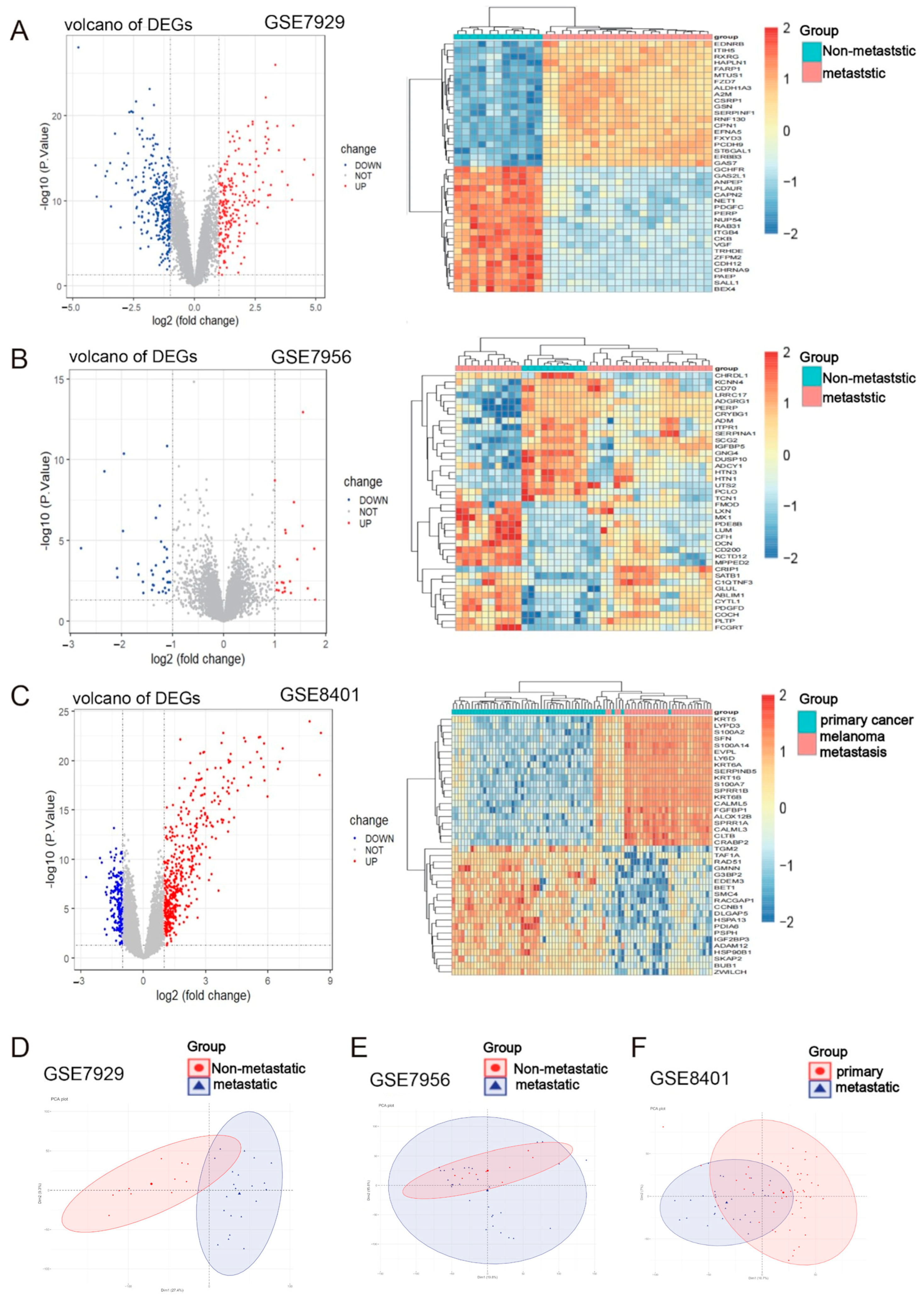
2.1. Identification of Metastasis-Associated Melanoma DEGs
2.2. The Roles Played by M1 Macrophages in Melanoma Development and Metastasis
2.3. Functional Analysis of Key Genes and Identification of MMIMs
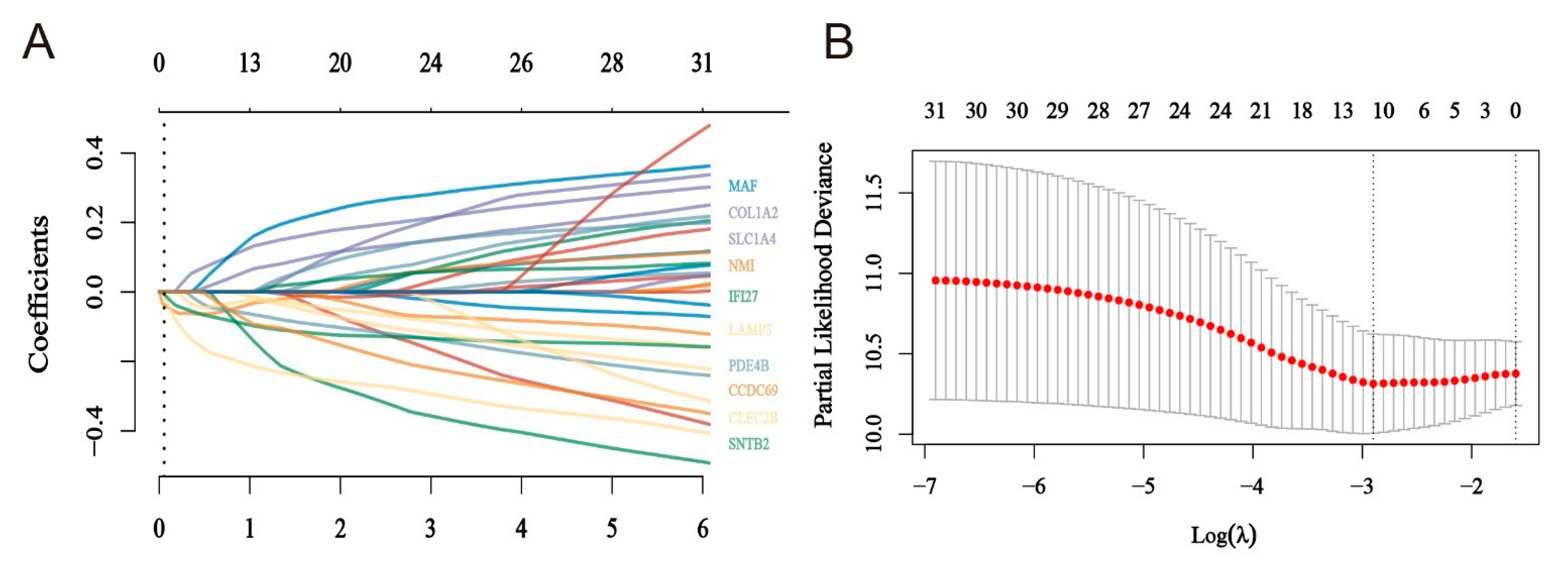
2.4. Construction of a Risk Model for Macrophage Immunomarker Molecules of MMIMs
2.5. Prognostic Models Constructed Based on MMIMs Molecules Have Better Prognostic Effect and Specificity in Melanoma Metastatic Cancer
2.6. Clinical Evaluation of the Model
2.7. Analysis of Immune Correlation in Targeted Model
2.8. The TF-Gene-miRNA Co-Regulatory Network
2.9. Protein-Drug Interactions Predict Potential Drugs
3. Discussion
4. Materials and Methods
4.1. Data Source
4.2. Sample Immunoinfiltration Analysis
4.3. Differentially Expressed Gene (DEGs) Selection and Identification of Key Immune-Related Molecules
4.4. Functional and KEGG Enrichment Analysis of DEGs
4.5. Lasso Method to Determine the Risk Model and Test
4.6. Group Comparison of Risk Models
4.7. Re-Validation of the Model
4.8. Immunocorrelation Analysis of Model
4.9. TF-Gene-miRNA Interaction Network Based on MMIMs
4.10. The Pro-Drug Interaction Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Holzel, M.; Tuting, T. Inflammation-Induced Plasticity in Melanoma Therapy and Metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef]
- Yu, R.; Jin, L.; Li, F.; Fujimoto, M.; Wei, Q.; Lin, Z.; Ren, X.; Jin, Q.; Li, H.; Meng, F.; et al. Dihydroartemisinin inhibits melanoma by regulating CTL/Treg anti-tumor immunity and STAT3-mediated apoptosis via IL-10 dependent manner. J. Dermatol. Sci. 2020, 99, 193–202. [Google Scholar] [CrossRef]
- Zhang, Q.; Jin, L.; Jin, Q.; Wei, Q.; Sun, M.; Yue, Q.; Liu, H.; Li, F.; Li, H.; Ren, X.; et al. Inhibitory Effect of Dihydroartemisinin on the Proliferation and Migration of Melanoma Cells and Experimental Lung Metastasis From Melanoma in Mice. Front. Pharmacol. 2021, 12, 727275. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The Role of CXC Chemokine Receptors 1-4 on Immune Cells in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2159. [Google Scholar] [CrossRef]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-beta/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef]
- Fan, Z.; Duan, J.; Luo, P.; Shao, L.; Chen, Q.; Tan, X.; Zhang, L.; Xu, X. SLC25A38 as a novel biomarker for metastasis and clinical outcome in uveal melanoma. Cell Death Dis. 2022, 13, 330. [Google Scholar] [CrossRef]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Zietek, M.; Matkowski, R.; Nowak, D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 2020, 21, 8359. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, W.; Wang, S.; Yang, T.; Zhang, G.; Wang, D.; Ju, R.; Lu, Y.; Wang, H.; Wang, L. Tumor microenvironment remodeling and tumor therapy based on M2-like tumor associated macrophage-targeting nano-complexes. Theranostics 2021, 11, 2892–2916. [Google Scholar] [CrossRef] [PubMed]
- Falleni, M.; Savi, F.; Tosi, D.; Agape, E.; Cerri, A.; Moneghini, L.; Bulfamante, G.P. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017, 27, 200–210. [Google Scholar] [CrossRef]
- Zhu, M.-H.; John, S.; Berg, M.; Leonard, W.J. Functional Association of Nmi with Stat5 and Stat1 in IL-2- and IFN γ-Mediated Signaling. Cell 1999, 96, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, R.A.; Mitra, A.; Xi, Y.; Ju, J.; Scammell, J.; Shevde, L.A.; Samant, R.S. Nmi (N-Myc interactor) inhibits Wnt/beta-catenin signaling and retards tumor growth. Int. J. Cancer 2009, 125, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zou, K.; Feng, X.; Chen, M.; Li, C.; Tang, R.; Xuan, Y.; Luo, M.; Chen, W.; Qiu, H.; et al. Downregulation of NMI promotes tumor growth and predicts poor prognosis in human lung adenocarcinomas. Mol. Cancer 2017, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Murata, T.; Shimizu, K.; Morita, H.; Inui, M.; Tagawa, T. Phosphodiesterase 4 regulates the migration of B16-F10 melanoma cells. Exp. Ther. Med. 2012, 4, 205–210. [Google Scholar] [CrossRef]
- Narita, M.; Murata, T.; Shimizu, K.; Nakagawa, T.; Sugiyama, T.; Inui, M.; Hiramoto, K.; Tagawa, T. A role for cyclic nucleotide phosphodiesterase 4 in regulation of the growth of human malignant melanoma cells. Oncol. Rep. 2007, 17, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Marquette, A.; Andre, J.; Bagot, M.; Bensussan, A.; Dumaz, N. ERK and PDE4 cooperate to induce RAF isoform switching in melanoma. Nat. Struct. Mol. Biol. 2011, 18, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Inoue, K. Platelets and cancer-associated thrombosis: Focusing on the platelet activation receptor CLEC-2 and podoplanin. Blood 2019, 134, 1912–1918. [Google Scholar] [CrossRef]
- Shirai, T.; Inoue, O.; Tamura, S.; Tsukiji, N.; Sasaki, T.; Endo, H.; Satoh, K.; Osada, M.; Sato-Uchida, H.; Fujii, H.; et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J. Thromb. Haemost. 2017, 15, 513–525. [Google Scholar] [CrossRef]
- Xu, M.; Wang, X.; Pan, Y.; Zhao, X.; Yan, B.; Ruan, C.; Xia, L.; Zhao, Y. Blocking podoplanin suppresses growth and pulmonary metastasis of human malignant melanoma. BMC Cancer 2019, 19, 599. [Google Scholar] [CrossRef]
- Cheriyath, V.; Leaman, D.W.; Borden, E.C. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J. Interferon Cytokine Res. 2011, 31, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Mallardo, D.; Simeone, E.; Vanella, V.; Vitale, M.G.; Palla, M.; Scarpato, L.; Paone, M.; De Cristofaro, T.; Borzillo, V.; Cortellini, A.; et al. Concomitant medication of cetirizine in advanced melanoma could enhance anti-PD-1 efficacy by promoting M1 macrophages polarization. J. Transl. Med. 2022, 20, 436. [Google Scholar] [CrossRef]
- Fujiwara, S.; Nagai, H.; Jimbo, H.; Jimbo, N.; Tanaka, T.; Inoie, M.; Nishigori, C. Gene Expression and Methylation Analysis in Melanomas and Melanocytes From the Same Patient: Loss of NPM2 Expression Is a Potential Immunohistochemical Marker for Melanoma. Front. Oncol. 2018, 8, 675. [Google Scholar] [CrossRef] [PubMed]
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. 2018, 74, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Henin, C.; Maurizio, J.; Imbratta, C.; Bourdely, P.; Buferne, M.; Baitsch, L.; Vanhille, L.; Sieweke, M.H.; Speiser, D.E.; et al. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J. 2015, 34, 2042–2058. [Google Scholar] [CrossRef] [PubMed]
- Verdeil, G. MAF drives CD8+ T-cell exhaustion. Oncoimmunology 2016, 5, e1082707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Guo, L.; Zhou, Z.; An, R.; Wang, J. Identification and validation of a prognostic model for melanoma patients with 9 ferroptosis-related gene signature. BMC Genom. 2022, 23, 245. [Google Scholar] [CrossRef]
- Morales, L.; Simpson, D.; Ferguson, R.; Cadley, J.; Esteva, E.; Monson, K.; Chat, V.; Martinez, C.; Weber, J.; Osman, I.; et al. Tumor immunogenomic signatures improve a prognostic model of melanoma survival. J. Transl. Med. 2021, 19, 78. [Google Scholar] [CrossRef]
- Wan, Q.; Tang, J.; Lu, J.; Jin, L.; Su, Y.; Wang, S.; Cheng, Y.; Liu, Y.; Li, C.; Wang, Z. Six-gene-based prognostic model predicts overall survival in patients with uveal melanoma. Cancer Biomark. 2020, 27, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Botti, G.; Cerrone, M.; Scognamiglio, G.; Anniciello, A.; Ascierto, P.A.; Cantile, M. Microenvironment and tumor progression of melanoma: New therapeutic prospectives. J. Immunotoxicol. 2013, 10, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Boecke, A.; Carstens, A.C.; Neacsu, C.D.; Baschuk, N.; Haubert, D.; Kashkar, H.; Utermohlen, O.; Pongratz, C.; Kronke, M. TNF-receptor-1 adaptor protein FAN mediates TNF-induced B16 melanoma motility and invasion. Br. J. Cancer 2013, 109, 422–432. [Google Scholar] [CrossRef][Green Version]
- Zaidi, M.R.; Davis, S.; Noonan, F.P.; Graff-Cherry, C.; Hawley, T.S.; Walker, R.L.; Feigenbaum, L.; Fuchs, E.; Lyakh, L.; Young, H.A.; et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature 2011, 469, 548–553. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.G.; Gelbard, A.; Davies, M.A.; Sano, D.; Ekmekcioglu, S.; Kwon, J.; Hailemichael, Y.; Jayaraman, P.; Myers, J.N.; Grimm, E.A.; et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin. Cancer Res. 2010, 16, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Annamalai, R.T.; Turner, P.A.; Carson, W.F.T.; Levi, B.; Kunkel, S.; Stegemann, J.P. Harnessing macrophage-mediated degradation of gelatin microspheres for spatiotemporal control of BMP2 release. Biomaterials 2018, 161, 216–227. [Google Scholar] [CrossRef]
- Hao, S.; Meng, J.; Zhang, Y.; Liu, J.; Nie, X.; Wu, F.; Yang, Y.; Wang, C.; Gu, N.; Xu, H. Macrophage phenotypic mechanomodulation of enhancing bone regeneration by superparamagnetic scaffold upon magnetization. Biomaterials 2017, 140, 16–25. [Google Scholar] [CrossRef]
- Bruns, H.; Buttner, M.; Fabri, M.; Mougiakakos, D.; Bittenbring, J.T.; Hoffmann, M.H.; Beier, F.; Pasemann, S.; Jitschin, R.; Hofmann, A.D.; et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci. Transl. Med. 2015, 7, 282ra247. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source Database | Data Type | Data Number Information | Data Grouping Information |
|---|---|---|---|
| GEO | Transcriptome information | GSE7929 | Poorly metastatic A375 parental cell line (11) and highly metastatic A375 melanoma metastases (21) |
| GEO | Transcriptome information | GSE7956 | Poorly metastatic A375 parental cell line (10) and highly metastatic A375 melanoma metastases (29) |
| GEO | Transcriptome information | GSE8401 | Clinical primary melanoma (31) and melanoma metastasis (52) |
| GEO | Transcriptome information | GSE114445 | Normal skin (5) and primary cutaneous melanoma (16) |
| TCGA | Transcriptome information | SKCM | 471 patients with melanoma, and 167 of those patients had metastases |
| MSigDB | Gene Set Collection | GSE11864_1087_200_DN and GSE5099_3808_200_UP | 349 differentially expressed genes in M1- and M2-type macrophages |
| Id | Label (Genes) | Degree | Betweenness |
|---|---|---|---|
| 6645 | NMI | 104 | 30,695.18 |
| 4094 | COL1A2 | 56 | 15,718.36 |
| 26112 | SLC1A4 | 56 | 15,309.81 |
| 6509 | LAMP3 | 51 | 17,738.41 |
| 1278 | MAF | 30 | 9543.11 |
| 9111 | PDE4B | 27 | 9550.66 |
| 5142 | CLEC2B | 13 | 4370.97 |
| 27074 | IFI27 | 8 | 1965.45 |
| 3429 | CCDC69 | 6 | 1620 |
| 9976 | SNTB2 | 4 | 545.04 |
| Id | Label (miRNAs) | Degree | Betweenness |
|---|---|---|---|
| MIMAT0000083 | hsa-mir-26b-5p | 4 | 3041.04 |
| MIMAT0000081 | hsa-mir-25-3p | 2 | 6248.14 |
| MIMAT0022720 | hsa-mir-1304-3p | 2 | 2174.33 |
| MIMAT0027681 | hsa-mir-6890-3p | 2 | 2174.33 |
| MIMAT0000449 | hsa-mir-146a-5p | 2 | 1926 |
| MIMAT0000086 | hsa-mir-29a-3p | 2 | 1813.46 |
| MIMAT0000765 | hsa-mir-335-5p | 2 | 1382.67 |
| MIMAT0000422 | hsa-mir-124-3p | 2 | 1295.13 |
| MIMAT0001532 | hsa-mir-448 | 2 | 1295.13 |
| MIMAT0004597 | hsa-mir-140-3p | 2 | 1295.13 |
| Id | Label (TFs) | Degree | Betweenness |
|---|---|---|---|
| 56897 | WRNIP1 | 3 | 3779.71 |
| 171017 | ZNF384 | 2 | 7894.6 |
| 5978 | REST | 2 | 2185.42 |
| 55929 | DMAP1 | 2 | 2185.42 |
| 467 | ATF3 | 2 | 2185.42 |
| 10346 | TRIM22 | 2 | 2174.33 |
| 51385 | ZNF589 | 2 | 1553.79 |
| 121340 | SP7 | 2 | 1553.79 |
| 865 | CBFB | 2 | 1382.67 |
| 1999 | ELF3 | 2 | 1295.13 |
| ID | Molecule and Drug | Drug Trial Phase | Clinical Drug Types | Indications |
|---|---|---|---|---|
| 5142 | PDE4B | |||
| DB00131 | Adenosine monophosphate | Clinical phase 2/3 merged into clinical phase 3 | AMP-activated protein kinase agonist | Type 2 diabetes and metabolic diseases |
| DB00201 | Caffeine | Approved | ADORA2A antagonist and ADORA1 antagonist | Respiratory disease and mental fatigue |
| DB00277 | Theophylline | Approved | Adenosine family receptor antagonist | Immune system diseases and respiratory system diseases |
| DB00651 | Dyphylline | Approved | cDMP-PDE inhibitor | Asthma and chronic bronchitis |
| DB00806 | Pentoxifylline | Approved | Calcium nervous system inhibitor | Cardiovascular disease and type 1 peripheral arterial occlusion xing |
| DB00824 | Enprofylline | - | - | - |
| DB00920 | Ketotifen | Clinical phase 2 | - | Immune system diseases, allergic conjunctivitis |
| DB01088 | Iloprost | Approved | Cycloprostenol targeting drug | Respiratory diseases and cardiovascular diseases |
| DB01113 | Papaverine | Approved | cGMP-PDE inhibitor | Neurological diseases and cardiovascular diseases |
| DB01412 | Theobromine | Approved | - | Cardiovascular diseases |
| DB01427 | Amrinone | Approved | cGMP-PDE inhibitor | Cardiovascular diseases |
| DB01647 | (R)-Mesopram | Clinical phase II termination | PDE4 inhibitor | Inflammation, multiple sclerosis |
| DB01656 | Roflumilast | Approved | PDE4 inhibitor | Psoriasis, seborrheic dermatitis |
| DB01791 | Piclamilast | Clinical phase 2 termination | PDE4 inhibitor | Asthma and dermatitis |
| DB01954 | Rolipram | Clinical phase 3 termination | PDE4 inhibitor | Neurological diseases and immune diseases |
| DB01959 | 3,5-Dimethyl-1-(3-nitrophenyl)-1H-pyrazole-4-carboxylic acid ethyl ester | - | - | - |
| DB02660 | Filaminast | Clinical phase 2 termination | Phospholipase A2 inhibitor | Immune system diseases and respiratory system diseases |
| DB03349 | 8-Bromo-adenosine-5′-monophosphate | - | - | - |
| DB03606 | (S)-Rolipram | Clinical phase 1 | PDE4 inhibitor | Huntington’s chorea |
| DB03807 | 1-(2-Chlorophenyl)-3,5-dimethyl-1H-pyrazole-4-carboxylic acid ethyl ester | - | - | - |
| DB03849 | Cilomilast | Clinical phase 3 Termination | PDE4 inhibitor | Asthma and dermatitis |
| DB04149 | (R)-Rolipram | Clinical phase 1 | Positron emission tomography enhancer | Neurological diseases |
| DB04530 | S,S-(2-Hydroxyethyl)thiocysteine | - | - | - |
| DB05219 | AN2728 | Approved | PDE4 inhibitor | Atopic dermatitis, psoriasis |
| DB05266 | Ibudilast | Approved | PDE4 inhibitor | Uveal melanoma, keratoblastoma, allergic conjunctivitis |
| DB05676 | Apremilast | Approved | PDE4 inhibitor | psoriasis and novel coronavirus pneumonia |
| DB06909 | 1-Ethyl-N-(phenylmethyl)-4-(tetrahydro-2H-pyran-4-ylamino)-1H-pyrazolo[3,4-b]pyridine-5-carboxamide | - | - | - |
| DB08299 | 4-[8-(3-Nitrophenyl)-1,7-naphthyridin-6-yl]benzoic acid | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Zhang, X.; Jin, Q.; Zhang, Q.; Yue, Q.; Fujimoto, M.; Jin, G. Development of a Macrophage-Related Risk Model for Metastatic Melanoma. Int. J. Mol. Sci. 2023, 24, 13752. https://doi.org/10.3390/ijms241813752
Li Z, Zhang X, Jin Q, Zhang Q, Yue Q, Fujimoto M, Jin G. Development of a Macrophage-Related Risk Model for Metastatic Melanoma. International Journal of Molecular Sciences. 2023; 24(18):13752. https://doi.org/10.3390/ijms241813752
Chicago/Turabian StyleLi, Zhaoxiang, Xinyuan Zhang, Quanxin Jin, Qi Zhang, Qi Yue, Manabu Fujimoto, and Guihua Jin. 2023. "Development of a Macrophage-Related Risk Model for Metastatic Melanoma" International Journal of Molecular Sciences 24, no. 18: 13752. https://doi.org/10.3390/ijms241813752
APA StyleLi, Z., Zhang, X., Jin, Q., Zhang, Q., Yue, Q., Fujimoto, M., & Jin, G. (2023). Development of a Macrophage-Related Risk Model for Metastatic Melanoma. International Journal of Molecular Sciences, 24(18), 13752. https://doi.org/10.3390/ijms241813752