Abstract
Leber congenital amaurosis (LCA) is the most severe form of inherited retinal dystrophy. RPGRIP1-related LCA accounts for 5–6% of LCA. We performed whole-exome sequencing and whole-genome sequencing (WGS) on 29 patients with clinically suspected LCA and examined ophthalmic findings in patients with biallelic pathogenic variants of RPGRIP1. In addition to five previously reported cases, we identified five cases from four families with compound heterozygous RPGRIP1 variants using WGS. Five patients had null variants comprising frameshift variants, an Alu insertion, and microdeletions. A previously reported 1339 bp deletion involving exon 18 was found in four cases, and the deletion was relatively prevalent in the Japanese population (allele frequency: 0.002). Microdeletions involving exon 1 were detected in four cases. In patients with RPGRIP1 variants, visual acuity remained low, ranging from light perception to 0.2, and showed no correlation with age. In optical coherence tomography images, the ellipsoid zone (EZ) length decreased with age in all but one case of unimpaired EZ. The retinal structure was relatively preserved in all cases; however, there were cases with great differences in visual function compared to their siblings and a 56-year-old patient who still had a faint EZ line. Structural abnormalities may be important genetic causes of RPGRIP1-related retinal dystrophy in Japanese patients, and WGS was useful for detecting them.
1. Introduction
Leber congenital amaurosis (LCA; MIM#204000) is the most severe form of inherited retinal dystrophy (IRD) and is characterized by nystagmus, a lack of pursuit, and a severely reduced or absent electroretinogram (ERG) from birth, accompanied by severe visual impairment [,]. Autosomal dominant forms have been reported; however, most are autosomal recessive, and 25 causative genes have been reported [,,]. RPGRIP1-related LCA (LCA6; #613826) reportedly accounts for 5–6% of LCA [,,,]. RPGRIP1 variants have been reported to cause IRD, including retinitis pigmentosa (RP; #268000), cone–rod dystrophy (CRD; #120970), LCA, and pigmented paravenous retinochoroidal atrophy (PPRCA), and genetic and clinical data have been accumulated [,,,,,]. RPGRIP1 plays a key role in connecting the cilia of photoreceptors, and RPGRIP1 deficiency makes it difficult to maintain light-sensitive outer segments [,]. In LCA6, although the retinal function is lost during childhood, the retinal structure is relatively retained until adulthood []. These characteristics are advantageous for gene therapy.
The yield of genetic diagnoses has improved owing to the increased availability of whole-genome sequencing (WGS) and whole-exome sequencing (WES), and also due to the reduction in cost and the rapid progress of various data analysis technologies. WGS can detect structural abnormalities, transposon insertions, and deep-intronic splicing variants that are impossible or difficult to detect using WES. In cases where causative variants cannot be detected via WES, the chance of detecting them increases when performing WGS [,,,,]. We believe WGS is effective for cases of suspected autosomal recessive disorders in which a likely pathogenic variant is found in one allele with no abnormality detected in another allele. Furthermore, the risk assessment system based on genetic counseling has improved due to enhancing databases of healthy individuals.
Previously, we examined five individuals from three families with biallelic RPGRIP1 variants and found that retinal function was severely impaired in childhood, whereas the retinal structure was relatively preserved [,]. By performing WES/WGS on clinically suspected LCA cases without systemic diseases other than psychomotor retardation, we identified LCA caused by RPGRIP1 variants in five cases from four families. In particular, a 1339 bp deletion encompassing exon 18 of RPGRIP1 was observed in three individuals from two families. This deletion is relatively prevalent in the Japanese population (allele frequency: 0.002) and possibly in Japanese patients with RPGRIP1-related retinal dystrophy. Furthermore, microdeletions involving exon 1 were observed in four cases. Because LCA is a rare disease, the number of subjects is too small to draw conclusions; nonetheless, structural abnormalities may be a major cause in RPGRIP1-related retinal dystrophy in the Japanese population. We also analyzed detailed clinical findings during the follow-up period in 10 patients, one of whom was 56 years old. This report provides important information for both genetic counseling and gene therapy.
2. Results
2.1. Pathogenic Variants in the Subjects
In this study, we performed WES on 29 patients with clinically suspected LCA without systemic symptoms other than psychomotor delay (see Section 4 for details). WES identified two patients with heterozygous frameshift variants in one RPGRIP1 allele (EYE170 and 345); however, no abnormalities were found in the other allele. Therefore, we performed WGS on 14 unsolved cases, including EYE170 and 345. Table 1 shows a summary of the patients with pathogenic variants of RPGRIP1 (see also Supplementary Table S1). We identified five cases from four families with biallelic pathogenic variants (Figure 1a). EYE170 had a heterozygous microdeletion involving exon 1 of RPGRIP1 (NM_020366.4), detected using two tools, Canvas [] and Manta []. Exon 1 of NM_020366.4 is a newly annotated non-coding exon that was absent in the previous version (NM_020366.3) and was not covered by the SureSelect Human All Exon V6 kit (Figure 1b). EYE345 had a heterozygous Alu insertion that was detected using TEMP2 []. The remaining three cases (JU0954, 0955, and 1556) from two families had compound heterozygous microdeletions involving one to three exons (Figure 1b). Notably, a 1339 bp deletion involving exon 18 was identified in three cases via Manta. We previously reported five cases from three families with biallelic RPGRIP1 variants [,] (Figure 1b), including one case with a deletion involving exon 18, suggesting that microdeletion may be a major genetic cause of LCA in Japanese patients.

Table 1.
List of patients with pathogenic variants of RPGRIP1(NM_020366.4).
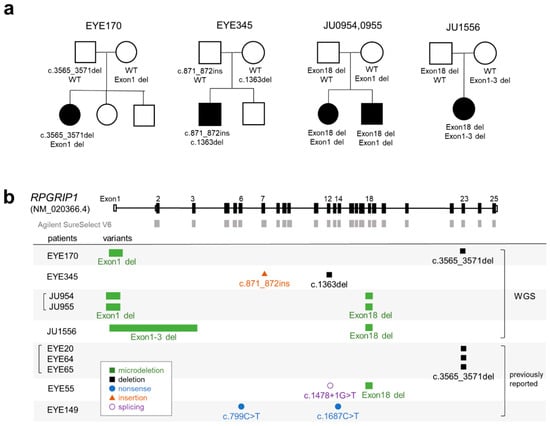
Figure 1.
Pedigrees and schematic representation of RPGRIP1 variants. (a) Pedigree of five patients with biallelic RPGRIP1 variants. The segregation of each variant is shown. (b) Schematic representation of RPGRIP1 transcript (NM_020366.4). The UTR and coding region are open and filled rectangles, respectively. Baits of Agilent SureSelect V6 are shown in gray boxes. RPGRIP1 variants in five patients diagnosed using WGS are shown together with variants in five patients from our previous report.
Next, we characterized the breakpoint sequences by de novo assembly using aberrantly aligned read pairs identified by manual inspection using an integrative genomics viewer (IGV), and confirmed these using breakpoint-specific polymerase chain reaction (PCR) (Supplementary Table S2). The microdeletion involving exon 18 was 1339 bp in size (Figure 2a; NC_000014.9: g.21326547_21327885del). Using breakpoint PCR and Sanger sequencing, we confirmed that the microdeletions were identical among four patients (EYE55, JU0954, 0955, and 1556) (Figure 2b). Microdeletions were not observed in gnomAD structural variants (SVs) v2.1; however, they were found in 1 of 444 alleles in JSV1 (long-read sequencing of Japanese, ID: JSV1_hg38_DEL.10214) and 33 of 16,760 alleles in 8.3KJPN-SV (short-read sequencing of Japanese, ID:111853). Therefore, microdeletions involving exon 18 are considered relatively prevalent in the Japanese population (allele frequency = 0.002). Breakpoints of a 4466 bp deletion (EYE170; NC_000014.9: g.21276834_21281300del) and a 4119 bp deletion (JU0954 and 0955; NC_000014.9: g.21276147_21280265del), both involving exon 1, were localized in the Alu sequence (Figure 2c). Breakpoints of a 19,934 bp deletion involving exons 1–3 (NC_000014.9: g.21277271_21297204delinsN [330]) were also localized in the Alu sequence (Figure 2d), suggesting that Alu-mediated recombination might be frequent in the vicinity of exon 1. Consistent with this, a 4421 bp deletion (chr14:21276854-21281275, ID:111850) was found in three of 16,760 alleles in 8.3KJPN-SV, suggesting that microdeletions involving exon 1 should be carefully examined. In EYE345, an Alu insertion was detected by TEMP2; the IGV showed an abrupt decrease in the depth of RPGRIP1 exon 7, which was unclear from the WES data (Figure 3a). De novo assembly and breakpoint PCR confirmed an approximate 400 bp AluY insertion with an 18 bp target site duplication (Figure 3b,c), suggesting that transposition occurred via LINE-1. In summary, structural abnormalities were found in six cases from five families, including microdeletions involving exon 1 or exon 18 and an Alu insertion.

Figure 2.
Microdeletions involving RPGRIP1 exons. (a) The IGV view of a microdeletion involving exon 18. The deletion interval is highlighted in pink. (b) Breakpoint sequences in three patients showing an identical 1399 bp deletion. An asterisk indicates a sequence match between the upper and lower nucleotides. (c) The IGV view of microdeletions involving exon 1 (left) and results of breakpoint PCR (right). (d) Breakpoint sequences in three patients suggesting Alu-mediated recombination in three cases. Alu sequences are highlighted in gray. An asterisk indicates a sequence match between the upper and lower nucleotides.
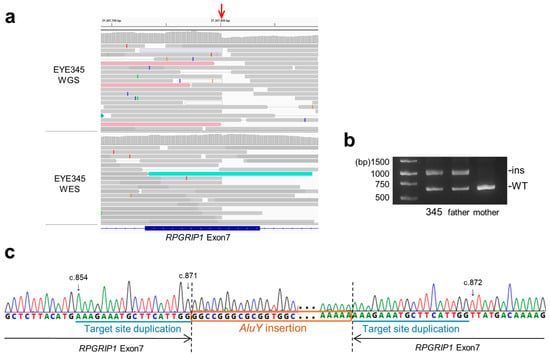
Figure 3.
Alu insertion in RPGRIP1 exon 7. (a) The IGV view shows an abrupt decrease (red arrow) in the depth of RPGRIP1 exon 7 in WGS data. However, such a decrease is subtle in WES data, suggesting that inserted DNA fragments were not efficiently captured. (b) Breakpoint PCR shows paternally transmitted insertion that is approximately 400 bp in size. (c) Breakpoint sequences show AluY insertion with 18 bp target site duplication.
2.2. Clinical Findings of Participants
Ten patients with biallelic pathogenic variants of RPGRIP1 ranged in age from 3 months to 50 years at their first visit and from 10 to 56 years at their last visit, with a follow-up period ranging from 6 to 20 years. Ophthalmological measurements of the patients, including best-collected visual acuity (BCVA), visual field, and electrophysiological findings, are summarized in Table 2.
Table 2.
Summary of ophthalmological measurements of patients with RPGRIP1 variants.
Decimal BCVA at the last visit varied from light perception to 0.2 (logMAR 0.7). The progression of the logMAR visual acuity in each case is shown in Figure 4. Although visual acuity improved a little in some cases until around age 12, visual acuity remained low and did not correlate with age. The visual field was unmeasurable in three cases, severely constricted at first examination in four cases (one of which progressed to unmeasurable), progressed from moderate to severe constriction in two cases, and mild constriction but relatively maintained in one case. Regarding ERG, all cases were non-recordable for the cone response at the first examination, but rod responses were reduced or subnormal in four cases and non-recordable in the remaining six cases. In the four cases in which the rod response remained, in three of the four cases the rod response disappeared on later re-examination, but in one case it remained. The retinal structure was evaluated for ellipsoid zone (EZ) length and central foveal retinal thickness. Figure 5a shows that the EZ length decreased with age (r = −0.728, p < 0.01) in 44 examinations of 18 eyes of nine patients, excluding one case with unimpaired EZ; however, it was maintained even in the case of the patient in their 50s. The central foveal retinal thickness of 19 eyes in 10 cases, excluding one eye with abnormal thickening due to traction by the epiretinal membrane, is shown in Figure 5b. Although the central retinal thickness varied from case to case, in individual cases, there seemed to be a trend of a slow decrease. The retinal structure was preserved over time relative to visual acuity in all cases.
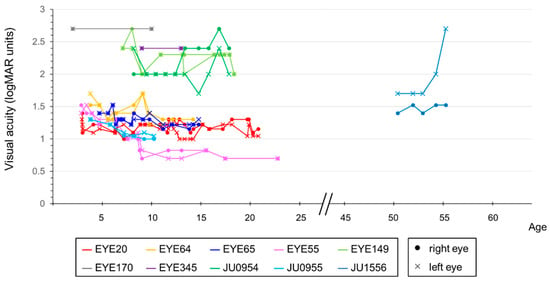
Figure 4.
Correlation between visual acuity and age in patients with RPGRIP1 variants. The graph compares the visual acuity values, in logMAR units, with age. Lines indicate data from the corresponding person and eye. Different colors are used to represent each person, with dots indicating data for the right eye and crosses indicating data for the left eye. Although visual acuity was variable and remained low, there was no significant correlation with age.
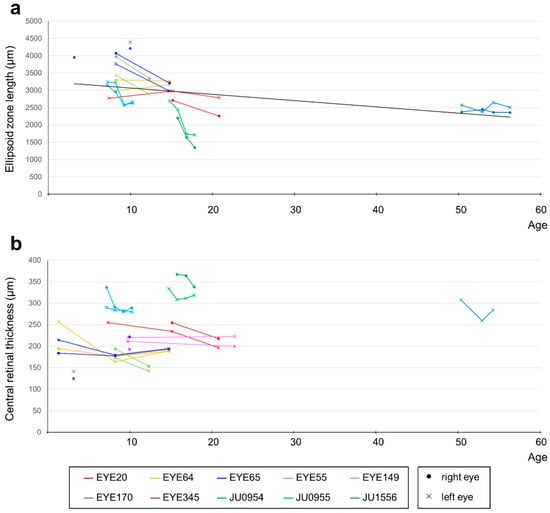
Figure 5.
Correlation between OCT findings and age in patients with RPGRIP1 variants. (a) The correlation between EZ length and age. Although EZ length showed a decreasing trend with age, it was maintained even after age 50. Different colors are used to represent each person, with dots indicating data for the right eye and crosses indicating data for the left eye. EZs that were maintained over the entire length of the imaging range were not plotted (EYE55 and parts of EYE64 and EYE65). (b) The correlation between central foveal retinal thickness and age. The right eye at all visits and the left eye at last visit of JU1556 were excluded because of abnormal thickening due to the epiretinal membrane. Although central retinal thickness varied from case to case, there seemed to be a trend of slow decrease with age in individual cases.
Two representative examples are provided: Figure 6a shows the case of a 17-year-old female (JU0954). At nine months, the patient was brought to a nearby university hospital with a chief complaint of nystagmus and was diagnosed with LCA. At 14 years old, the patient visited Jikei University School of Medicine. The patient’s visual acuity remained low thereafter, and, at the patient’s last visit at age 17, her corrected visual acuity was hand-motion vision in the right eye and 0.01 in the left eye. Figure 6b shows the case of a 54-year-old female (JU1556). Six months after birth, the parents of the patient noticed no pursuit with her eyes and she was subsequently diagnosed with IRD at another university hospital. The patient was followed-up regularly until age 21. The patient had a maximum visual acuity of 0.07 from age 6 to 12 and 0.1 from age 13 to 21. In elementary school, the patient attended a school for blind students. The patient had no history of visiting an ophthalmologist between the ages of 21 and 45. Around the age of 45 years, the patient visited an ophthalmologist at a local doctor’s office with the chief complaint of progressive night blindness. At 50, the patient was referred to Jikei University School of Medicine for a detailed examination. At her last visit, visual acuity was 0.03 in her right eye and hand-motion vision in her left eye. Additionally, epiretinal membrane formation was observed in her right eye at the first visit and in the left eye at the last visit. In both cases, JU0954 and JU1556, ERG was absent in both cones and rods.
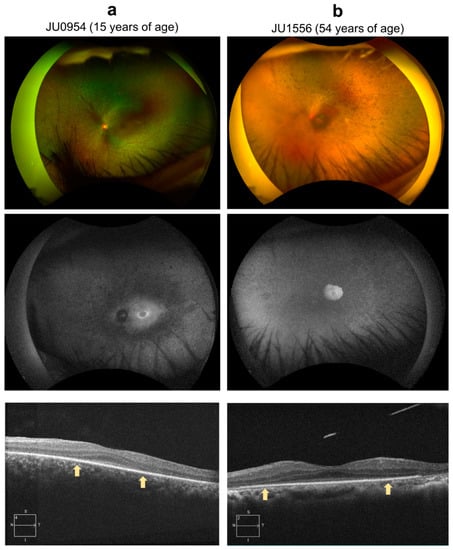
Figure 6.
Color fundus photography, fundus autofluorescence (FAF), and optic coherence tomography (OCT). (a) The left eye of case JU0954, a 15-year-old female at the time of examination. Fundus examination showed narrowing of retinal vessels, diffuse retinal degeneration, and bone-spicule pigmentation; FAF showed a hyperfluorescent ring around the fovea, and hypofluorescence in the midperiphery where retinal degeneration was shown in fundus photograph; and OCT showed thinning of the outer nuclear layer and disruption of the peripheral EZ (outside of arrows). However, the central foveal retinal thickness was preserved. (b) The left eye of case JU1556, a 54-year-old female. Fundus examination shows almost complete obliteration of retinal vessels and extensive pigmentations with superior retinal dominance, which were consistent with a peripheral visual field defect; FAF showed hyperfluorescence in the macula area surrounded by multiple hypofluorescent spots from the arcade to periphery; and OCT showed thinning of the outer nuclear layer and disruption of the peripheral EZ (outside of arrows). However, central foveal retinal thickness was maintained.
3. Discussion
This study successfully identified four copy number variations (CNVs) in four cases from three families using WGS (excluding the previously reported EYE55 with an exon 18 deletion). These CNVs involve only one to three exons. Therefore, detecting these CNVs using genome-wide CNV analysis of WES data may be difficult. To examine whether target CNV analysis for known disease-causing genes could detect these CNVs, we retrospectively examined CNV in RPGRIP1 using jNord []. Microdeletions involving only exon 1 were not detected (JU0954, JU0955, and EYE170), as there were no baits on exon 1 (Figure 1b and Supplementary Figure S1). In contrast, a microdeletion involving exons 1–3 was detected as a deletion of exon 3 (JU1556). Microdeletions involving exon 18 were detected in two out of three cases. Therefore, these findings strongly support the idea that WES-based CNV analysis increases diagnostic yield; however, WGS is more reliable for CNV/SV analysis. A recent WES analysis of 1210 Japanese pedigree patients with inherited retinal diseases revealed no pathogenic variants of RPGRIP1 []. CNVs involving RPGRIP1 may be one of the genetic causes of IRD that were not detected by the WES-based analysis. There have been reports of the detection of non-coding variants such as CNVs and deep intron variants of RPGRIP1 from cases unresolved via WES using WGS, indicating that non-coding variants may contribute significantly to RPGRIP1-related retinal dystrophy [,,,].
The causative genes of LCA, occurring at a frequency of approximately ≥10%, include GUCY2D, RPE65, CRB1, CEP290, and RDH12 [,,,]. Previous reports have shown that the frequency of the causative genes varies according to the patient’s ethnic background [,]. Although CEP290 is a common causative gene in patients of European ancestry, it was reported not to be as high in a large-scale study of patients of Chinese ancestry. In contrast, RPGRIP1 is a more common causative gene in patients of Chinese ancestry than those of European ancestry [,,]. In our previous study employing 34 Japanese families with LCA, most of the causative genes accounted for only one pedigree, except CRB1 (three families), NMNAT1 (three families), RPGRIP1 (three families), and GUCY2D (two families) []. Although it is difficult to demonstrate the accurate frequency of RPGRIP1 mutations in Japanese families with LCA because of the small number of cases and bias in the patient collection, we believe that RPGRIP1 is possibly a common causative gene in Japanese patients with LCA.
Common pathogenic variants are crucial for genetic counseling of recessive diseases. A previous report showed that c.1107del (25 patients) and c. 2480G>T (12 patients) mutations were frequently isolated from patients with LCA6 []. The c.1107del mutation was prevalent in Saudi Arabia [,,,]. Consistent with a previous report, an exon 18 deletion in RPGRIP1 was detected in six individuals from four families in Japan [,]. An exon 18 deletion was also identified in a recently published database of Japanese cohorts. Because the allele frequency is relatively high at 0.002%, it is considered a variant that should be noted in genetic counseling for LCA. Although examination by population, including Chinese and Koreans, is necessary, it is considered important for genetic counseling of patients of East Asian origin with LCA.
Gene-specific clinical trials using viral vectors are actively being conducted. In particular, the success of gene therapy for RPE65 abnormalities is expected to be a breakthrough in this extremely serious disease [,,]. Based on the experience of the long-term use of adeno-associated virus (AAV) vectors in Europe and the United States, a certain degree of safety has been ensured for the administration of viral vectors. It is a promising treatment due to its efficacy [,]. Gene therapy using AAV vectors in RPGRIP1-related murine and canine models has also been investigated [,]. Although LCA is a serious disease, it generally progresses slowly, and the patient’s visual function fluctuates daily, making it difficult to determine the therapeutic effect. The detailed clinical features were analyzed herein as basic data for future genetic therapy.
All participants had likely null variants such as splice site variants, nonsense variants, frameshift variants, and microdeletions; thus, we expected to show clinical trends. However, the results for visual function were variable. In our study, the visual field and refraction varied; visual acuity remained low and did not noticeably decline with age; and ERGs showed a loss of cone response and a decrease or loss of rod response in all cases.
The three siblings EYE20, 64, and 65 had marked nystagmus and photophobia from infancy and we diagnosed them with LCA. An ERG performed in the twin brothers at 15 months of age showed detectable rod responses []. However, we thought, since the twin brothers were under general anesthesia and at the young age of 1 year and 3 months, we could detect a significant response. When we re-examined them 13 years later, the rod responses were absent in all three patients. Biallelic variants of RPGRIP1 are known to cause RP, CRD, and PPRCA in addition to LCA, and cases of isolated cone dysfunction were recently reported []. There have been reports of severe retinal degeneration with reduced but measurable rod response when examined at very young ages, as in our cases, suggesting that more severely affected cones than rods are characteristic of RPGRIP1 [,].
Patient EYE55 first saw his previous doctor for marked nystagmus at age 7 months, but he was unable to undergo an ERG. He was followed up as LCA because his decimal BCVA did not increase beyond 0.1 until the age of 8 years. At age 9, ERG was performed under general anesthesia and the cone response was absent and rod response was decreased(subnormal) []. At age 20, the ERG was re-examined and the rod response remained. Other clinical findings were also better than in the other cases. Patient EYE55 was initially diagnosed with LCA, but long-term follow-up suggested a form of the disease similar to CRD.
The optical coherence tomography (OCT) findings of RPGRIP1-related retinal dystrophy confirmed that although EZ length and retinal thickness tended to decrease gradually, retinal structures were relatively preserved, and visual acuities were low but maintained, as in previous reports [,,,]. Interestingly, the retinal structure of the EZ was relatively preserved in a patient in her 50s with LCA. Based on an examination of the clinical picture, gene therapy, which greatly improves visual function in childhood, is promising.
A limitation of this study was the small number of cases, as LCA is an extremely rare disease. Regarding the examination of clinical features, there was a limit to the accuracy of ophthalmic examinations because most of the recruited patients were pediatric.
In summary, WGS is useful for detecting structural abnormalities and played a key role in the cohort examined in this study. Structural variants may be important genetic causes of RPGRIP1-related retinal dystrophy in Japanese patients. It is expected that the use of WGS will become mainstream in the future. Deletion of exon 18 is relatively common in the Japanese population, and genetic counseling for childhood IRDs requires careful attention.
4. Materials and Methods
4.1. Patient Recruitment
We performed WES on 29 participants from 28 families with clinically suspected LCA without systemic symptoms other than psychomotor delays. Among the 29 cases, 11 were newly recruited and 18 were unresolved cases from the previous target sequencing study. We identified causative variants in 12 cases using WES. We performed WGS for 14 unresolved cases. Among the sibling cases (JU0954 and 0955), we performed WGS in only one case (JU0955). The DNA samples in the remaining two cases were insufficient for WGS. Clinical data were collected from 10 Japanese patients with identified pathogenic variants of RPGRIP1 who visited the National Center for Child Health and Development (NCCHD) and Jikei University School of Medicine.
4.2. WES and WGS
For WES, genomic DNA was captured using a SureSelect Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a NextSeq500 (Illumina, San Diego, CA, USA) with 151 bp paired-end reads. Data processing, variant calling, annotation, and filtering were performed as previously described []. Retrospective WES-based CNV analysis was performed using jNord []. WGS was commissioned by BGI Japan Corp. (Kobe, Japan). Sequencing was performed using DNBSEQ (MGI TECH, Shenzhen, China) with 150 bp paired-end reads. Data processing, variant calling, and variant annotation were performed as previously described []. CNV, SV, and transposon detection were performed using Canvas v1.40.0 [], Manta v1.6.0 [], and TEMP2 v0.1.4 [], respectively. Alignments were visualized using the IGV v2.16.0 (downloaded from https://igv.org/). De novo assembly using aberrantly aligned read pairs, such as clipped reads, was performed using Megahit v1.2.9 []. The allele frequency of structural variation was examined using gnomAD SVs v2.1 (https://gnomad.broadinstitute.org/; accessed on 13 January 2023), 8.3KJPN-SV, based on short-read whole-genome sequencing of 8300 Japanese individuals, and JSV1, based on long-read whole-genome sequencing of 222 Japanese individuals (https://jmorp.megabank.tohoku.ac.jp/; accessed on 13 January 2023) [].
4.3. Clinical Examination
The affected participants were examined at the NCCHD and Jikei University School of Medicine and underwent ophthalmic examinations, including BCVA, refraction measurement, kinetic visual fields on the Goldmann perimeter, and full-field ERG. Additionally, slit-lamp biomicroscopy, ophthalmoscopy after pupillary dilation, fundus photography, fundus autofluorescence (FAF) (Optos California, Optos Plc, Dunfermline, UK), and spectral-domain OCT (Cirrus HD-OCT 5000, Carl Zeiss Meditec AG, Dublin, CA, USA) or swept-source OCT (DRI OCT-1, Topcon Corporation, Tokyo, Japan) were performed. Information on family history was obtained through interviews with participants or their family members. BCVA was measured as decimal visual acuity using a Landolt C chart and converted to logMAR units for analysis. A picture chart was used for patients whose condition was difficult to examine using the Landolt C-chart. Visual acuity values for counting fingers, hand motion, and light perception were extrapolated to logMAR 2.0, 2.4, and 2.7, respectively []. The refractive error was recorded as a spherical equivalent and analyzed for each participant. BCVA and OCT were examined on multiple occasions in all affected subjects, except for OCT in cases EYE170 and 345, and phenotypes were analyzed by plotting BCVA versus age, central foveal retinal thickness versus age, and EZ length versus age. All statistical analyses were performed using SPSS version 25 (IBM Corp., Armonk, NY, USA). Spearman’s rank correlation coefficient was used for simple correlation tests. Statistical significance was set at p < 0.05.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms241813678/s1.
Author Contributions
Conceptualization, K.T., S.N., N.A., T.H. and Y.H.; Methodology, H.S. and Y.H.; Formal Analysis, K.T., M.T., K.K. and H.S.; Investigation, K.T., S.N., H.M., K.M., M.T., N.T., A.H., M.S., T.H. and H.S.; Resources, S.N., H.M., K.M. and T.H.; Writing—Original Draft Preparation, K.T., H.S. and Y.H.; Writing—Review and Editing, S.N., T.N., M.F., N.A., T.H., H.S. and Y.H.; Supervision, Y.H.; Funding Acquisition, S.N., M.F., T.H. and Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (C), grant number JP20K09825 to Y.H., 21K09756 to T.H., 23K09041 to K.T.; a grant from the Initiative on Rare and Undiagnosed Diseases, No. JP23ek0109549 to Y.H. from the Japan Agency for Medical Research and Development (AMED); the Health and Labour Sciences Research Grant of Research on Intractable Diseases, No. 23FC1055 to S.N.; and the National Center for Child Health and Development grant, No. 2022A-1 to M.F. and S.N.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board for Human Genetic and Genome Research at the Hamamatsu University School of Medicine (permit no. 14-040), the National Centre for Child Health and Development (permit no. 518), and Jikei University School of Medicine (permit no. 24-232 6997).
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors thank the participants of this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Koenekoop, R.K. An overview of Leber congenital amaurosis: A model to understand human retinal development. Surv. Ophthalmol. 2004, 49, 379–398. [Google Scholar] [CrossRef] [PubMed]
- Schappert-Kimmijser, J.; Henkes, H.E.; Van Den Bosch, J. Amaurosis congenita (Leber). AMA Arch. Ophthalmol. 1959, 61, 211–218. [Google Scholar] [CrossRef] [PubMed]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Huang, C.H.; Yang, C.M.; Yang, C.H.; Hou, Y.C.; Chen, T.C. Leber’s Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes 2021, 12, 1261. [Google Scholar] [CrossRef]
- Dryja, T.P.; Adams, S.M.; Grimsby, J.L.; McGee, T.L.; Hong, D.H.; Li, T.; Andréasson, S.; Berson, E.L. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet. 2001, 68, 1295–1298. [Google Scholar] [CrossRef]
- Jamshidi, F.; Place, E.M.; Mehrotra, S.; Navarro-Gomez, D.; Maher, M.; Branham, K.E.; Valkanas, E.; Cherry, T.J.; Lek, M.; MacArthur, D.; et al. Contribution of noncoding pathogenic variants to RPGRIP1-mediated inherited retinal degeneration. Genet. Med. 2019, 21, 694–704. [Google Scholar] [CrossRef]
- Beryozkin, A.; Aweidah, H.; Carrero Valenzuela, R.D.; Berman, M.; Iguzquiza, O.; Cremers, F.P.M.; Khan, M.I.; Swaroop, A.; Amer, R.; Khateb, S.; et al. Retinal Degeneration Associated with RPGRIP1: A Review of Natural History, Mutation Spectrum, and Genotype-Phenotype Correlation in 228 Patients. Front. Cell Dev. Biol. 2021, 9, 746781. [Google Scholar] [CrossRef]
- Zou, G.; Zhang, T.; Cheng, X.; Igelman, A.D.; Wang, J.; Qian, X.; Fu, S.; Wang, K.; Koenekoop, R.K.; Fishman, G.A.; et al. Noncoding mutation in RPGRIP1 contributes to inherited retinal degenerations. Mol. Vis. 2021, 27, 95–106. [Google Scholar]
- Mao, Y.; Long, Y.; Liu, B.; Cao, Q.; Li, Y.; Li, S.; Wang, G.; Meng, X.; Li, S. Ocular Characteristics of Patients with Leber Congenital Amaurosis 6 Caused by Pathogenic RPGRIP1 Gene Variation in a Chinese Cohort. J. Ophthalmol. 2021, 2021, 9966427. [Google Scholar] [CrossRef]
- Perrault, I.; Hanein, S.; Gerard, X.; Mounguengue, N.; Bouyakoub, R.; Zarhrate, M.; Fourrage, C.; Jabot-Hanin, F.; Bocquet, B.; Meunier, I.; et al. Whole Locus Sequencing Identifies a Prevalent Founder Deep Intronic RPGRIP1 Pathologic Variant in the French Leber Congenital Amaurosis Cohort. Genes 2021, 12, 287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, H.; He, X.; Tao, D.; Li, L. Identifying two pathogenic variants in a patient with pigmented paravenous retinochoroidal atrophy. Open Life Sci. 2023, 18, 20220532. [Google Scholar] [CrossRef]
- Li, T. Leber congenital amaurosis caused by mutations in RPGRIP1. Cold Spring Harb. Perspect. Med. 2014, 5, a017384. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Gifford, E.; Smith, R.S.; Yi, H.; Ferreira, P.A.; Hicks, W.L.; Li, T.; Naggert, J.K.; Nishina, P.M. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum. Mol. Genet. 2009, 18, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Fadaie, Z.; Whelan, L.; Ben-Yosef, T.; Dockery, A.; Corradi, Z.; Gilissen, C.; Haer-Wigman, L.; Corominas, J.; Astuti, G.D.N.; de Rooij, L.; et al. Whole genome sequencing and in vitro splice assays reveal genetic causes for inherited retinal diseases. NPJ Genom. Med. 2021, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Hussain, H.M.J.; Wang, M.; Huang, A.; Schmidt, R.; Qian, X.; Yang, P.; Marra, M.; Li, Y.; Pennesi, M.E.; Chen, R. Novel Pathogenic Mutations Identified from Whole-Genome Sequencing in Unsolved Cases of Patients Affected with Inherited Retinal Diseases. Genes 2023, 14, 447. [Google Scholar] [CrossRef]
- Wen, S.; Wang, M.; Qian, X.; Li, Y.; Wang, K.; Choi, J.; Pennesi, M.E.; Yang, P.; Marra, M.; Koenekoop, R.K.; et al. Systematic assessment of the contribution of structural variants to inherited retinal diseases. Hum. Mol. Genet. 2023, 32, 2005–2015. [Google Scholar] [CrossRef]
- Hosono, K.; Nishina, S.; Yokoi, T.; Katagiri, S.; Saitsu, H.; Kurata, K.; Miyamichi, D.; Hikoya, A.; Mizobuchi, K.; Nakano, T.; et al. Molecular Diagnosis of 34 Japanese Families with Leber Congenital Amaurosis Using Targeted Next Generation Sequencing. Sci. Rep. 2018, 8, 8279. [Google Scholar] [CrossRef]
- Miyamichi, D.; Nishina, S.; Hosono, K.; Yokoi, T.; Kurata, K.; Sato, M.; Hotta, Y.; Azuma, N. Retinal structure in Leber’s congenital amaurosis caused by RPGRIP1 mutations. Hum. Genome Var. 2019, 6, 32. [Google Scholar] [CrossRef]
- Roller, E.; Ivakhno, S.; Lee, S.; Royce, T.; Tanner, S. Canvas: Versatile and scalable detection of copy number variants. Bioinformatics 2016, 32, 2375–2377. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Yu, T.; Huang, X.; Dou, S.; Tang, X.; Luo, S.; Theurkauf, W.E.; Lu, J.; Weng, Z. A benchmark and an algorithm for detecting germline transposon insertions and measuring de novo transposon insertion frequencies. Nucleic Acids Res. 2021, 49, e44. [Google Scholar] [CrossRef]
- Seong, M.W.; Kim, S.Y.; Yu, Y.S.; Hwang, J.M.; Kim, J.Y.; Park, S.S. Molecular characterization of Leber congenital amaurosis in Koreans. Mol. Vis. 2008, 14, 1429–1436. [Google Scholar]
- Suzuki, T.; Fujimaki, T.; Yanagawa, A.; Arai, E.; Fujiki, K.; Wada, Y.; Murakami, A. A novel exon 17 deletion mutation of RPGRIP1 gene in two siblings with Leber congenital amaurosis. Jpn. J. Ophthalmol. 2014, 58, 528–535. [Google Scholar] [CrossRef]
- Li, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Guo, X.; Jiao, X.; Zhang, Q.; Hejtmancik, J.F. Detection of variants in 15 genes in 87 unrelated Chinese patients with Leber congenital amaurosis. PLoS ONE 2011, 6, e19458. [Google Scholar] [CrossRef]
- Uchiyama, Y.; Yamaguchi, D.; Iwama, K.; Miyatake, S.; Hamanaka, K.; Tsuchida, N.; Aoi, H.; Azuma, Y.; Itai, T.; Saida, K.; et al. Efficient detection of copy-number variations using exome data: Batch- and sex-based analyses. Hum. Mutat. 2021, 42, 50–65. [Google Scholar] [CrossRef]
- Suga, A.; Yoshitake, K.; Minematsu, N.; Tsunoda, K.; Fujinami, K.; Miyake, Y.; Kuniyoshi, K.; Hayashi, T.; Mizobuchi, K.; Ueno, S.; et al. Genetic characterization of 1210 Japanese pedigrees with inherited retinal diseases by whole-exome sequencing. Hum. Mutat. 2022, 43, 2251–2264. [Google Scholar] [CrossRef]
- Sweeney, M.O.; McGee, T.L.; Berson, E.L.; Dryja, T.P. Low prevalence of lecithin retinol acyltransferase mutations in patients with Leber congenital amaurosis and autosomal recessive retinitis pigmentosa. Mol. Vis. 2007, 13, 588–593. [Google Scholar]
- Wang, H.; Wang, X.; Zou, X.; Xu, S.; Li, H.; Soens, Z.T.; Wang, K.; Li, Y.; Dong, F.; Chen, R.; et al. Comprehensive Molecular Diagnosis of a Large Chinese Leber Congenital Amaurosis Cohort. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3642–3655. [Google Scholar] [CrossRef]
- Coppieters, F.; Casteels, I.; Meire, F.; De Jaegere, S.; Hooghe, S.; van Regemorter, N.; Van Esch, H.; Matuleviciene, A.; Nunes, L.; Meersschaut, V.; et al. Genetic screening of LCA in Belgium: Predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum. Mutat. 2010, 31, E1709–E1766. [Google Scholar] [CrossRef]
- Astuti, G.D.; Bertelsen, M.; Preising, M.N.; Ajmal, M.; Lorenz, B.; Faradz, S.M.; Qamar, R.; Collin, R.W.; Rosenberg, T.; Cremers, F.P. Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. Eur. J. Hum. Genet. 2016, 24, 1071–1079. [Google Scholar] [CrossRef]
- Khan, A.O.; Abu-Safieh, L.; Eisenberger, T.; Bolz, H.J.; Alkuraya, F.S. The RPGRIP1-related retinal phenotype in children. Br. J. Ophthalmol. 2013, 97, 760–764. [Google Scholar] [CrossRef]
- Khan, A.O.; Al-Mesfer, S.; Al-Turkmani, S.; Bergmann, C.; Bolz, H.J. Genetic analysis of strictly defined Leber congenital amaurosis with (and without) neurodevelopmental delay. Br. J. Ophthalmol. 2014, 98, 1724–1728. [Google Scholar] [CrossRef]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef]
- Weleber, R.G.; Pennesi, M.E.; Wilson, D.J.; Kaushal, S.; Erker, L.R.; Jensen, L.; McBride, M.T.; Flotte, T.R.; Humphries, M.; Calcedo, R.; et al. Results at 2 Years after Gene Therapy for RPE65-Deficient Leber Congenital Amaurosis and Severe Early-Childhood-Onset Retinal Dystrophy. Ophthalmology 2016, 123, 1606–1620. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef]
- Lorenz, B.; Tavares, J.; van den Born, L.I.; Marques, J.P.; Pilotto, E.; Stingl, K.; Charbel Issa, P.; Leroux, D.; Dollfus, H.; Scholl, H.P.N. Current Management of Inherited Retinal Degeneration Patients in Europe: Results of a 2-Year Follow-Up Multinational Survey by the European Vision Institute Clinical Research Network—EVICR.net. Ophthalmic Res. 2023, 66, 550–568. [Google Scholar] [CrossRef]
- Timmers, A.M.; Newmark, J.A.; Turunen, H.T.; Farivar, T.; Liu, J.; Song, C.; Ye, G.J.; Pennock, S.; Gaskin, C.; Knop, D.R.; et al. Ocular Inflammatory Response to Intravitreal Injection of Adeno-Associated Virus Vector: Relative Contribution of Genome and Capsid. Hum. Gene Ther. 2020, 31, 80–89. [Google Scholar] [CrossRef]
- Bucher, K.; Rodríguez-Bocanegra, E.; Dauletbekov, D.; Fischer, M.D. Immune responses to retinal gene therapy using adeno-associated viral vectors—Implications for treatment success and safety. Prog. Retin. Eye Res. 2021, 83, 100915. [Google Scholar] [CrossRef]
- Pawlyk, B.S.; Bulgakov, O.V.; Liu, X.; Xu, X.; Adamian, M.; Sun, X.; Khani, S.C.; Berson, E.L.; Sandberg, M.A.; Li, T. Replacement gene therapy with a human RPGRIP1 sequence slows photoreceptor degeneration in a murine model of Leber congenital amaurosis. Hum. Gene Ther. 2010, 21, 993–1004. [Google Scholar] [CrossRef]
- Lhériteau, E.; Petit, L.; Weber, M.; Le Meur, G.; Deschamps, J.Y.; Libeau, L.; Mendes-Madeira, A.; Guihal, C.; François, A.; Guyon, R.; et al. Successful gene therapy in the RPGRIP1-deficient dog: A large model of cone-rod dystrophy. Mol. Ther. 2014, 22, 265–277. [Google Scholar] [CrossRef]
- Khan, A.O. RPGRIP1-related retinal disease presenting as isolated cone dysfunction. Ophthalmic Genet. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Tachibana, N.; Hosono, K.; Nomura, S.; Arai, S.; Torii, K.; Kurata, K.; Sato, M.; Shimakawa, S.; Azuma, N.; Ogata, T.; et al. Maternal Uniparental Isodisomy of Chromosome 4 and 8 in Patients with Retinal Dystrophy: SRD5A3-Congenital Disorders of Glycosylation and RP1-Related Retinitis Pigmentosa. Genes 2022, 13, 359. [Google Scholar] [CrossRef]
- Hiraide, T.; Shimizu, K.; Miyamoto, S.; Aoto, K.; Nakashima, M.; Yamaguchi, T.; Kosho, T.; Ogata, T.; Saitsu, H. Genome sequencing and RNA sequencing of urinary cells reveal an intronic FBN1 variant causing aberrant splicing. J. Hum. Genet. 2022, 67, 387–392. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Tadaka, S.; Hishinuma, E.; Komaki, S.; Motoike, I.N.; Kawashima, J.; Saigusa, D.; Inoue, J.; Takayama, J.; Okamura, Y.; Aoki, Y.; et al. jMorp updates in 2020: Large enhancement of multi-omics data resources on the general Japanese population. Nucleic Acids Res. 2021, 49, D536–D544. [Google Scholar] [CrossRef]
- Lange, C.; Feltgen, N.; Junker, B.; Schulze-Bonsel, K.; Bach, M. Resolving the clinical acuity categories “hand motion” and “counting fingers” using the Freiburg Visual Acuity Test (FrACT). Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 137–142. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).