
Association of Phosphorylated Pyruvate Dehydrogenase with Pyruvate Kinase M2 Promotes PKM2 Stability in Response to Insulin

 , ,
, ,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
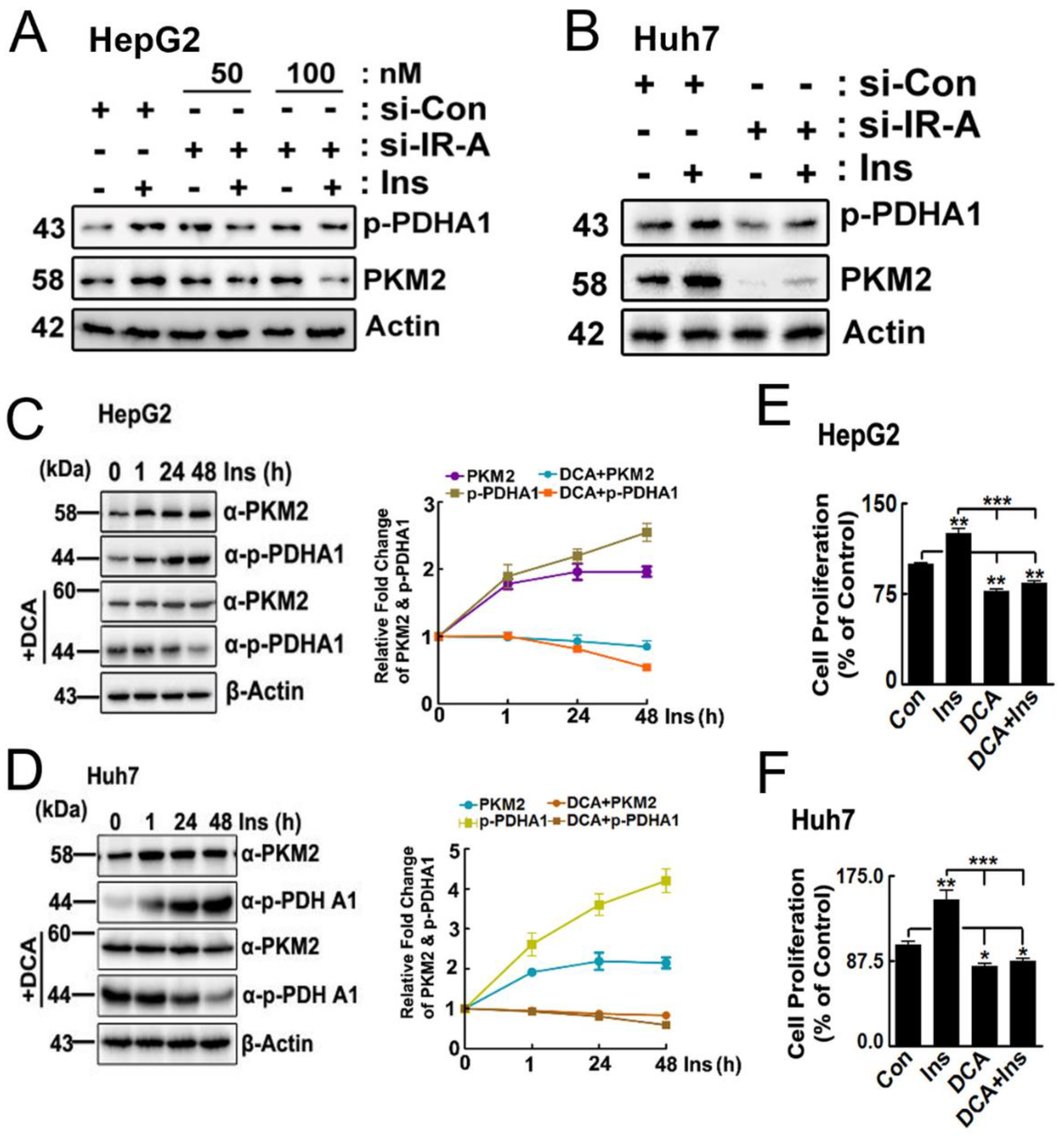
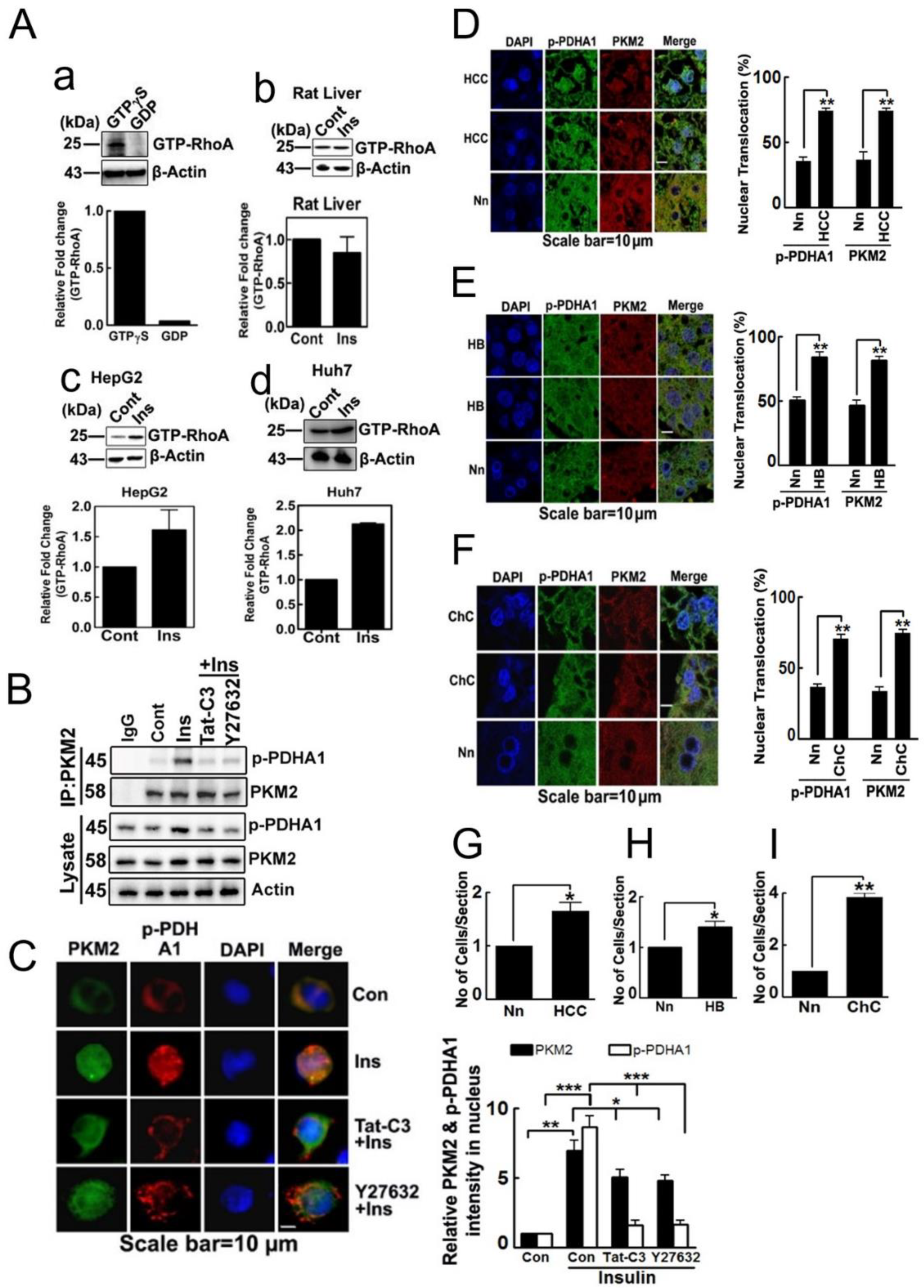
2.1. p-PDHA1 Interacts with PKM2
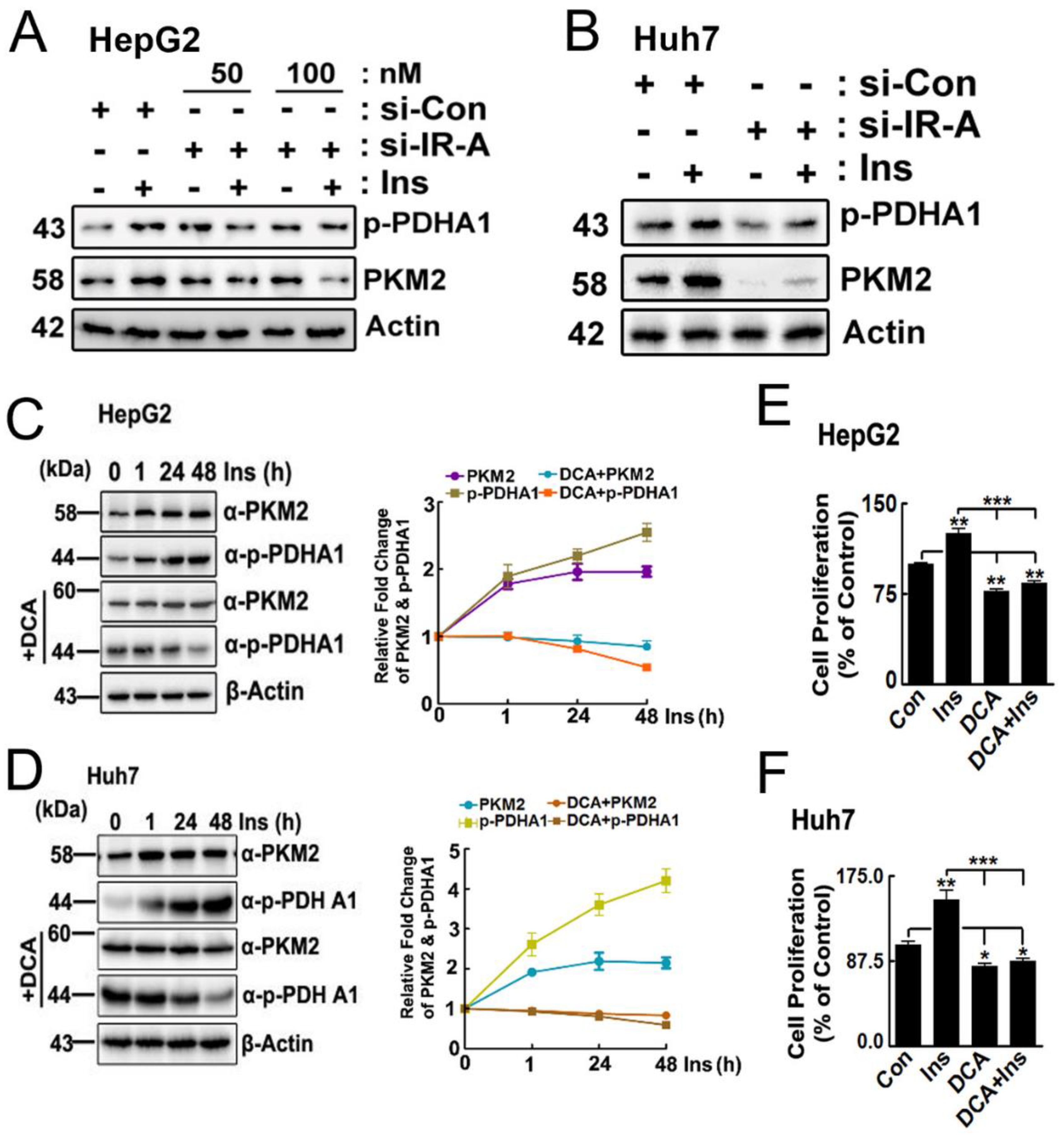
2.2. DCA Reduces PKM2 Levels
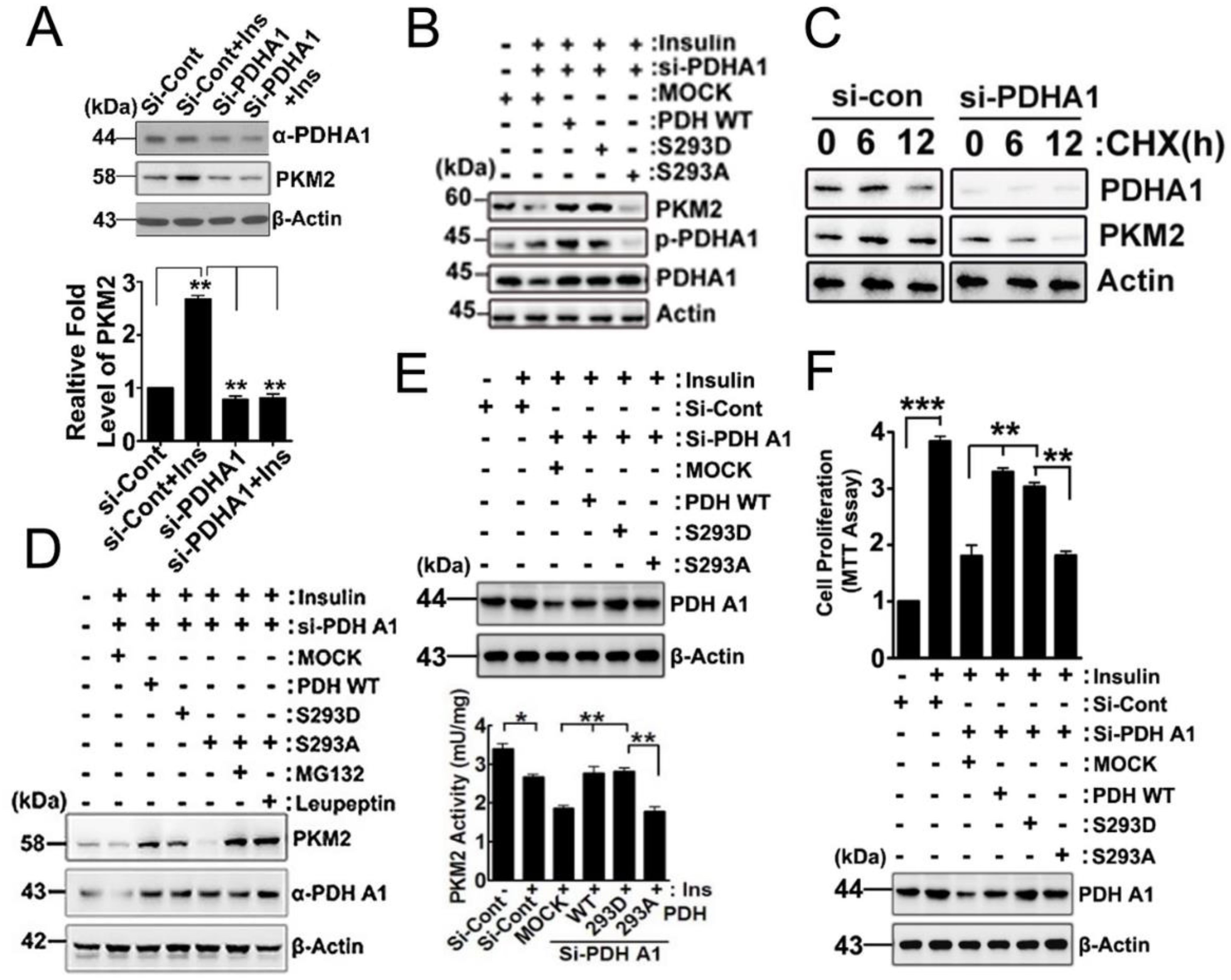
2.3. Effects of p-PDHA1 on PKM2
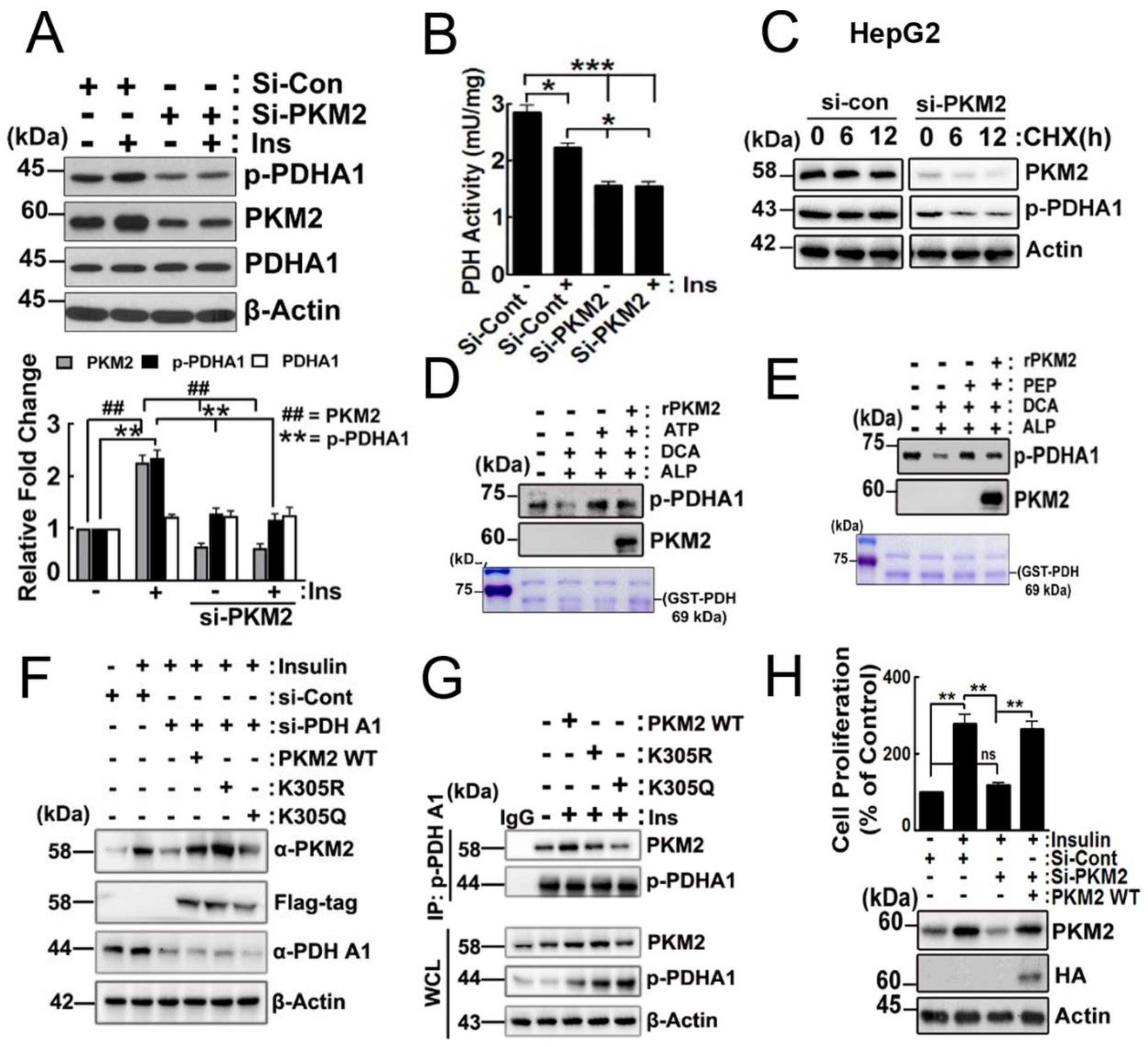
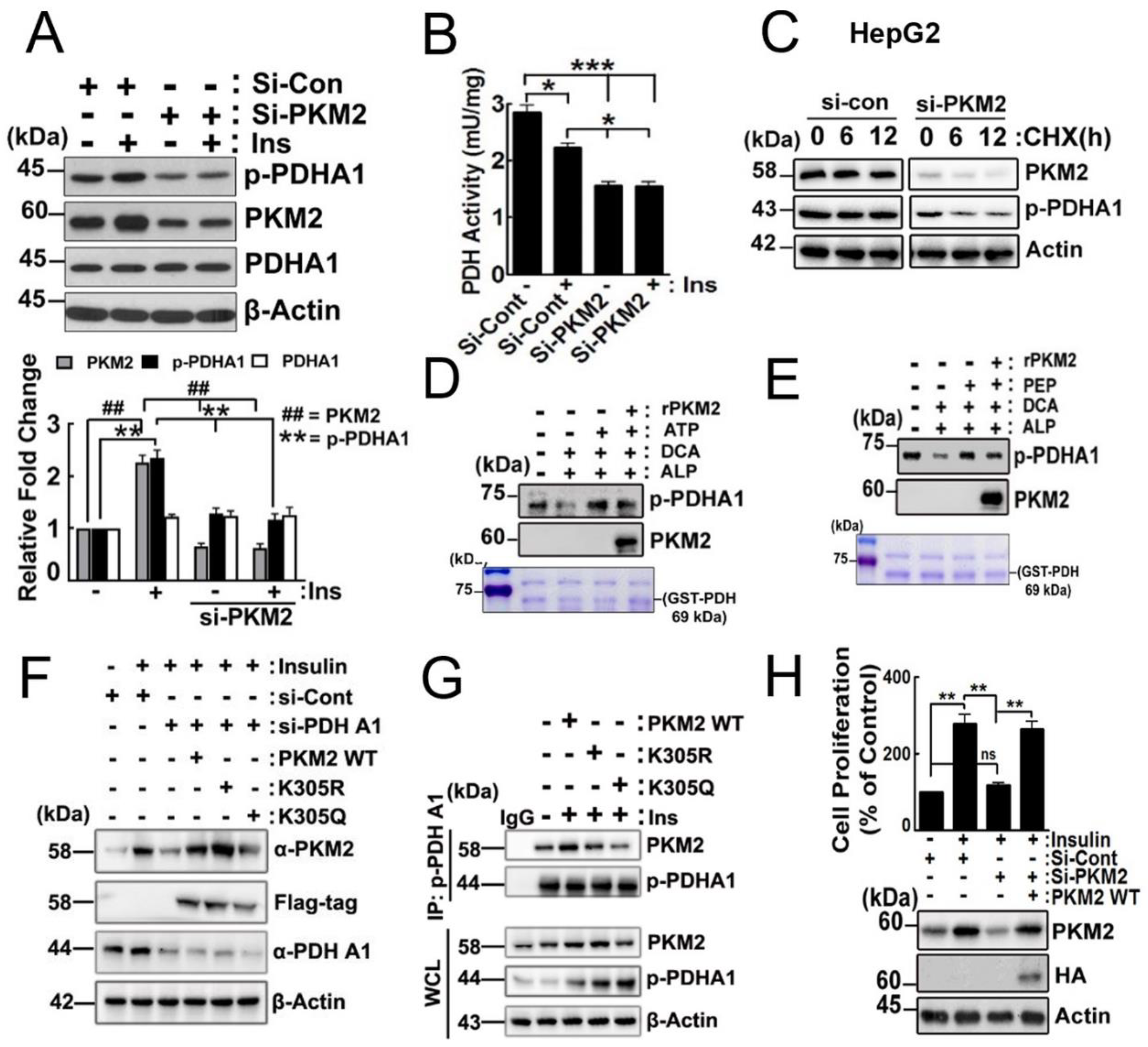
2.4. Effects of PKM2 on p-PDHA1
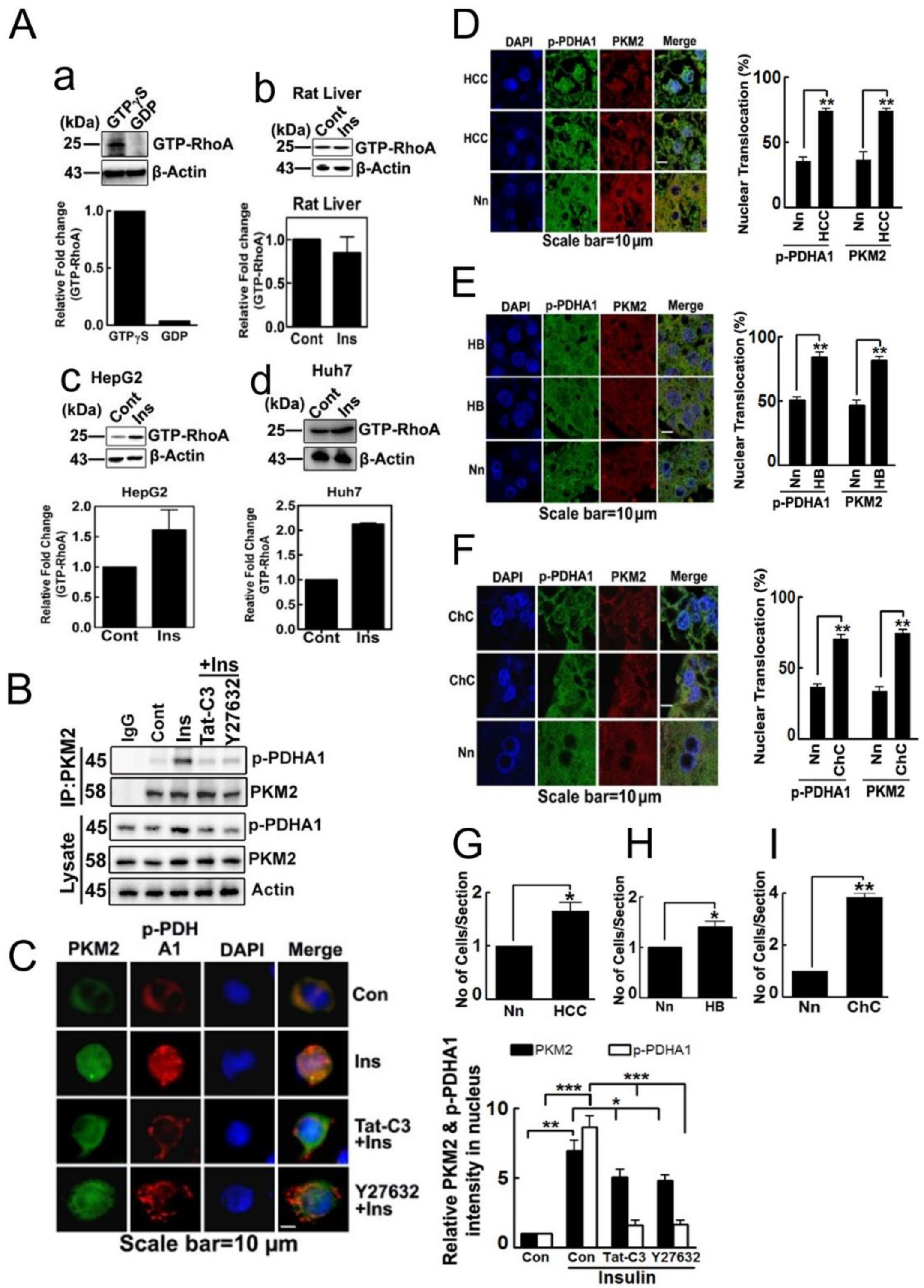
2.5. Nuclear Localization of p-PDHA1 and PKM2 in Human Hepatocellular Carcinoma Cells and Tissues
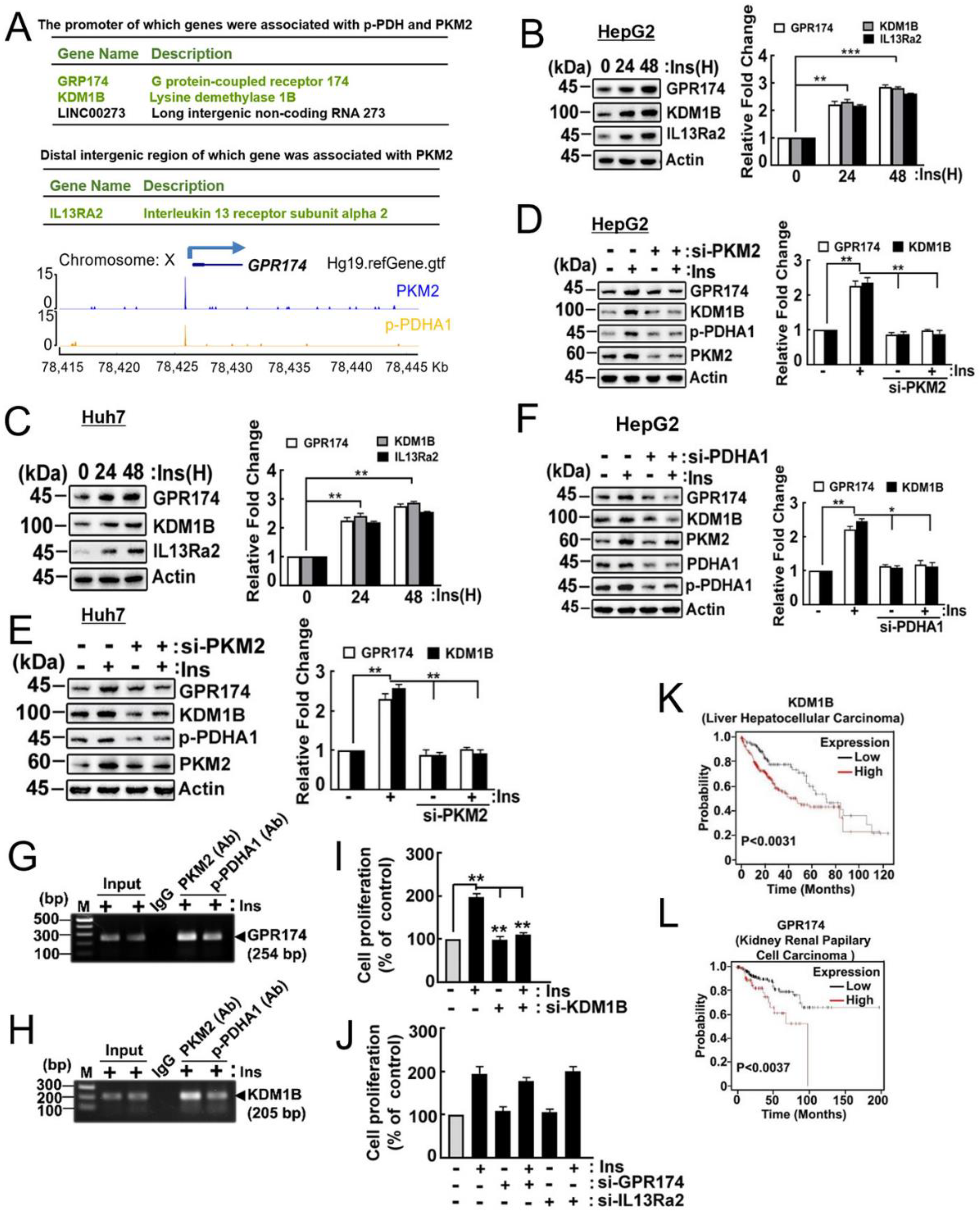
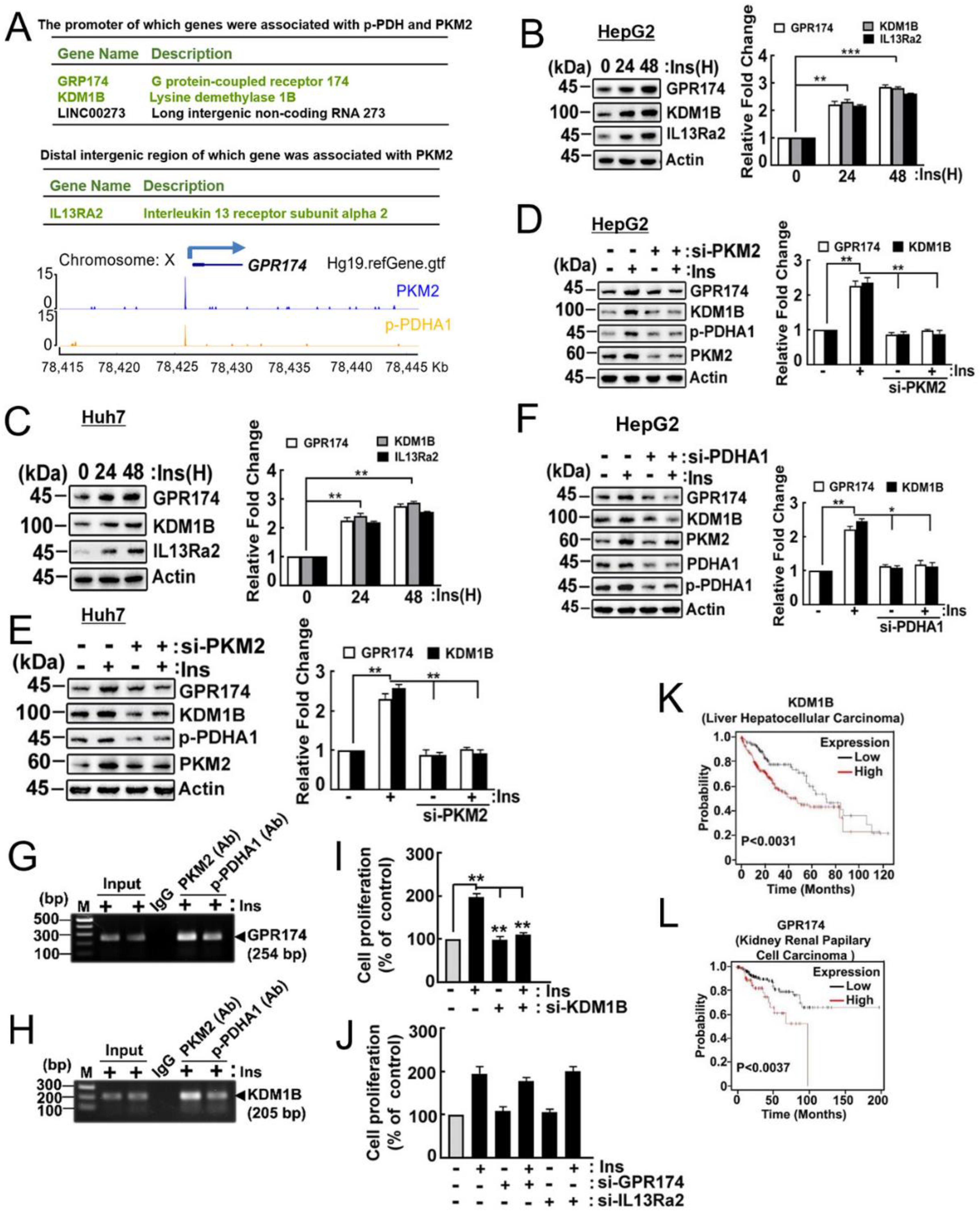
2.6. Target Genes of p-PDHA1 and PKM2
3. Discussion
4. Materials and Methods
4.1. Materials
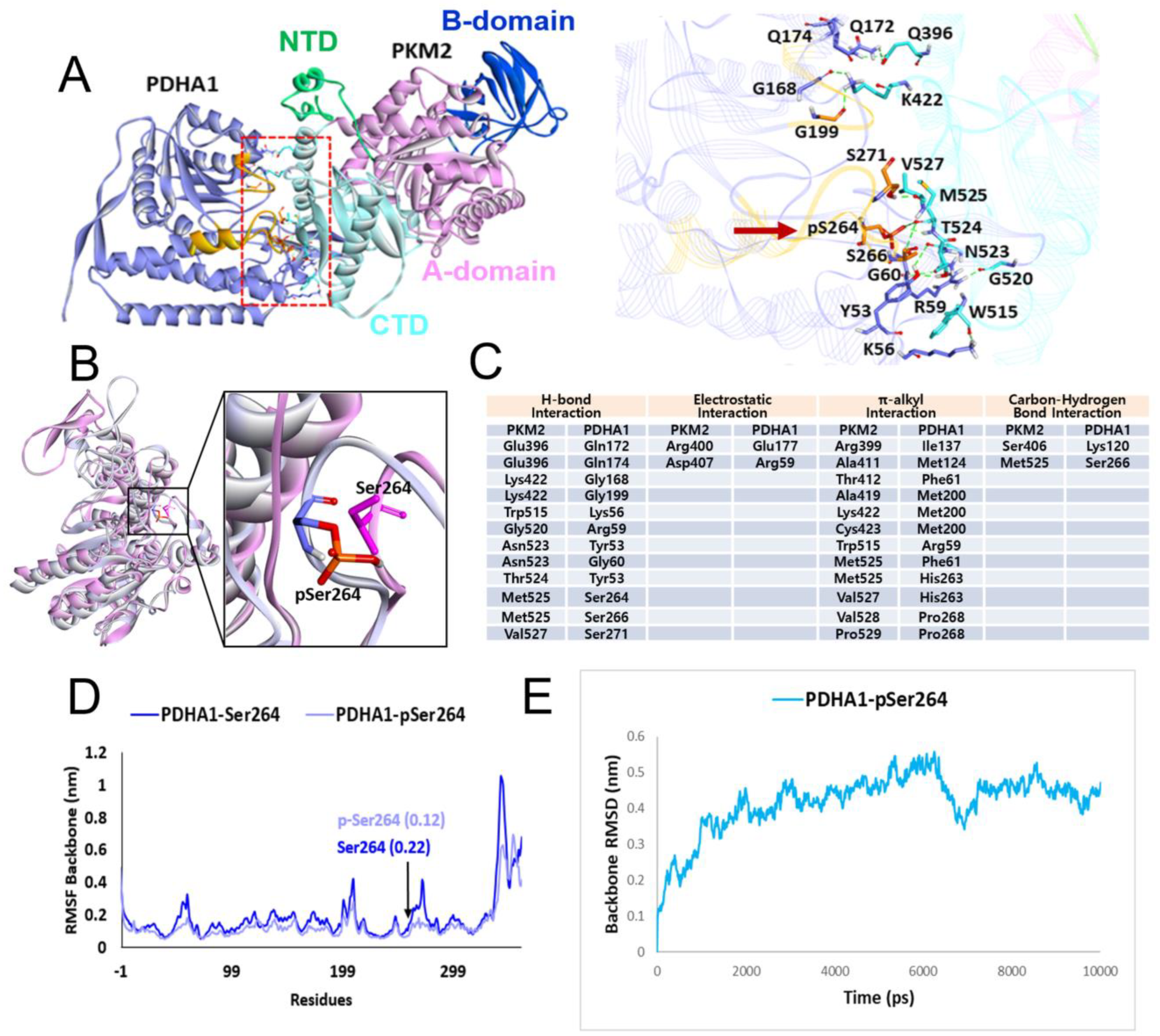
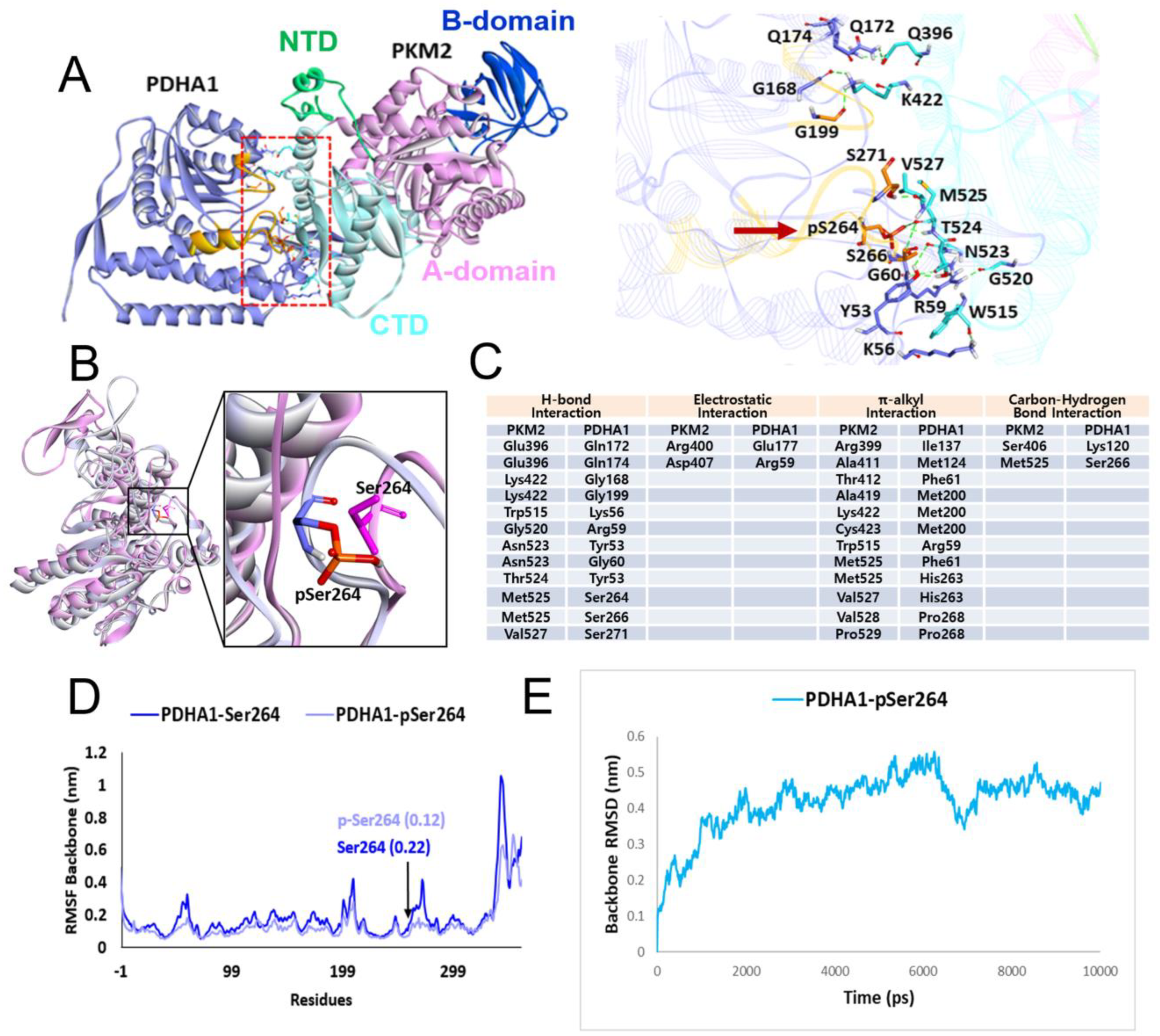
4.2. Molecular Docking Study between p-PDHA1 and PKM2
4.3. Cell Culture
4.4. Western Blot Analysis
4.5. Immunoprecipitation (Co-IP)
4.6. GTP-RhoA Pull-Down Assay
4.7. In Vitro Kinase Assay
4.8. GST Fusion Protein Purification
4.9. Cell Proliferation Assay
4.10. Plasmid DNA and si-RNA Transfection
4.11. Site-Directed Mutagenesis
4.12. Pyruvate Kinase M2 Activity Assay
4.13. PDH Activity Assay
4.14. Fluorescence Microscopy Assay of Liver Cancer Tissues and Cells
4.15. Chromatin Immunoprecipitation (ChIP) Sequencing
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Choi, E.; Bai, X.C. The Activation Mechanism of the Insulin Receptor: A Structural Perspective. Annu. Rev. Biochem. 2023, 92, 247–272. [Google Scholar] [CrossRef]
- Baumann, C.A.; Saltiel, A.R. Spatial compartmentalization of signal transduction in insulin action. Bioessays 2001, 23, 215–222. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Noel, A.; Barrier, L.; Rinaldi, F.; Hubert, C.; Fauconneau, B.; Ingrand, S. Lithium chloride and staurosporine potentiate the accumulation of phosphorylated glycogen synthase kinase 3beta/Tyr216, resulting in glycogen synthase kinase 3beta activation in SH-SY5Y human neuroblastoma cell lines. J. Neurosci. Res. 2011, 89, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R. Insulin receptor and cancer. Endocr. Relat. Cancer 2011, 18, R125–R147. [Google Scholar] [CrossRef]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef]
- Islam, R.; Kim, J.G.; Park, Y.; Cho, J.Y.; Cap, K.C.; Kho, A.R.; Chung, W.S.; Suh, S.W.; Park, J.B. Insulin induces phosphorylation of pyruvate dehydrogenase through RhoA activation pathway in HepG2 cells. FASEB J. 2019, 33, 2072–2083. [Google Scholar] [CrossRef]
- Hossain, A.J.; Islam, R.; Kim, J.G.; Dogsom, O.; Cap, K.C.; Park, J.B. Pyruvate Dehydrogenase A1 Phosphorylated by Insulin Associates with Pyruvate Kinase M2 and Induces LINC00273 through Histone Acetylation. Biomedicines 2022, 10, 1256. [Google Scholar] [CrossRef]
- Li, Z.; Yang, P.; Li, Z. The multifaceted regulation and functions of PKM2 in tumor progression. Biochim. Biophys. Acta 2014, 1846, 285–296. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell. 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Lv, W.W.; Liu, D.; Liu, X.C.; Feng, T.N.; Li, L.; Qian, B.Y.; Li, W.X. Effects of PKM2 on global metabolic changes and prognosis in hepatocellular carcinoma: From gene expression to drug discovery. BMC Cancer 2018, 18, 1150. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Wang, H.; Ni, M.; Wang, Z.; Wang, Z.; Wei, S.; Liu, M.; Wang, P.; Qiu, J.; Zhang, L.; et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut 2022, 71, 2539–2550. [Google Scholar] [CrossRef]
- Qin, Y.; Verdegaal, E.M.; Siderius, M.; Bebelman, J.P.; Smit, M.J.; Leurs, R.; Willemze, R.; Tensen, C.P.; Osanto, S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target. Pigment. Cell Melanoma. Res. 2011, 24, 207–218. [Google Scholar] [CrossRef]
- Chen, L.; Vasilatos, S.N.; Qin, Y.; Katz, T.A.; Cao, C.; Wu, H.; Tasdemir, N.; Levine, K.M.; Oesterreich, S.; Davidson, N.E.; et al. Functional characterization of lysine-specific demethylase 2 (LSD2/KDM1B) in breast cancer progression. Oncotarget 2017, 8, 81737–81753. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Wang, J.; Zeng, W.; Cheng, X.; Liu, L.; Li, W. Lysine-specific histone demethylase 1B (LSD2/KDM1B) represses p53 expression to promote proliferation and inhibit apoptosis in colorectal cancer through LSD2-mediated H3K4me2 demethylation. Aging 2020, 12, 14990–15001. [Google Scholar] [CrossRef]
- Katz, T.A.; Vasilatos, S.N.; Harrington, E.; Oesterreich, S.; Davidson, N.E.; Huang, Y. Inhibition of histone demethylase, LSD2 (KDM1B), attenuates DNA methylation and increases sensitivity to DNMT inhibitor-induced apoptosis in breast cancer cells. Breast. Cancer Res. Treat. 2014, 146, 99–108. [Google Scholar] [CrossRef]
- Fang, R.; Barbera, A.J.; Xu, Y.; Rutenberg, M.; Leonor, T.; Bi, Q.; Lan, F.; Mei, P.; Yuan, G.C.; Lian, C.; et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell. 2010, 39, 222–233. [Google Scholar] [CrossRef]
- Fang, R.; Chen, F.; Dong, Z.; Hu, D.; Barbera, A.J.; Clark, E.A.; Fang, J.; Yang, Y.; Mei, P.; Rutenberg, M.; et al. LSD2/KDM1B and its cofactor NPAC/GLYR1 endow a structural and molecular model for regulation of H3K4 demethylation. Mol. Cell. 2013, 49, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, R.; Sciacca, L.; Vigneri, P. Rethinking the Relationship between Insulin and Cancer. Trends Endocrinol. Metab. 2020, 31, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, D.; De Marchi, U.; Wollheim, C.B.; Wiederkehr, A. Pyruvate dehydrogenase E1alpha phosphorylation is induced by glucose but does not control metabolism-secretion coupling in INS-1E clonal beta-cells. Biochim. Biophys. Acta 2012, 1823, 1815–1824. [Google Scholar] [CrossRef]
- Park, J.; Kim, J.S.; Jung, K.C.; Lee, H.J.; Kim, J.I.; Kim, J.; Lee, J.Y.; Park, J.B.; Choi, S.Y. Exoenzyme Tat-C3 inhibits association of zymosan particles, phagocytosis, adhesion, and complement binding in macrophage cells. Mol. Cells 2003, 16, 216–223. [Google Scholar]
- Kim, J.S.; Diebold, B.A.; Kim, J.I.; Kim, J.; Lee, J.Y.; Park, J.B. Rho is involved in superoxide formation during phagocytosis of opsonized zymosans. J. Biol. Chem. 2004, 279, 21589–21597. [Google Scholar] [CrossRef]
- Kato, M.; Wynn, R.M.; Chuang, J.L.; Tso, S.C.; Machius, M.; Li, J.; Chuang, D.T. Structural basis for inactivation of the human pyruvate dehydrogenase complex by phosphorylation: Role of disordered phosphorylation loops. Structure 2008, 16, 1849–1859. [Google Scholar] [CrossRef]
- Dombrauckas, J.D.; Santarsiero, B.D.; Mesecar, A.D. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005, 44, 9417–9429. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Swetha, R.G.; Ramaiah, S.; Anbarasu, A. R521C and R521H mutations in FUS result in weak binding with Karyopherinbeta2 leading to Amyotrophic lateral sclerosis: A molecular docking and dynamics study. J. Biomol. Struct. Dyn. 2017, 35, 2169–2185. [Google Scholar] [CrossRef]
- Parate, S.; Rampogu, S.; Lee, G.; Hong, J.C.; Lee, K.W. Exploring the Binding Interaction of Raf Kinase Inhibitory Protein with the N-Terminal of C-Raf through Molecular Docking and Molecular Dynamics Simulation. Front. Mol. Biosci. 2021, 8, 655035. [Google Scholar] [CrossRef]
- Kim, J.G.; Kwon, H.J.; Wu, G.; Park, Y.; Lee, J.Y.; Kim, J.; Kim, S.C.; Choe, M.; Kang, S.G.; Seo, G.Y.; et al. RhoA GTPase oxidation stimulates cell proliferation via nuclear factor-kappaB activation. Free Radic. Biol. Med. 2017, 103, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, J.G.; Moon, M.Y.; Jeon, C.Y.; Won, H.Y.; Kim, H.J.; Jeon, Y.J.; Seo, J.Y.; Kim, J.I.; Kim, J.; et al. Transforming growth factor-beta1 regulates macrophage migration via RhoA. Blood 2006, 108, 1821–1829. [Google Scholar] [CrossRef]
- Ren, X.D.; Schwartz, M.A. Determination of GTP loading on Rho. Methods. Enzymol. 2000, 325, 264–272. [Google Scholar] [PubMed]
- Brierley, G.V.; Macaulay, S.L.; Forbes, B.E.; Wallace, J.C.; Cosgrove, L.J.; Macaulay, V.M. Silencing of the insulin receptor isoform A favors formation of type 1 insulin-like growth factor receptor (IGF-IR) homodimers and enhances ligand-induced IGF-IR activation and viability of human colon carcinoma cells. Endocrinology 2010, 151, 1418–1427. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.G.; Moon, M.Y.; Park, S.H.; Park, J.B. IkappaB kinase gamma/nuclear factor-kappaB-essential modulator (IKKgamma/NEMO) facilitates RhoA GTPase activation, which, in turn, activates Rho-associated KINASE (ROCK) to phosphorylate IKKbeta in response to transforming growth factor (TGF)-beta1. J. Biol. Chem. 2014, 289, 1429–1440. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, A.J.; Islam, R.; Seo, J.-B.; Park, H.-S.; Kim, J.-I.; Kumar, V.; Lee, K.W.; Park, J.-B. Association of Phosphorylated Pyruvate Dehydrogenase with Pyruvate Kinase M2 Promotes PKM2 Stability in Response to Insulin. Int. J. Mol. Sci. 2023, 24, 13697. https://doi.org/10.3390/ijms241813697
Hossain AJ, Islam R, Seo J-B, Park H-S, Kim J-I, Kumar V, Lee KW, Park J-B. Association of Phosphorylated Pyruvate Dehydrogenase with Pyruvate Kinase M2 Promotes PKM2 Stability in Response to Insulin. International Journal of Molecular Sciences. 2023; 24(18):13697. https://doi.org/10.3390/ijms241813697
Chicago/Turabian StyleHossain, Abu Jubayer, Rokibul Islam, Jong-Bok Seo, Hwee-Seon Park, Jong-Il Kim, Vikas Kumar, Keun Woo Lee, and Jae-Bong Park. 2023. "Association of Phosphorylated Pyruvate Dehydrogenase with Pyruvate Kinase M2 Promotes PKM2 Stability in Response to Insulin" International Journal of Molecular Sciences 24, no. 18: 13697. https://doi.org/10.3390/ijms241813697
APA StyleHossain, A. J., Islam, R., Seo, J.-B., Park, H.-S., Kim, J.-I., Kumar, V., Lee, K. W., & Park, J.-B. (2023). Association of Phosphorylated Pyruvate Dehydrogenase with Pyruvate Kinase M2 Promotes PKM2 Stability in Response to Insulin. International Journal of Molecular Sciences, 24(18), 13697. https://doi.org/10.3390/ijms241813697