
Glioma Stem Cells Are Sensitized to BCL-2 Family Inhibition by Compromising Histone Deacetylases

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
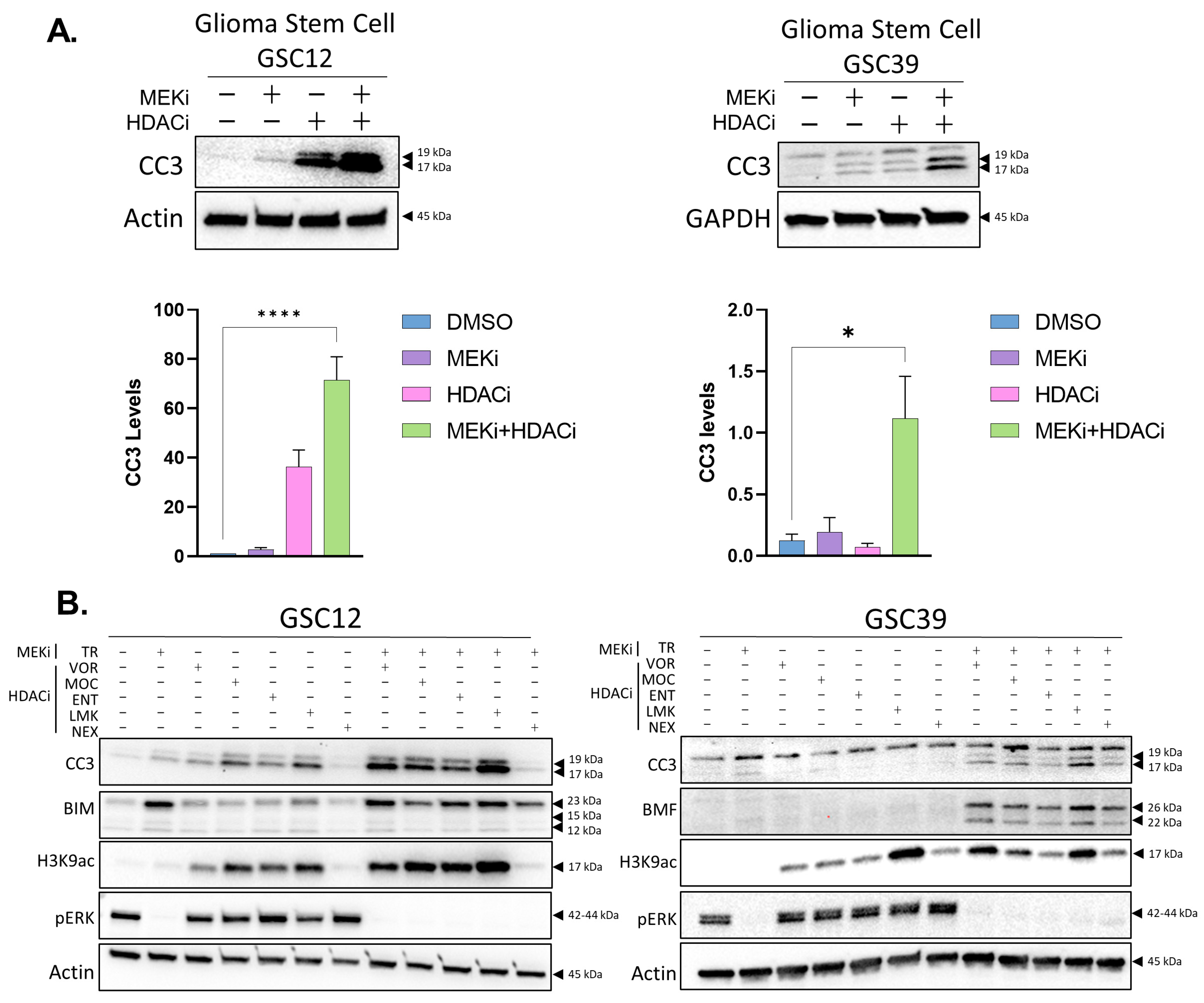
2.1. Inhibition of Histone Deacetylases Enhances MEKi-Induced Apoptosis in GBM
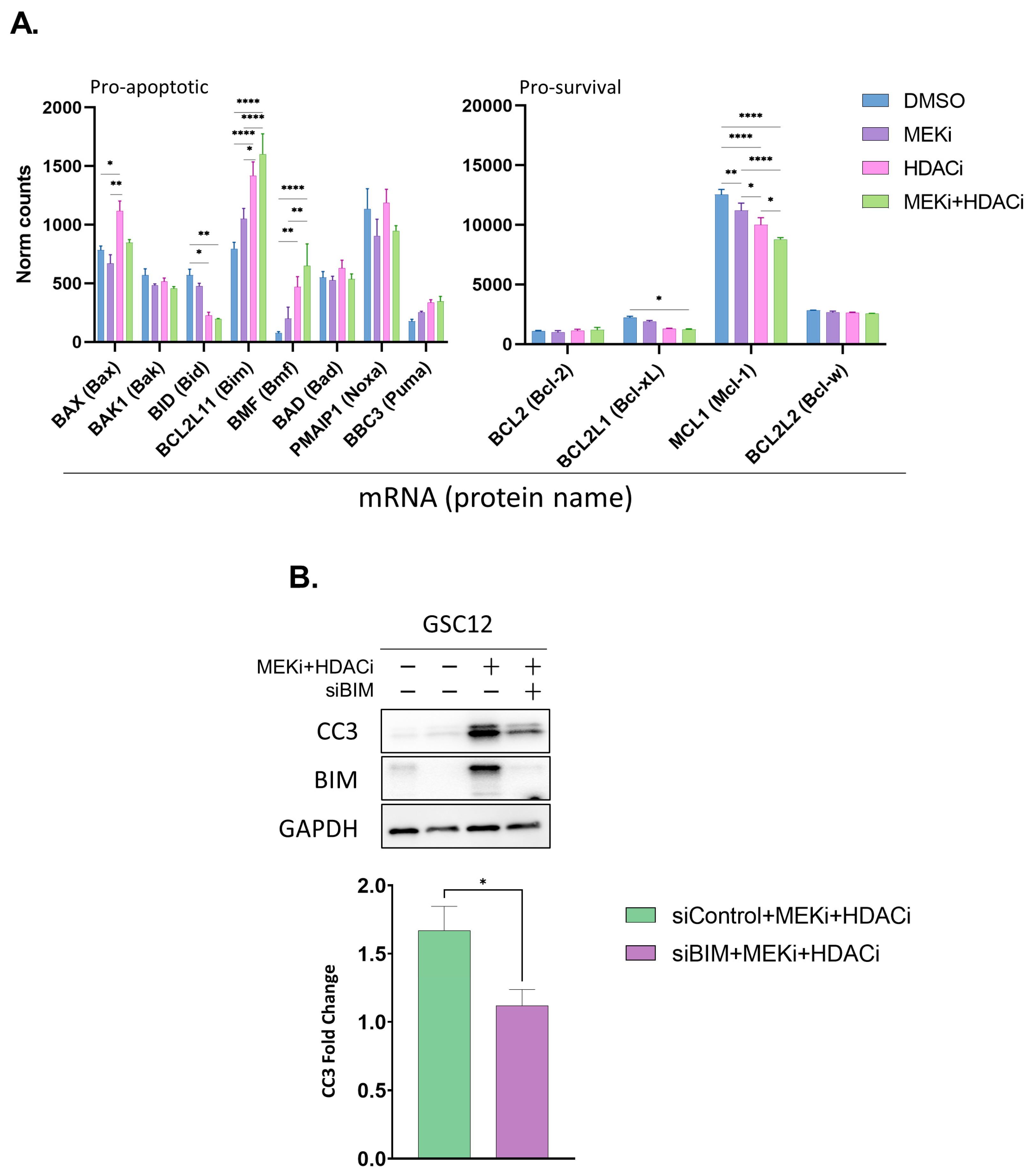
2.2. MEKi+HDACi Rescues BIM and BMF Expression and Represses MCL1 and BCL-XL in GSCs
2.3. MEKi+HDACi Disrupts the Cell Cycle in GSCs
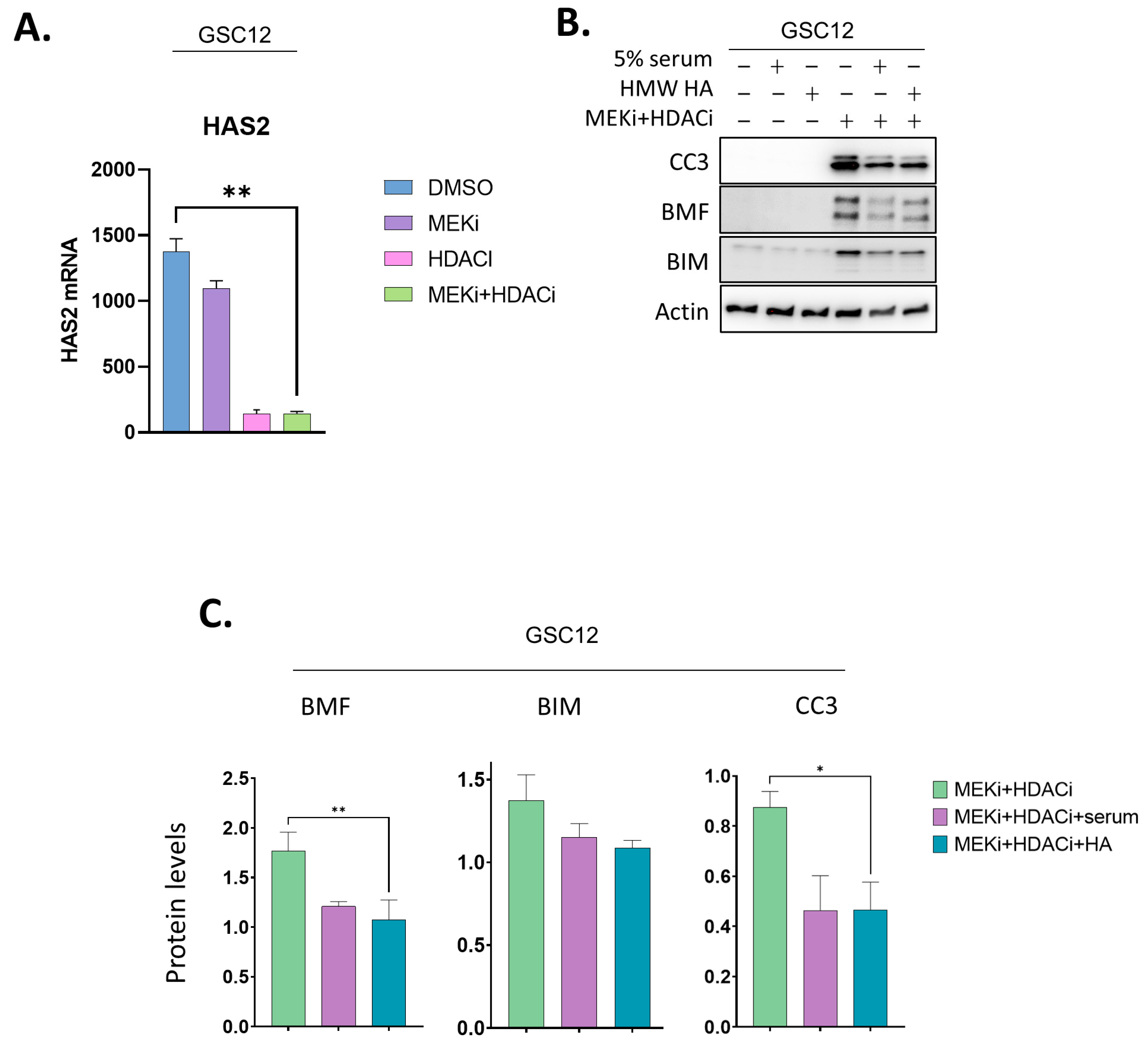
2.4. Addition of a CD44 Ligand Partially Represses BMF and BIM and Reverses Apoptosis in GSCs
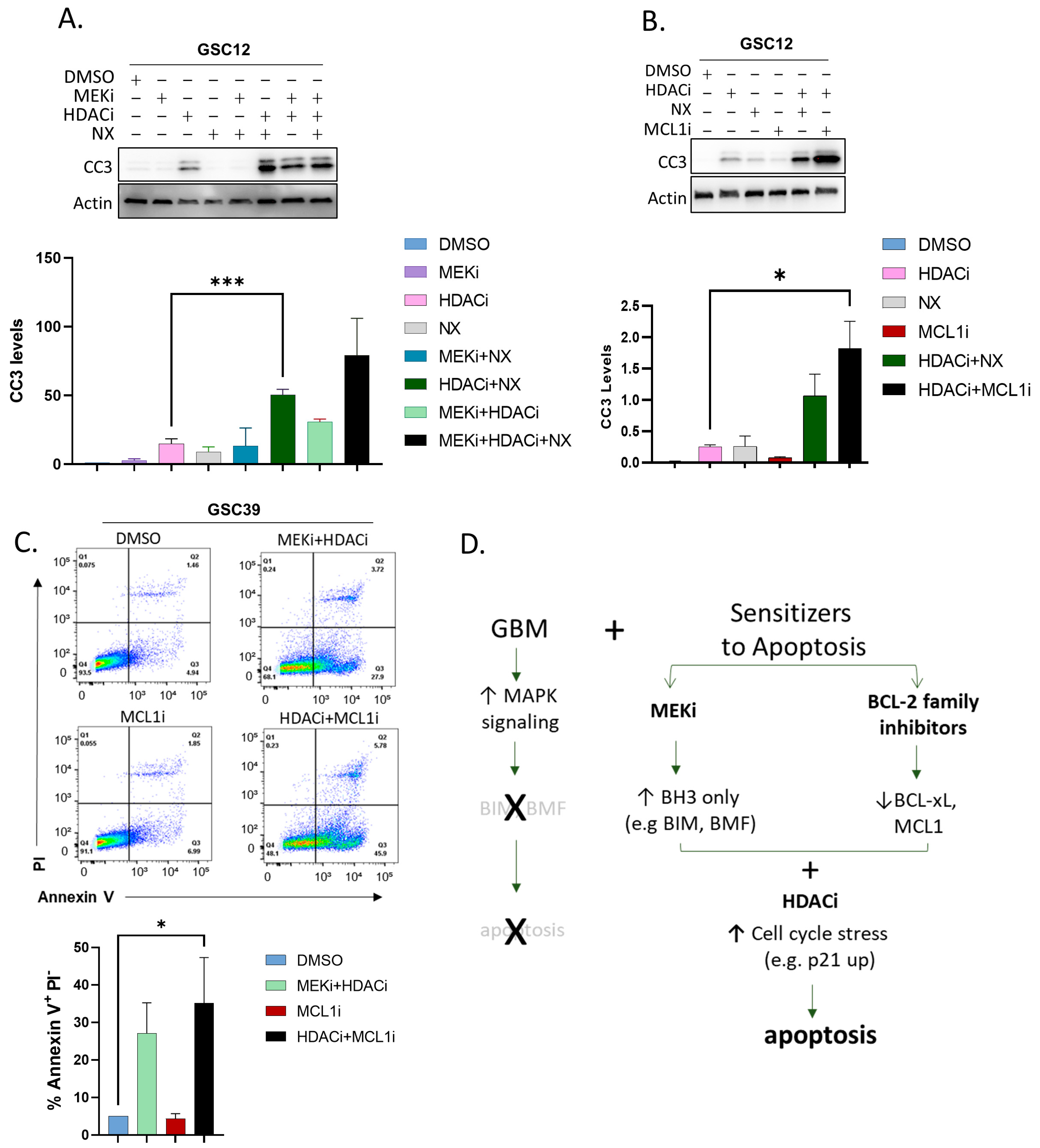
2.5. Substituting BCL-2 Family Inhibitors for MEKi Induces Apoptosis in GSCs
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Cultures
4.2. Cell Lysis and Immunoblots
4.3. Gene Knockdown Using Small Interfering RNA (siRNA)
4.4. Growth and Cytotoxicity Curves
4.5. Annexin V Flow Cytometry
4.6. Cell Cycle Analysis by Flow Cytometry
4.7. RNAseq Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2010–2014. Neuro Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef]
- Kamiya-Matsuoka, C.; Gilbert, M.R. Treating Recurrent Glioblastoma: An Update. CNS Oncol. 2015, 4, 91–104. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Kumar, R. TERT Promoter Mutations in Telomere Biology. Mutat. Res. Rev. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- D’Angelo, F.; Ceccarelli, M.; Tala; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; et al. The Molecular Landscape of Glioma in Patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The Integrated Landscape of Driver Genomic Alterations in Glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma Intrinsic Gene Expression Subtype Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting Glioma Stem Cells through Combined BMI1 and EZH2 Inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, D.; Chen, Y.J.; Xie, X.; Shi, Y.; Tabar, V.; Brennan, C.W.; Bale, T.A.; Jayewickreme, C.D.; Laks, D.R.; et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell 2020, 38, 366–379.e8. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma Stem-like Cells: At the Root of Tumor Recurrence and a Therapeutic Target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.P.; Laks, D.R.; Sun, D.; Ganbold, M.; Wang, Z.; Pedraza, A.M.; Bale, T.; Tabar, V.; Brennan, C.; Zhou, X.; et al. Quiescent Human Glioblastoma Cancer Stem Cells Drive Tumor Initiation, Expansion, and Recurrence Following Chemotherapy. Dev. Cell 2022, 57, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, T.; Sengupta, S.; Glantz, M.J.; Green, R.M.; Yu, A.; Aregawi, D.; Chaudhary, R.; Chen, R.; Zuccarello, M.; Lu-Emerson, C.; et al. Cancer Stem Cell Assay-Guided Chemotherapy Improves Survival of Patients with Recurrent Glioblastoma in a Randomized Trial. Cell Rep. Med. 2023, 4, 101025. [Google Scholar] [CrossRef]
- Lauko, A.; Volovetz, J.; Turaga, S.M.; Bayik, D.; Silver, D.J.; Mitchell, K.; Mulkearns-Hubert, E.E.; Watson, D.C.; Desai, K.; Midha, M.; et al. SerpinB3 Drives Cancer Stem Cell Survival in Glioblastoma. Cell Rep. 2022, 40, 111348. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-ΚB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020, 30, 3383–3396.e7. [Google Scholar] [CrossRef] [PubMed]
- Telles, E.; Seto, E. Modulation of Cell Cycle Regulators by HDACs. Front. Biosci. (Schol. Ed.) 2012, 4, 831. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Westhoff, M.-A.; Karpel-Massler, G.; Siegelin, M.D.; Nguyen, T.T.T.; Westhoff, M.-A.; Karpel-Massler, G.; Siegelin, M.D. Targeting Super-Enhancers Reprograms Glioblastoma Central Carbon Metabolism. Oncotarget 2021, 12, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Asklund, T.; Kvarnbrink, S.; Holmlund, C.; Wibom, C.; Bergenheim, T.; Henriksson, R.; Hedman, H. Synergistic Killing of Glioblastoma Stem-like Cells by Bortezomib and HDAC Inhibitors. Anticancer Res. 2012, 32, 2407–2413. [Google Scholar]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC Inhibitors Elicit Metabolic Reprogramming by Targeting Super-Enhancers in Glioblastoma Models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR Signaling Networks in Glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and Metabolomic Characterization of Human Glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef]
- Wykosky, J.; Hu, J.; Gomez, G.G.; Taylor, T.; Villa, G.R.; Pizzo, D.; Van Den Berg, S.R.; Thorne, A.H.; Chen, C.C.; Mischel, P.S.; et al. A Urokinase Receptor-Bim Signaling Axis Emerges During EGFR Inhibitor Resistance in Mutant EGFR Glioblastoma. Cancer Res. 2015, 75, 394–404. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Gielniewski, B.; Wojtas, B.; Kaminska, B.; Mieczkowski, J. EGFR/FOXO3a/BIM Signaling Pathway Determines Chemosensitivity of BMP4-Differentiated Glioma Stem Cells to Temozolomide. Exp. Mol. Med. 2020, 52, 1326–1340. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Meise, R.; Aasland, D.; Berte, N.; Kitzinger, R.; Krämer, O.H.; Kaina, B.; Christmann, M.; Tomicic, M.T.; Meise, R.; et al. Apoptosis Induced by Temozolomide and Nimustine in Glioblastoma Cells Is Supported by JNK/c-Jun-Mediated Induction of the BH3-Only Protein BIM. Oncotarget 2015, 6, 33755–33768. [Google Scholar] [CrossRef]
- Acquaviva, J.; Jun, H.G.; Lessard, J.; Ruiz, R.; Zhu, H.; Donovan, M.; Woolfenden, S.; Boskovitz, A.; Raval, A.; Bronson, R.T.; et al. Chronic Activation of Wild-Type Epidermal Growth Factor Receptor and Loss of Cdkn2a Cause Mouse Glioblastoma Formation. Cancer Res. 2011, 71, 7198–7206. [Google Scholar] [CrossRef]
- Shao, Y.; Aplin, A.E. ERK2 Phosphorylation of Serine 77 Regulates Bmf Pro-Apoptotic Activity. Cell Death Dis. 2012, 3, e253. [Google Scholar] [CrossRef]
- Montero, J.; Letai, A. Why Do BCL-2 Inhibitorswork and Where Should We Use Them in the Clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor Microenvironment in Glioblastoma: Current and Emerging Concepts. Neurooncol. Adv. 2023, 5, 1–16. [Google Scholar] [CrossRef]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the Glioblastoma Immune Microenvironment as Basis for the Development of New Immunotherapeutic Strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the Glioblastoma Microenvironment: Shaping the Phenotype of Cancer Stem-like Cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P. Hyaluronan-CD44 Interactions in Cancer: Paradoxes and Possibilities. Clin. Cancer Res. 2009, 15, 7462–7468. [Google Scholar] [CrossRef]
- Pibuel, M.A.; Poodts, D.; Díaz, M.; Hajos, S.E.; Lompardía, S.L. The Scrambled Story between Hyaluronan and Glioblastoma. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef]
- Hartheimer, J.S.; Park, S.; Rao, S.S.; Kim, Y. Targeting Hyaluronan Interactions for Glioblastoma Stem Cell Therapy. Cancer Microenviron. 2019, 12, 47–56. [Google Scholar] [CrossRef]
- Ward, J.A.; Huang, L.; Guo, H.; Ghatak, S.; Toole, B.P. Perturbation of Hyaluronan Interactions Inhibits Malignant Properties of Glioma Cells. Am. J. Pathol. 2003, 162, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Witham, T.; Villa, L.; Erff, M.; Attanucci, J.; Watkins, S.; Kondziolka, D.; Okada, H.; Pollack, I.; Chambers, W. Glioma-Associated Hyaluronan Induces Apoptosis in Dendritic Cells via Inducible Nitric Oxide Synthase: Implications for the Use of Dendritic Cells for Therapy of Gliomas. Cancer Res. 2002, 62, 2583–2591. [Google Scholar] [PubMed]
- Xu, Y.; Stamenkovic, I.; Yu, Q. CD44 Attenuates Activation of the Hippo Signaling Pathway and Is a Prime Therapeutic Target for Glioblastoma. Cancer Res. 2010, 70, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone Deacetylase Inhibitors Trigger a G2 Checkpoint in Normal Cells That Is Defective in Tumor Cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone Deacetylase Inhibitor Induces DNA Damage, Which Normal but Not Transformed Cells Can Repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Pucci, B.; Kasten, M.; Giordano, A. Cell Cycle and Apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef]
- Marenco-Hillembrand, L.; Wijesekera, O.; Suarez-Meade, P.; Mampre, D.; Jackson, C.; Peterson, J.; Trifiletti, D.; Hammack, J.; Ortiz, K.; Lesser, E.; et al. Trends in Glioblastoma: Outcomes over Time and Type of Intervention: A Systematic Evidence Based Analysis. J. Neurooncol. 2020, 147, 297–307. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The Application of Histone Deacetylases Inhibitors in Glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Everix, L.; Seane, E.N.; Ebenhan, T.; Goethals, I.; Bolcaen, J. Introducing HDAC-Targeting Radiopharmaceuticals for Glioblastoma Imaging and Therapy. Pharmaceuticals 2023, 16, 227. [Google Scholar] [CrossRef]
- Zhang, C.; Richon, V.; Ni, X.; Talpur, R.; Duvic, M. Selective Induction of Apoptosis by Histone Deacetylase Inhibitor SAHA in Cutaneous T-Cell Lymphoma Cells: Relevance to Mechanism of Therapeutic Action. J. Investig. Dermatol. 2005, 125, 1045–1052. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally Defined Therapeutic Targets in Diffuse Intrinsic Pontine Glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.S.; et al. Suberoylanilide Hydroxamic Acid, a Histone Deacetylase Inhibitor, Ameliorates Motor Deficits in a Mouse Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Eyüpoglu, I.Y.; Hahnen, E.; Buslei, R.; Siebzehnrübl, F.A.; Savaskan, N.E.; Lüders, M.; Tränkle, C.; Wick, W.; Weller, M.; Fahlbusch, R.; et al. Suberoylanilide Hydroxamic Acid (SAHA) Has Potent Anti-Glioma Properties in Vitro, Ex Vivo and in Vivo. J. Neurochem. 2005, 93, 992–999. [Google Scholar] [CrossRef]
- Choi, M.A.; Park, S.Y.; Chae, H.Y.; Song, Y.; Sharma, C.; Seo, Y.H. Design, Synthesis and Biological Evaluation of a Series of CNS Penetrant HDAC Inhibitors Structurally Derived from Amyloid-β Probes. Sci. Rep. 2019, 9, 13187. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of Histone Deacetylation Potentiates the Evolution of Acquired Temozolomide Resistance Linked to MGMT Upregulation in Glioblastoma Xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A.; Patel, S.J.; et al. Mechanisms and Clinical Significance of Histone Deacetylase Inhibitors: Epigenetic Glioblastoma Therapy. Anticancer Res. 2015, 35, 615–625. [Google Scholar]
- Lo Cascio, C.; McNamara, J.B.; Melendez, E.L.; Lewis, E.M.; Dufault, M.E.; Sanai, N.; Plaisier, C.L.; Mehta, S. Nonredundant, Isoform-Specific Roles of HDAC1 in Glioma Stem Cells. JCI Insight 2021, 6, e149232. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Lucas, D.M.; Davis, M.E.; Parthun, M.R.; Mone, A.P.; Kitada, S.; Cunningham, K.D.; Flax, E.L.; Wickham, J.; Reed, J.C.; Byrd, J.C.; et al. The Histone Deacetylase Inhibitor MS-275 Induces Caspase-Dependent Apoptosis in B-Cell Chronic Lymphocytic Leukemia Cells. Leukemia 2004, 18, 1207–1214. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone Deacetylase Inhibitors: Molecular Mechanisms of Action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Kaya-Aksoy, E.; Cingoz, A.; Senbabaoglu, F.; Seker, F.; Sur-Erdem, I.; Kayabolen, A.; Lokumcu, T.; Sahin, G.N.; Karahuseyinoglu, S.; Bagci-Onder, T. The Pro-Apoptotic Bcl-2 Family Member Harakiri (HRK) Induces Cell Death in Glioblastoma Multiforme. Cell Death Discov. 2019, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.; Zhang, Y.; Shu, C.; Ishida, C.T.; Bianchetti, E.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. Dual Inhibition of Bcl-2/Bcl-XL and XPO1 Is Synthetically Lethal in Glioblastoma Model Systems. Sci. Rep. 2018, 8, 15383. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Southon, A.G.; Saleh, E.; Brinkmann, K.; Ke, F.; Iliopoulos, M.; Cross, R.S.; Jenkins, M.R.; Nhu, D.; Wang, Z.; et al. BH3 Mimetic Drugs Cooperate with Temozolomide, JQ1 and Inducers of Ferroptosis in Killing Glioblastoma Multiforme Cells. Cell Death Differ. 2022, 29, 1335–1348. [Google Scholar] [CrossRef]
- Vera, M.B.; Morris-Hanon, O.; Nogueiras, G.I.; Ripari, L.B.; Esquivel, M.I.; Perez-Castro, C.; Romorini, L.; Sevlever, G.E.; Scassa, M.E.; Videla-Richardson, G.A. Noxa and Mcl-1 Expression Influence the Sensitivity to BH3-Mimetics That Target Bcl-XL in Patient-Derived Glioma Stem Cells. Sci. Rep. 2022, 12, 17729. [Google Scholar] [CrossRef]
- Berghauser Pont, L.M.E.; Spoor, J.K.H.; Venkatesan, S.; Swagemakers, S.; Kloezeman, J.J.; Dirven, C.M.F.; Van Der Spek, P.J.; Lamfers, M.L.M.; Leenstra, S. The Bcl-2 Inhibitor Obatoclax Overcomes Resistance to Histone Deacetylase Inhibitors SAHA and LBH589 as Radiosensitizers in Patient-Derived Glioblastoma Stem-like Cells. Genes Cancer 2014, 5, 445–459. [Google Scholar] [CrossRef]
- Koessinger, A.L.; Cloix, C.; Koessinger, D.; Heiland, D.H.; Bock, F.J.; Strathdee, K.; Kinch, K.; Martínez-Escardó, L.; Paul, N.R.; Nixon, C.; et al. Increased Apoptotic Sensitivity of Glioblastoma Enables Therapeutic Targeting by BH3-Mimetics. Cell Death Differ. 2022, 29, 2089–2104. [Google Scholar] [CrossRef]
- Ploumaki, I.; Triantafyllou, E.; Koumprentziotis, I.A.; Karampinos, K.; Drougkas, K.; Karavolias, I.; Trontzas, I.; Kotteas, E.A. Bcl-2 Pathway Inhibition in Solid Tumors: A Review of Clinical Trials. Clin. Transl. Oncol. 2023, 25, 1554–1578. [Google Scholar] [CrossRef] [PubMed]
- Faião-Flores, F.; Emmons, M.F.; Durante, M.A.; Kinose, F.; Saha, B.; Fang, B.; Koomen, J.M.; Chellappan, S.P.; Maria-Engler, S.S.; Rix, U.; et al. HDAC Inhibition Enhances the In Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. Clin. Cancer Res. 2019, 25, 5686–5701. [Google Scholar] [CrossRef] [PubMed]
- Carson, R.; Celtikci, B.; Fenning, C.; Javadi, A.; Crawford, N.; Perez-Carbonell, L.; Lawler, M.; Longley, D.B.; Johnston, P.G.; Van Schaeybroeck, S. HDAC Inhibition Overcomes Acute Resistance to MEK Inhibition in BRAF-Mutant Colorectal Cancer by Downregulation of c-FLIPL. Clin. Cancer Res. 2015, 21, 3230–3240. [Google Scholar] [CrossRef]
- Moustafa-Kamal, M.; Gamache, I.; Lu, Y.; Li, S.; Teodoro, J.G. BimEL Is Phosphorylated at Mitosis by Aurora A and Targeted for Degradation by ΒTrCP1. Cell Death Differ. 2013, 20, 1393–1403. [Google Scholar] [CrossRef]
- Essien, E.I.; Hofer, T.P.; Atkinson, M.J.; Anastasov, N. Combining HDAC and MEK Inhibitors with Radiation against Glioblastoma-Derived Spheres. Cells 2022, 11, 775. [Google Scholar] [CrossRef]
- Abatangelo, G.; Vindigni, V.; Avruscio, G.; Pandis, L.; Brun, P. Hyaluronic Acid: Redefining Its Role. Cells 2020, 9, 1743. [Google Scholar] [CrossRef] [PubMed]
- Piñon, J.D.; Labi, V.; Egle, A.; Villunger, A. Bim and Bmf in Tissue Homeostasis and Malignant Disease. Oncogene 2008, 27 (Suppl. S1), S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Z.; Ouyang, Z.; Ren, Y.; Cheng, Y.; Liu, P.; Wen, Y.; Shao, Y. Non-Canonical Phosphorylation of Bmf by P38 MAPK Promotes Its Apoptotic Activity in Anoikis. Cell Death Differ. 2022, 29, 323–336. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK Phosphorylation of Bim-Related Members of the Bcl2 Family Induces Bax-Dependent Apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed]
- Tolg, C.; Hamilton, S.R.; Morningstar, L.; Zhang, J.; Zhang, S.; Esguerra, K.V.; Telmer, P.G.; Luyt, L.G.; Harrison, R.; McCarthy, J.B.; et al. RHAMM Promotes Interphase Microtubule Instability and Mitotic Spindle Integrity through MEK1/ERK1/2 Activity. J. Biol. Chem. 2010, 285, 26461–26474. [Google Scholar] [CrossRef]
- Tosi, U.; Kommidi, H.; Adeuyan, O.; Guo, H.; Maachani, U.B.; Chen, N.; Su, T.; Zhang, G.; Pisapia, D.J.; Dahmane, N.; et al. PET, Image-Guided HDAC Inhibition of Pediatric Diffuse Midline Glioma Improves Survival in Murine Models. Sci. Adv. 2020, 6, eabb4105. [Google Scholar] [CrossRef]
- Denk, F.; Huang, W.; Sidders, B.; Bithell, A.; Crow, M.; Grist, J.; Sharma, S.; Ziemek, D.; Rice, A.S.C.; Buckley, N.J.; et al. HDAC Inhibitors Attenuate the Development of Hypersensitivity in Models of Neuropathic Pain. Pain 2013, 154, 1668–1679. [Google Scholar] [CrossRef]
- Tagscherer, K.E.; Fassl, A.; Campos, B.; Farhadi, M.; Kraemer, A.; Böck, B.C.; Macher-Goeppinger, S.; Radlwimmer, B.; Wiestler, O.D.; Herold-Mende, C.; et al. Apoptosis-Based Treatment of Glioblastomas with ABT-737, a Novel Small Molecule Inhibitor of Bcl-2 Family Proteins. Oncogene 2008, 27, 6646–6656. [Google Scholar] [CrossRef]
- Vaubel, R.A.; Tian, S.; Remonde, D.; Schroeder, M.A.; Mladek, A.C.; Kitange, G.J.; Caron, A.; Kollmeyer, T.M.; Grove, R.; Peng, S.; et al. Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clin. Cancer Res. 2020, 26, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merati, A.; Kotian, S.; Acton, A.; Placzek, W.; Smithberger, E.; Shelton, A.K.; Miller, C.R.; Stern, J.L. Glioma Stem Cells Are Sensitized to BCL-2 Family Inhibition by Compromising Histone Deacetylases. Int. J. Mol. Sci. 2023, 24, 13688. https://doi.org/10.3390/ijms241813688
Merati A, Kotian S, Acton A, Placzek W, Smithberger E, Shelton AK, Miller CR, Stern JL. Glioma Stem Cells Are Sensitized to BCL-2 Family Inhibition by Compromising Histone Deacetylases. International Journal of Molecular Sciences. 2023; 24(18):13688. https://doi.org/10.3390/ijms241813688
Chicago/Turabian StyleMerati, Aran, Spandana Kotian, Alexus Acton, William Placzek, Erin Smithberger, Abigail K. Shelton, C. Ryan Miller, and Josh L. Stern. 2023. "Glioma Stem Cells Are Sensitized to BCL-2 Family Inhibition by Compromising Histone Deacetylases" International Journal of Molecular Sciences 24, no. 18: 13688. https://doi.org/10.3390/ijms241813688
APA StyleMerati, A., Kotian, S., Acton, A., Placzek, W., Smithberger, E., Shelton, A. K., Miller, C. R., & Stern, J. L. (2023). Glioma Stem Cells Are Sensitized to BCL-2 Family Inhibition by Compromising Histone Deacetylases. International Journal of Molecular Sciences, 24(18), 13688. https://doi.org/10.3390/ijms241813688