
Mitochondrial Factors in the Cell Nucleus



Abstract
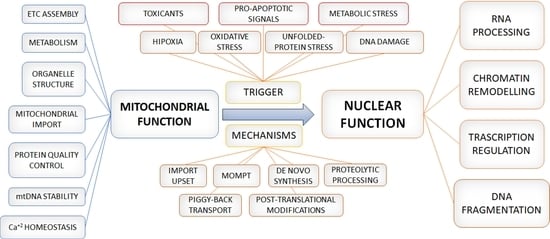
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mito-Nuclear Crosstalk: An Outcome of Evolution
3. From Crosstalk to Integral Signalling
3.1. Adaptation to Oxygen Levels and Mitochondrial Regulation of Nuclear-Gene Transcription
3.1.1. Response Mediated by Cytoplasmic and Nuclear Factors: HIF1α
3.1.2. Mitochondrial Factors and Responses to Oxygen Levels
3.2. Dealing with Oxidative Stress
3.3. The Mitochondrial Unfolded-Protein Stress Response Targets Nuclear Genes
3.4. Mitochondrial Factors in the Response to Nuclear DNA Damage
3.4.1. AIF and DNA damage
3.4.2. ENDOG
3.4.3. Fumarase
3.4.4. Hexokinase 2
3.4.5. HIGD1A and DNA Damage
3.4.6. B-Cell Lymphoma 2 Proteins and DDR
3.4.7. Cytochrome c
3.4.8. Dynamin-Related Protein 1
3.4.9. CR6-Interacting Factor 1
3.5. Mitochondrial Metabolites and Proteins Affecting Chromatin Remodelling
3.5.1. Mitochondrial Metabolites and Chromatin Condensation State
α-Ketoglutarate, Succinate, and Fumarateple
Flavin Adenine Dinucleotide
3.5.2. Mitochondrial Proteins and Chromatin Remodelling
Mitochondrial Matrix Dehydrogenases
Transcription Factor A
Clock 1/Coenzyme Q7 Homolog
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef]
- Maier, K.P.; Helbig, C.; Hoppe-Seyler, G.; Talke, H.; Fröhlich, J.; Schollmeyer, P.; Gerok, W. Extractability and intracellular localisation of urea cycle enzym es from rat liver. Clin. Chem. Lab. Med. 1974, 12, 524–529. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sano, S.; Inoue, S.; Tanabe, Y.; Sumiya, C.; Koike, S. Significance of mitochondria for porphyrin and heme biosynthesis. Science 1959, 129, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Muhlenhoff, U. Iron-sulfur-protein biogenesis in eukaryotes. Trends Biochem. Sci. 2005, 30, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Mitochondrial specificity of the early steps in steroidogenesis. J. Steroid. Biochem. Mol. Biol. 1995, 55, 607–616. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-carbon metabolism in health and disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Jones, M.E. Pyrimidine nucleotide biosynthesis in animals: Genes, enzymes, and regulation of ump biosynthesis. Annu. Rev. Biochem. Annu. Rev. 1980, 49, 253–279. [Google Scholar] [CrossRef]
- Warburg, O. Über den stoffwechsel der carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Nicholls, D.G. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem. J. 1978, 176, 463–474. [Google Scholar] [CrossRef]
- Kretsinger, R.H. Calcium-binding proteins. Annu. Rev. Biochem. 1976, 45, 239–266. [Google Scholar] [CrossRef]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Zamzami, N.; Castedo, M.; Hirsch, T.; Marchetti, P.; Macho, A.; Daugas, E.; Geuskens, M.; Kroemer, G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996, 184, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Hirsch, T.; Zamzami, N.; Castedo, M.; Decaudin, D.; Susin, S.A.; Masse, B.; Kroemer, G. Mitochondrial permeability transition triggers lymphocyte apoptosis. J. Immunol. 1996, 157, 4830–4836. [Google Scholar] [CrossRef]
- Chandel, N.S. Evolution of mitochondria as signaling organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. Mitocarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shan, G. Mitochondria encoded non-coding RNAs in cell physiology. Front. Cell Dev. Biol. 2021, 9, 713729. [Google Scholar] [CrossRef]
- Kim, S.J.; Xiao, J.; Wan, J.; Cohen, P.; Yen, K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017, 595, 6613–6621. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The origin and diversification of mitochondria. Curr. Biol. CB 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Gabaldón, T.; Huynen, M.A. From endosymbiont to host-controlled organelle: The hijacking of mitochondrial protein synthesis and metabolism. PLoS Comput. Biol. 2007, 3, e219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, M. Phylogenomic reconstruction indicates mitochondrial ancestor was an energy parasite. PLoS ONE 2014, 9, e110685. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response—synchronizing genomes. Curr. Opin. Cell Biol. 2015, 33, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-coa and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. Atp-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Liu, Y.; Makarova, K.S.; Huang, W.-C.; Wolf, Y.I.; Nikolskaya, A.N.; Zhang, X.; Cai, M.; Zhang, C.-J.; Xu, W.; Luo, Z.; et al. Expanded diversity of asgard archaea and their relationships with eukaryotes. Nature 2021, 593, 553–557. [Google Scholar] [CrossRef]
- Takahashi, H.; McCaffery, J.M.; Irizarry, R.A.; Boeke, J.D. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell 2006, 23, 207–217. [Google Scholar] [CrossRef]
- Huang, K.-Y. Metabolic activity of the trench fever rickettsia, Rickettsia quintana. J. Bacteriol. 1967, 93, 853–859. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Hou, T.; Wang, X.; Ma, Q.; Cheng, H. Mitochondrial flashes: New insights into mitochondrial ROS signalling and beyond. J. Physiol. 2014, 592, 3703–3713. [Google Scholar] [CrossRef]
- Espinós, C.; Galindo, M.I.; García-Gimeno, M.A.; Ibáñez-Cabellos, J.S.; Martínez-Rubio, D.; Millán, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative stress, a crossroad between rare diseases and neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef]
- Diebold, L.P.; Gil, H.J.; Gao, P.; Martinez, C.A.; Weinberg, S.E.; Chandel, N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019, 1, 158–171. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as signaling organelles control mammalian stem cell fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef]
- Zhang, F.; Pracheil, T.; Thornton, J.; Liu, Z. Adenosine triphosphate (atp) is a candidate signaling molecule in the mitochondria-to-nucleus retrograde response pathway. Genes 2013, 4, 86–100. [Google Scholar] [CrossRef]
- Miravet-Verde, S.; Ferrar, T.; Espadas-Garcia, G.; Mazzolini, R.; Gharrab, A.; Sabido, E.; Serrano, L.; Lluch-Senar, M. Unraveling the hidden universe of small proteins in bacterial genomes. Mol. Syst. Biol. 2019, 15, e8290. [Google Scholar] [CrossRef]
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef]
- Jazwinski, S.M. The retrograde response: When mitochondrial quality control is not enough. Biochim. Et Biophys. Acta (BBA)–Mol. Cell Res. 2013, 1833, 400–409. [Google Scholar] [CrossRef]
- Liu, Z.; Spírek, M.; Thornton, J.; Butow, R.A. A novel degron-mediated degradation of the rtg pathway regulator, mks1p, by scfgrr1. Mol. Biol. Cell 2005, 16, 4893–4904. [Google Scholar] [CrossRef] [PubMed]
- Pray-Grant, M.G.; Schieltz, D.; McMahon, S.J.; Wood, J.M.; Kennedy, E.L.; Cook, R.G.; Workman, J.L.; Yates, J.R.; Grant, P.A. The novel slik histone acetyltransferase complex functions in the yeast retrograde response pathway. Mol. Cell. Biol. 2002, 22, 8774–8786. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca2+/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef]
- Sabbir, M.G.; Taylor, C.G.; Zahradka, P. CAMKK2 regulates mitochondrial function by controlling succinate dehydrogenase expression, post-translational modification, megacomplex assembly, and activity in a cell-type-specific manner. Cell Commun. Signal. 2021, 19, 98. [Google Scholar] [CrossRef]
- Cardamone, M.D.; Tanasa, B.; Cederquist, C.T.; Huang, J.; Mahdaviani, K.; Li, W.; Rosenfeld, M.G.; Liesa, M.; Perissi, V. Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation. Mol. Cell 2018, 69, 757–772.e7. [Google Scholar] [CrossRef]
- Venteclef, N.; Jakobsson, T.; Ehrlund, A.; Damdimopoulos, A.; Mikkonen, L.; Ellis, E.; Nilsson, L.-M.; Parini, P.; Jänne, O.A.; Gustafsson, J.-Å.; et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRβ in the hepatic acute phase response. Genes Dev. 2010, 24, 381–395. [Google Scholar] [CrossRef]
- Cederquist, C.T.; Lentucci, C.; Martinez-Calejman, C.; Hayashi, V.; Orofino, J.; Guertin, D.; Fried, S.K.; Lee, M.-J.; Cardamone, M.D.; Perissi, V. Systemic insulin sensitivity is regulated by GPS2 inhibition of AKT ubiquitination and activation in adipose tissue. Mol. Metab. 2017, 6, 125–137. [Google Scholar] [CrossRef]
- Jakobsson, T.; Venteclef, N.; Toresson, G.; Damdimopoulos, A.E.; Ehrlund, A.; Lou, X.; Sanyal, S.; Steffensen, K.R.; Gustafsson, J.-Å.; Treuter, E. GPS2 Is Required for Cholesterol Efflux by Triggering Histone Demethylation, LXR Recruitment, and Coregulator Assembly at the ABCG1 Locus. Mol. Cell 2009, 34, 510–518. [Google Scholar] [CrossRef]
- Burgener, A.-V.; Bantug, G.R.; Meyer, B.J.; Higgins, R.; Ghosh, A.; Bignucolo, O.; Ma, E.H.; Loeliger, J.; Unterstab, G.; Geigges, M.; et al. SDHA gain-of-function engages inflammatory mitochondrial retrograde signaling via KEAP1–Nrf2. Nat. Immunol. 2019, 20, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Kleine, T.; Leister, D. Retrograde signaling: Organelles go networking. Biochim. Et Biophys. Acta (BBA)–Bioenerg. 2016, 1857, 1313–1325. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Ali, M.; Boosi Narayana Rao, K.; Majumder, P.; Sarkar, R.; Mapa, K. Alterations in inter-organelle crosstalk and Ca2+ signaling through mitochondria during proteotoxic stresses. Mitochondrion 2021, 57, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Gohel, D.; Singh, R. Mitohormesis; Potential implications in neurodegenerative diseases. Mitochondrion 2021, 56, 40–46. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Meyer, J.N.; Hartman, J.H.; Mello, D.F. Mitochondrial Toxicity. Toxicol. Sci. 2018, 162, 15–23. [Google Scholar] [CrossRef]
- Ferrer, P.E.; Frederick, P.; Gulbis, J.M.; Dewson, G.; Kluck, R.M. Translocation of a Bak C-terminus mutant from cytosol to mitochondria to mediate cytochrome C release: Implications for Bak and Bax apoptotic function. PLoS ONE 2012, 7, e31510. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Wu, J.; Back, S.H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef]
- Kaufman, D.M.; Crowder, C.M. Mitochondrial Proteostatic Collapse Leads to Hypoxic Injury. Curr. Biol. 2015, 25, 2171–2176. [Google Scholar] [CrossRef]
- Peña, S.; Sherman, T.; Brookes, P.S.; Nehrke, K. The Mitochondrial Unfolded Protein Response Protects against Anoxia in Caenorhabditis elegans. PLoS ONE 2016, 11, e0159989. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213. [Google Scholar] [PubMed]
- Agnieszka, L.; Alicja, J.; Jozef, D. HIF-1 and HIF-2 transcription factors–similar but not identical. Mol. Cells. 05/31 Ed Korean Soc. Mol. Cell. Biol. 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y.-P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Briston, T.; Yang, J.; Ashcroft, M. HIF-1α localization with mitochondria. Cell Cycle 2011, 10, 4170–4171. [Google Scholar] [CrossRef]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: Potential role in the regulation of aging process. Cell. Mol. Life Sci. 2015, 72, 3897–3914. [Google Scholar] [CrossRef]
- Hirsilä, M.; Koivunen, P.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the Human Prolyl 4-Hydroxylases That Modify the Hypoxia-inducible Factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef]
- Semenza, G.L.; Agani, F.; Booth, G.; Forsythe, J.; Iyer, N.; Jiang, B.H.; Leung, S.; Roe, R.; Wiener, C.; Yu, A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997, 51, 553–555. [Google Scholar] [CrossRef]
- Pescador, N.; Cuevas, Y.; Naranjo, S.; Alcaide, M.; Villar, D.; Landázuri, M.O.; del Peso, L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005, 390, 189–197. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsilä, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef]
- Sang, N.; Fang, J.; Srinivas, V.; Leshchinsky, I.; Caro, J. Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1 alpha is governed by a von Hippel-Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol. Cell Biol. 2002, 22, 2984–2992. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Kasper, L.H.; Boussouar, F.; Boyd, K.; Xu, W.; Biesen, M.; Rehg, J.; Baudino, T.A.; Cleveland, J.L.; Brindle, P.K. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005, 24, 3846–3858. [Google Scholar] [CrossRef]
- Freedman, S.J.; Sun, Z.Y.; Poy, F.; Kung, A.L.; Livingston, D.M.; Wagner, G.; Eck, M.J. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc. Natl. Acad. Sci. USA 2002, 99, 5367–5372. [Google Scholar] [CrossRef]
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J. Biol. Chem. 2004, 279, 41966–41974. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2021, 50, D687–D692. [Google Scholar] [CrossRef]
- Makino, Y.; Uenishi, R.; Okamoto, K.; Isoe, T.; Hosono, O.; Tanaka, H.; Kanopka, A.; Poellinger, L.; Haneda, M.; Morimoto, C. Transcriptional Up-regulation of Inhibitory PAS Domain protein gene expression by hypoxia-inducible factor 1 (HIF-1): A negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. J. Biol. Chem. 2007, 282, 14073–14082. [Google Scholar] [CrossRef]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha Locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef]
- Bakker, W.J.; Harris, I.S.; Mak, T.W. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol. Cell 2007, 28, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 2017, 543, 447–451. [Google Scholar] [CrossRef]
- Peng, X.; Gao, H.; Xu, R.; Wang, H.; Mei, J.; Liu, C. The interplay between HIF-1α and noncoding RNAs in cancer. J. Exp. Clin. Cancer Res. 2020, 39, 27. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.N.; Chen, H.J.; Liu, J.Q.; Li, W.T. Long non-coding RNA DLEU1 promotes malignancy of breast cancer by acting as an indispensable coactivator for HIF-1α-induced transcription of CKAP2. Cell Death Dis. 2022, 13, 625. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 617–626. [Google Scholar] [CrossRef]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef]
- Hausinger, R.P. Fe(II)/α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Asano, Y.; Shintani, Y.; Aoyama, H.; Kioka, H.; Tsukamoto, O.; Hikita, M.; Shinzawa-Itoh, K.; Takafuji, K.; Higo, S.; et al. Higd1a is a positive regulator of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Vukotic, M.; Oeljeklaus, S.; Wiese, S.; Vogtle, F.N.; Meisinger, C.; Meyer, H.E.; Zieseniss, A.; Katschinski, D.M.; Jans, D.C.; Jakobs, S.; et al. Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. 2012, 15, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.; Garlich, J.; Stuart, R.A.; Ugalde, C.; Barrientos, A. Distinct Roles of Mitochondrial HIGD1A and HIGD2A in Respiratory Complex and Supercomplex Biogenesis. Cell Rep. 2020, 31, 107607. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.O.; Eshoo, M.W.; del Rio, G.; Rao, R.; Chen, D.; Simon, R.P.; Greenberg, D.A. cDNA microarray analysis of changes in gene expression induced by neuronal hypoxia in vitro. Neurochem. Res. 2002, 27, 1105–1112. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Y.; Chen, Y.; Chen, Y.; Gardner, P.; Steiner, D.F. Pancreatic beta cells lack a low glucose and O2-inducible mitochondrial protein that augments cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 10636–10641. [Google Scholar] [CrossRef]
- Kasper, L.H.; Brindle, P.K. Mammalian Gene Expression Program Resiliency: The Roles of Multiple Coactivator Mechanisms in Hypoxia–Responsive Transcription. Cell Cycle 2006, 5, 142–146. [Google Scholar] [CrossRef]
- Ameri, K.; Jahangiri, A.; Rajah, A.M.; Tormos, K.V.; Nagarajan, R.; Pekmezci, M.; Nguyen, V.; Wheeler, M.L.; Murphy, M.P.; Sanders, T.A.; et al. HIGD1A regulates oxygen consumption, ROS production, and AMPK activity during glucose deprivation to modulate cell survival and tumor growth. Cell Rep. 2015, 10, 891–899. [Google Scholar] [CrossRef]
- An, H.-J.; Shin, H.; Jo, S.-G.; Kim, Y.J.; Lee, J.-O.; Paik, S.-G.; Lee, H. The survival effect of mitochondrial Higd-1a is associated with suppression of cytochrome C release and prevention of caspase activation. Biochim. Et Biophys. Acta (BBA)–Mol. Cell Res. 2011, 1813, 2088–2098. [Google Scholar] [CrossRef][Green Version]
- Moreno-Beltran, B.; Guerra-Castellano, A.; Diaz-Quintana, A.; Del Conte, R.; Garcia-Maurino, S.M.; Diaz-Moreno, S.; Gonzalez-Arzola, K.; Santos-Ocana, C.; Velazquez-Campoy, A.; De la Rosa, M.A.; et al. Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48. Proc. Natl. Acad. Sci. USA 2017, 114, E3041–E3050. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Diaz-Quintana, A.; Perez-Mejias, G.; Elena-Real, C.A.; Gonzalez-Arzola, K.; Garcia-Maurino, S.M.; De la Rosa, M.A.; Diaz-Moreno, I. Oxidative stress is tightly regulated by cytochrome c phosphorylation and respirasome factors in mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 7955–7960. [Google Scholar] [CrossRef]
- Baughman, J.M.; Nilsson, R.; Gohil, V.M.; Arlow, D.H.; Gauhar, Z.; Mootha, V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009, 5, e1000590. [Google Scholar] [CrossRef]
- Modjtahedi, N.; Tokatlidis, K.; Dessen, P.; Kroemer, G. Mitochondrial proteins containing coiled-coil-helix-coiled-coil-helix (CHCH) domains in health and disease. Trends Biochem. Sci. 2016, 41, 245–260. [Google Scholar] [CrossRef]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Hüttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 32056–32065. [Google Scholar] [CrossRef]
- Aras, S.; Arrabi, H.; Purandare, N.; Hüttemann, M.; Kamholz, J.; Züchner, S.; Grossman, L.I. Abl2 kinase phosphorylates Bi-organellar regulator MNRR1 in mitochondria, stimulating respiration. Biochim. Et Biophys. Acta (BBA)–Mol. Cell Res. 2017, 1864, 440–448. [Google Scholar] [CrossRef]
- Huang, X.; Wu, B.P.; Nguyen, D.; Liu, Y.-T.; Marani, M.; Hench, J.; Bénit, P.; Kozjak-Pavlovic, V.; Rustin, P.; Frank, S.; et al. CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet. 2018, 27, 3881–3900. [Google Scholar] [CrossRef]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Hüttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef]
- Liu, W.; Duan, X.; Xu, L.; Shang, W.; Zhao, J.; Wang, L.; Li, J.-C.; Chen, C.-H.; Liu, J.-P.; Tong, C. Chchd2 regulates mitochondrial morphology by modulating the levels of Opa1. Cell Death Differ. 2020, 27, 2014–2029. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Liu, J.; Grossman, L.I. Transcription of mammalian cytochrome c oxidase subunit IV-2 is controlled by a novel conserved oxygen responsive element. FEBS J. 2007, 274, 5737–5748. [Google Scholar] [CrossRef]
- Ruan, Y.; Hu, J.; Che, Y.; Liu, Y.; Luo, Z.; Cheng, J.; Han, Q.; He, H.; Zhou, Q. CHCHD2 and CHCHD10 regulate mitochondrial dynamics and integrated stress response. Cell Death Dis. 2022, 13, 156. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef]
- Genin, E.C.; Bannwarth, S.; Lespinasse, F.; Ortega-Vila, B.; Fragaki, K.; Itoh, K.; Villa, E.; Lacas-Gervais, S.; Jokela, M.; Auranen, M.; et al. Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol. Dis. 2018, 119, 159–171. [Google Scholar] [CrossRef]
- Anderson, C.J.; Bredvik, K.; Burstein, S.R.; Davis, C.; Meadows, S.M.; Dash, J.; Case, L.; Milner, T.A.; Kawamata, H.; Zuberi, A.; et al. ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 2019, 138, 103–121. [Google Scholar] [CrossRef]
- Cavallaro, G. Genome-wide analysis of eukaryotic twin CX9C proteins. Mol. Biosyst. 2010, 6, 2459–2470. [Google Scholar] [CrossRef]
- Burstein, S.R.; Valsecchi, F.; Kawamata, H.; Bourens, M.; Zeng, R.; Zuberi, A.; Milner, T.A.; Cloonan, S.M.; Lutz, C.; Barrientos, A.; et al. In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet. 2018, 27, 160–177. [Google Scholar] [CrossRef]
- Pfanner, N.; van der Laan, M.; Amati, P.; Capaldi, R.A.; Caudy, A.A.; Chacinska, A.; Darshi, M.; Deckers, M.; Hoppins, S.; Icho, T.; et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 2014, 204, 1083–1086. [Google Scholar] [CrossRef]
- Woo, J.-A.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef]
- Purandare, N.; Somayajulu, M.; Hüttemann, M.; Grossman, L.I.; Aras, S. The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): Partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem. 2018, 293, 6517–6529. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef]
- Liu, T.; Woo, J.-A.A.; Bukhari, M.Z.; Wang, X.; Yan, Y.; Buosi, S.C.; Ermekbaeva, A.; Sista, A.; Kotsiviras, P.; LePochat, P.; et al. Modulation of synaptic plasticity, motor unit physiology, and TDP-43 pathology by CHCHD10. Acta Neuropathol. Commun. 2022, 10, 95. [Google Scholar] [CrossRef]
- Orrenius, S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab. Rev. 2007, 39, 443–455. [Google Scholar] [CrossRef]
- Go, Y.-M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef]
- Ischiropoulos, H. Protein tyrosine nitration-An update. Arch. Biochem. Biophys. 2009, 484, 117–121. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Ramallo Guevara, C.; Philipp, O.; Hamann, A.; Werner, A.; Osiewacz, H.D.; Rexroth, S.; Rögner, M.; Poetsch, A. Global Protein Oxidation Profiling Suggests Efficient Mitochondrial Proteome Homeostasis During Aging. Mol. Cell. Proteom. 2016, 15, 1692–1709. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, C.; Chai, J.; Shi, Y.; Xue, D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 2002, 298, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Gaume, B.; Karbowski, M.; Sharpe, J.C.; Cecconi, F.; Youle, R.J. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003, 22, 4385–4399. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prévost, M.C.; et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000, 192, 571–580. [Google Scholar] [CrossRef]
- Sevrioukova, I.F. Apoptosis-inducing factor: Structure, function, and redox regulation. Antioxid. Redox Signal. 2011, 14, 2545–2579. [Google Scholar] [CrossRef]
- Hisatomi, T.; Sakamoto, T.; Murata, T.; Yamanaka, I.; Oshima, Y.; Hata, Y.; Ishibashi, T.; Inomata, H.; Susin, S.A.; Kroemer, G. Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am. J. Pathol. 2001, 158, 1271–1278. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Graham, S.H.; Du, L.; Kochanek, P.M.; Draviam, R.; Guo, F.; Nathaniel, P.D.; Szabó, C.; Watkins, S.C.; et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J. Neurochem. 2002, 82, 181–191. [Google Scholar] [CrossRef]
- Plesnila, N.; Zhu, C.; Culmsee, C.; Gröger, M.; Moskowitz, M.A.; Blomgren, K. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2004, 24, 458–466. [Google Scholar] [CrossRef]
- Braun, J.S.; Novak, R.; Murray, P.J.; Eischen, C.M.; Susin, S.A.; Kroemer, G.; Halle, A.; Weber, J.R.; Tuomanen, E.I.; Cleveland, J.L. Apoptosis-inducing factor mediates microglial and neuronal apoptosis caused by pneumococcus. J. Infect. Dis. 2001, 184, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Cregan, S.P.; Fortin, A.; MacLaurin, J.G.; Callaghan, S.M.; Cecconi, F.; Yu, S.W.; Dawson, T.M.; Dawson, V.L.; Park, D.S.; Kroemer, G.; et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 2002, 158, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ohsakaya, S.; Nagaura, Z.-I.; Ishihara, N.; Mihara, K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005, 24, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Vahsen, N.; Candé, C.; Brière, J.J.; Bénit, P.; Joza, N.; Larochette, N.; Mastroberardino, P.G.; Pequignot, M.O.; Casares, N.; Lazar, V.; et al. AIF deficiency compromises oxidative phosphorylation. Embo J. 2004, 23, 4679–4689. [Google Scholar] [CrossRef]
- Apostolova, N.; Cervera, A.M.; Victor, V.M.; Cadenas, S.; Sanjuan-Pla, A.; Alvarez-Barrientos, A.; Esplugues, J.V.; McCreath, K.J. Loss of apoptosis-inducing factor leads to an increase in reactive oxygen species, and an impairment of respiration that can be reversed by antioxidants. Cell Death Differ. 2006, 13, 354–357. [Google Scholar] [CrossRef]
- Klein, J.A.; Longo-Guess, C.M.; Rossmann, M.P.; Seburn, K.L.; Hurd, R.E.; Frankel, W.N.; Bronson, R.T.; Ackerman, S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002, 419, 367–374. [Google Scholar] [CrossRef]
- Meyer, K.; Buettner, S.; Ghezzi, D.; Zeviani, M.; Bano, D.; Nicotera, P. Loss of apoptosis-inducing factor critically affects MIA40 function. Cell Death Dis. 2015, 6, e1814. [Google Scholar] [CrossRef]
- Bano, D.; Prehn, J.H.M. Apoptosis-inducing factor (AIF) in physiology and disease: The tale of a repented natural born killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef]
- Reinhardt, C.; Arena, G.; Nedara, K.; Edwards, R.; Brenner, C.; Tokatlidis, K.; Modjtahedi, N. AIF meets the CHCHD4/Mia40-dependent mitochondrial import pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165746. [Google Scholar] [CrossRef]
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol. 2002, 9, 680–684. [Google Scholar] [CrossRef]
- Candé, C.l.; Cecconi, F.; Dessen, P.; Kroemer, G. Apoptosis-inducing factor (AIF): Key to the conserved caspase-independent pathways of cell death? J. Cell Sci. 2002, 115, 4727–4734. [Google Scholar] [CrossRef] [PubMed]
- Artus, C.; Boujrad, H.; Bouharrour, A.; Brunelle, M.N.; Hoos, S.; Yuste, V.J.; Lenormand, P.; Rousselle, J.C.; Namane, A.; England, P.; et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. Embo J. 2010, 29, 1585–1599. [Google Scholar] [CrossRef] [PubMed]
- Candé, C.; Vahsen, N.; Kouranti, I.; Schmitt, E.; Daugas, E.; Spahr, C.; Luban, J.; Kroemer, R.T.; Giordanetto, F.; Garrido, C.; et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 2004, 23, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Seo, T.W.; Yi, J.H.; Shin, K.S.; Yoo, S.J. CHIP has a protective role against oxidative stress-induced cell death through specific regulation of Endonuclease G. Cell Death Dis. 2013, 4, e666. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, P.; Scholz, S.R.; Gimadutdinow, O.; Cymerman, I.A.; Bujnicki, J.M.; Ruiz-Carrillo, A.; Pingoud, A.; Meiss, G. Structural and Functional Characterization of Mitochondrial EndoG, a Sugar Non-specific Nuclease which Plays an Important Role During Apoptosis. J. Mol. Biol. 2004, 338, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Côté, J.; Ruiz-Carrillo, A. Primers for mitochondrial DNA replication generated by endonuclease G. Science 1993, 261, 765–769. [Google Scholar] [CrossRef]
- Wiehe, R.S.; Gole, B.; Chatre, L.; Walther, P.; Calzia, E.; Ricchetti, M.; Wiesmüller, L. Endonuclease G promotes mitochondrial genome cleavage and replication. Oncotarget 2018, 9, 18309–18326. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, H.; Li, H.; Nakagawa, A.; Lin, J.L.; Lee, E.S.; Harry, B.L.; Skeen-Gaar, R.R.; Suehiro, Y.; William, D.; et al. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science 2016, 353, 394–399. [Google Scholar] [CrossRef]
- David, K.K.; Sasaki, M.; Yu, S.W.; Dawson, T.M.; Dawson, V.L. EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ. 2006, 13, 1147–1155. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, J.; Altafaj, A.; Cardona, M.; Bahi, N.; Llovera, M.; Cañas, X.; Cook, S.A.; Comella, J.X.; Sanchis, D. EndoG Links Bnip3-Induced Mitochondrial Damage and Caspase-Independent DNA Fragmentation in Ischemic Cardiomyocytes. PLoS ONE 2011, 6, e17998. [Google Scholar] [CrossRef]
- van Loo, G.; Schotte, P.; van Gurp, M.; Demol, H.; Hoorelbeke, B.; Gevaert, K.; Rodriguez, I.; Ruiz-Carrillo, A.; Vandekerckhove, J.; Declercq, W.; et al. Endonuclease G: A mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001, 8, 1136–1142. [Google Scholar] [CrossRef]
- Choi, Y.N.; Seo, T.W.; Lee, Y.T.; Jeong, D.H.; Yoo, S.J. Nuclear endonuclease G controls cell proliferation in ovarian cancer. FEBS Open Bio 2023, 13, 655–669. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Tan, J.; Wang, M.; Yang, J.; Zhang, Z.M.; Li, C.; Basnakian, A.G.; Tang, H.W.; Perrimon, N.; et al. Endonuclease G promotes autophagy by suppressing mTOR signaling and activating the DNA damage response. Nat. Commun. 2021, 12, 476. [Google Scholar] [CrossRef]
- Pennington, K.L.; Chan, T.Y.; Torres, M.P.; Andersen, J.L. The dynamic and stress-adaptive signaling hub of 14-3-3: Emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene 2018, 37, 5587–5604. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Poloyac, S.M.; Tyurin, V.A.; Kapralov, A.A.; Jiang, J.; Anthonymuthu, T.S.; Kapralova, V.I.; Vikulina, A.S.; Jung, M.Y.; Epperly, M.W.; et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat. Chem. 2014, 6, 542–552. [Google Scholar] [CrossRef]
- Nietzel, T.; Mostertz, J.; Hochgräfe, F.; Schwarzländer, M. Redox regulation of mitochondrial proteins and proteomes by cysteine thiol switches. Mitochondrion 2017, 33, 72–83. [Google Scholar] [CrossRef]
- Wright, G.; Terada, K.; Yano, M.; Sergeev, I.; Mori, M. Oxidative Stress Inhibits the Mitochondrial Import of Preproteins and Leads to Their Degradation. Exp. Cell Res. 2001, 263, 107–117. [Google Scholar] [CrossRef]
- Wright, G.; Reichenbecher, V.; Green, T.; Wright, G.L.; Wang, S. Paraquat Inhibits the Processing of Human Manganese-Dependent Superoxide Dismutase by SF-9 Insect Cell Mitochondria. Exp. Cell Res. 1997, 234, 78–84. [Google Scholar] [CrossRef]
- von Stedingk, E.M.; Pavlov, P.F.; Grinkevich, V.A.; Glaser, E. Mitochondrial protein import: Modification of sulfhydryl groups of the inner mitochondrial membrane import machinery in Solanum tuberosum inhibits protein import. Plant Mol. Biol. 1997, 35, 809–820. [Google Scholar] [CrossRef]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Manicki, M.; Aydin, H.; Abriata, L.A.; Overmyer, K.A.; Guerra, R.M.; Coon, J.J.; Dal Peraro, M.; Frost, A.; Pagliarini, D.J. Structure and functionality of a multimeric human COQ7:COQ9 complex. Mol. Cell 2022, 82, 4307–4323.e10. [Google Scholar] [CrossRef]
- Jiang, N.; Levavasseur, F.; McCright, B.; Shoubridge, E.A.; Hekimi, S. Mouse CLK-1 is imported into mitochondria by an unusual process that requires a leader sequence but no membrane potential. J. Biol. Chem. 2001, 276, 29218–29225. [Google Scholar] [CrossRef]
- Padilla, S.; Jonassen, T.; Jimenez-Hidalgo, M.A.; Fernandez-Ayala, D.J.; Lopez-Lluch, G.; Marbois, B.; Navas, P.; Clarke, C.F.; Santos-Ocana, C. Demethoxy-Q, an intermediate of coenzyme Q biosynthesis, fails to support respiration in Saccharomyces cerevisiae and lacks antioxidant activity. J. Biol. Chem. 2004, 279, 25995–26004. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A. CLK-1 protein has DNA binding activity specific to OL region of mitochondrial DNA. FEBS Lett. 2002, 516, 279–284. [Google Scholar] [CrossRef]
- Kirby, C.S.; Patel, M.R. Elevated mitochondrial DNA copy number found in ubiquinone-deficient clk-1 mutants is not rescued by ubiquinone precursor 2-4-dihydroxybenzoate. Mitochondrion 2021, 58, 38–48. [Google Scholar] [CrossRef]
- Monaghan, R.M.; Barnes, R.G.; Fisher, K.; Andreou, T.; Rooney, N.; Poulin, G.B.; Whitmarsh, A.J. A nuclear role for the respiratory enzyme CLK-1 in regulating mitochondrial stress responses and longevity. Nat. Cell Biol. 2015, 17, 782–792. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2011, 40, D261–D270. [Google Scholar] [CrossRef]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef]
- González-Mariscal, I.; Martin-Montalvo, A.; Vazquez-Fonseca, L.; Pomares-Viciana, T.; Sánchez-Cuesta, A.; Fernández-Ayala, D.J.; Navas, P.; Santos-Ocana, C. The mitochondrial phosphatase PPTC7 orchestrates mitochondrial metabolism regulating coenzyme Q10 biosynthesis. Biochim. Et Biophys. Acta (BBA)–Bioenerg. 2018, 1859, 1235–1248. [Google Scholar] [CrossRef]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef]
- Zemanovic, S.; Ivanov, M.V.; Ivanova, L.V.; Bhatnagar, A.; Michalkiewicz, T.; Teng, R.-J.; Kumar, S.; Rathore, R.; Pritchard, K.A.; Konduri, G.G.; et al. Dynamic Phosphorylation of the C Terminus of Hsp70 Regulates the Mitochondrial Import of SOD2 and Redox Balance. Cell Rep. 2018, 25, 2605–2616.e7. [Google Scholar] [CrossRef] [PubMed]
- Leong, A.Z.-X.; Lee, P.Y.; Mohtar, M.A.; Syafruddin, S.E.; Pung, Y.-F.; Low, T.Y. Short open reading frames (sORFs) and microproteins: An update on their identification and validation measures. J. Biomed. Sci. 2022, 29, 19. [Google Scholar] [CrossRef]
- Pozzi, A.; Dowling, D.K. Small mitochondrial RNAs as mediators of nuclear gene regulation, and potential implications for human health. BioEssays 2021, 43, 2000265. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ito, Y.; Niikura, T.; Shao, Z.; Hata, M.; Oyama, F.; Nishimoto, I. Mechanisms of Neuroprotection by a Novel Rescue Factor Humanin from Swedish Mutant Amyloid Precursor Protein. Biochem. Biophys. Res. Commun. 2001, 283, 460–468. [Google Scholar] [CrossRef]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.-J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Ramanjaneya, M.; Bettahi, I.; Jerobin, J.; Chandra, P.; Abi Khalil, C.; Skarulis, M.; Atkin, S.L.; Abou-Samra, A.-B. Mitochondrial-Derived peptides are down regulated in diabetes subjects. Front. Endocrinol. 2019, 10, 331. [Google Scholar] [CrossRef]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018, 28, 516–524.e57. [Google Scholar] [CrossRef]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 2022, 291, 120111. [Google Scholar] [CrossRef]
- Tsuji, Y.; Torti, S.V.; Torti, F.M. Activation of the Ferritin H Enhancer, FER-1, by the Cooperative Action of Members of the AP1 and Sp1 Transcription Factor Families. J. Biol. Chem. 1998, 273, 2984–2992. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Young, L.; Leonhard, K.; Tatsuta, T.; Trowsdale, J.; Langer, T. Role of the ABC transporter Mdl1 in peptide export from mitochondria. Science 2001, 291, 2135–2138. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and Nuclear Accumulation of the Transcription Factor ATFS-1 Promotes OXPHOS Recovery during the UPRmt. Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef]
- Seiferling, D.; Szczepanowska, K.; Becker, C.; Senft, K.; Hermans, S.; Maiti, P.; König, T.; Kukat, A.; Trifunovic, A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. 2016, 17, 953–964. [Google Scholar] [CrossRef]
- Fessler, E.; Eckl, E.-M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef]
- Persengiev, S.P.; Devireddy, L.R.; Green, M.R. Inhibition of apoptosis by ATFx: A novel role for a member of the ATF/CREB family of mammalian bZIP transcription factors. Genes Dev. 2002, 16, 1806–1814. [Google Scholar] [CrossRef]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- Tan, K.; Fujimoto, M.; Takii, R.; Takaki, E.; Hayashida, N.; Nakai, A. Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat. Commun. 2015, 6, 6580. [Google Scholar] [CrossRef]
- Amir, M.; Mohammad, T.; Dohare, R.; Islam, A.; Ahmad, F.; Imtaiyaz Hassan, M. Structure, function and therapeutic implications of OB-fold proteins: A lesson from past to present. Brief. Funct. Genom. 2020, 19, 377–389. [Google Scholar] [CrossRef]
- Wanrooij, S.; Falkenberg, M. The human mitochondrial replication fork in health and disease. Biochim. Biophys. Acta 2010, 1797, 1378–1388. [Google Scholar] [CrossRef]
- Katiyar, A.; Fujimoto, M.; Tan, K.; Kurashima, A.; Srivastava, P.; Okada, M.; Takii, R.; Nakai, A. HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio 2020, 10, 1135–1148. [Google Scholar] [CrossRef]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage—How and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef]
- Tiwari, V.; Wilson, D.M., 3rd. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Rouse, J.; Jackson, S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002, 297, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.C.; Haber, J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet. 2006, 40, 209–235. [Google Scholar] [CrossRef]
- Basu, B.; Yap, T.A.; Molife, L.R.; De Bono, J.S. Targeting the DNA damage response in oncology: Past, present and future perspectives. Curr. Opin. Oncol. 2012, 24, 316–324. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Boss, M.K.; Allen, C.P.; LaRue, S.M. Translational research in radiation-induced DNA damage signaling and repair. Transl. Cancer Res. 2017, 6, S875–S891. [Google Scholar] [CrossRef]
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.C.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Ramamoorthy, M.; Sykora, P.; Maynard, S.; Lin, P.C.; Minor, R.K.; Wilson, D.M., 3rd; Cooper, M.; Spencer, R.; de Cabo, R.; et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012, 209, 855–869. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Bohr, V.A. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy 2014, 10, 1468–1469. [Google Scholar] [CrossRef]
- Park, S.-H.; Kim, S.; Lee, H.S.; Shin, I. Real-time spatial and temporal analysis of the translocation of the apoptosis-inducing factor in cells. ACS Chem. Biol. 2021, 16, 2462–2471. [Google Scholar] [CrossRef]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jäättelä, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef]
- Guida, M.; Zanon, A.; Montibeller, L.; Lavdas, A.A.; Ladurner, J.; Pischedda, F.; Rakovic, A.; Domingues, F.S.; Piccoli, G.; Klein, C.; et al. Parkin Interacts with Apoptosis-Inducing Factor and Interferes with Its Translocation to the Nucleus in Neuronal Cells. Int. J. Mol. Sci. 2019, 20, 748. [Google Scholar] [CrossRef]
- Rodríguez-Vargas, J.M.; Ruiz-Magaña, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodríguez, M.I.; Muñoz-Gámez, J.A.; de Almodóvar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar] [CrossRef]
- Chao, T.; Shih, H.-T.; Hsu, S.-C.; Chen, P.-J.; Fan, Y.-S.; Jeng, Y.-M.; Shen, Z.-Q.; Tsai, T.-F.; Chang, Z.-F. Autophagy restricts mitochondrial DNA damage-induced release of ENDOG (endonuclease G) to regulate genome stability. Autophagy 2021, 17, 3444–3460. [Google Scholar] [CrossRef]
- Yogev, O.; Yogev, O.; Singer, E.; Shaulian, E.; Goldberg, M.; Fox, T.D.; Pines, O. Fumarase: A Mitochondrial Metabolic Enzyme and a Cytosolic/Nuclear Component of the DNA Damage Response. PLoS Biol. 2010, 8, e1000328. [Google Scholar] [CrossRef]
- Ravdin, R.G.; Crandall, D.I. The enzymatic conversion of homogentisic acid to 4-fumarylacetoacetic acid. J. Biol. Chem. 1951, 189, 137–149. [Google Scholar] [CrossRef]
- Ratner, S.; Petrack, B.; Rochovansky, O. Biosynthesis of urea: V. isolation and properties of argininosuccinic acid. J. Biol. Chem. 1953, 204, 95–113. [Google Scholar] [CrossRef]
- Yogev, O.; Pines, O. Dual targeting of mitochondrial proteins: Mechanism, regulation and function. Biochim. Et Biophys. Acta (BBA)–Biomembr. 2011, 1808, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Sass, E.; Karniely, S.; Pines, O. Folding of Fumarase during Mitochondrial Import Determines its Dual Targeting in Yeast. J. Biol. Chem. 2003, 278, 45109–45116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Qian, X.; Shen, J.; Wang, Y.; Li, X.; Liu, R.; Xia, Y.; Chen, Q.; Peng, G.; Lin, S.Y.; et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat. Cell Biol. 2015, 17, 1158–1168. [Google Scholar] [CrossRef]
- Saatchi, F.; Kirchmaier, A.L. Tolerance of DNA Replication Stress Is Promoted by Fumarate Through Modulation of Histone Demethylation and Enhancement of Replicative Intermediate Processing in Saccharomyces cerevisiae. Genetics 2019, 212, 631–654. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.M.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Tomlinson, I.P.M. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Sudarshan, S.; Linehan, W.M.; Neckers, L. HIF and fumarate hydratase in renal cancer. Br. J. Cancer 2007, 96, 403–407. [Google Scholar] [CrossRef]
- Raimundo, N.; Ahtinen, J.; Fumić, K.; Barić, I.; Remes, A.M.; Renkonen, R.; Lapatto, R.; Suomalainen, A. Differential metabolic consequences of fumarate hydratase and respiratory chain defects. Biochim. Et Biophys. Acta (BBA)–Mol. Basis Dis. 2008, 1782, 287–294. [Google Scholar] [CrossRef]
- Johnson, T.I.; Costa, A.S.H.; Ferguson, A.N.; Frezza, C. Fumarate hydratase loss promotes mitotic entry in the presence of DNA damage after ionising radiation. Cell Death Dis. 2018, 9, 913. [Google Scholar] [CrossRef]
- Garcia, S.N.; Guedes, R.C.; Marques, M.M. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr. Med. Chem. 2019, 26, 7285–7322. [Google Scholar] [CrossRef]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef]
- Thomas, G.E.; Egan, G.; García-Prat, L.; Botham, A.; Voisin, V.; Patel, P.S.; Hoff, F.W.; Chin, J.; Nachmias, B.; Kaufmann, K.B.; et al. The metabolic enzyme hexokinase 2 localizes to the nucleus in AML and normal haematopoietic stem and progenitor cells to maintain stemness. Nat. Cell Biol. 2022, 24, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Hurlin, P.J. Control of vertebrate development by MYC. Cold Spring Harb. Perspect. Med. 2013, 3, a014332. [Google Scholar] [CrossRef] [PubMed]
- Ameri, K.; Rajah, A.M.; Nguyen, V.; Sanders, T.A.; Jahangiri, A.; DeLay, M.; Donne, M.; Choi, H.J.; Tormos, K.V.; Yeghiazarians, Y.; et al. Nuclear localization of the mitochondrial factor HIGD1A during metabolic stress. PLoS ONE 2013, 8, e62758. [Google Scholar] [CrossRef]
- Chen, B.; Xu, F.; Gao, Y.; Hu, G.; Zhu, K.; Lu, H.; Xu, A.; Chen, S.; Wu, L.; Zhao, G. DNA damage-induced translocation of mitochondrial factor HIGD1A into the nucleus regulates homologous recombination and radio/chemo-sensitivity. Oncogene 2022, 41, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Shivji, M.K.; Chen, C.; Kolodner, R.; Wood, R.D.; Dutta, A. The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J. Biol. Chem. 1998, 273, 1453–1461. [Google Scholar] [CrossRef]
- Douiev, L.; Miller, C.; Keller, G.; Benyamini, H.; Abu-Libdeh, B.; Saada, A. Replicative Stress Coincides with Impaired Nuclear DNA Damage Response in COX4-1 Deficiency. Int. J. Mol. Sci. 2022, 23, 4149. [Google Scholar] [CrossRef]
- Jia, Y.-Z.; Liu, J.; Wang, G.-Q.; Pan, H.; Huang, T.-Z.; Liu, R.; Zhang, Y. HIG1 domain family member 1A is a crucial regulator of disorders associated with hypoxia. Mitochondrion 2023, 69, 171–182. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Laulier, C.; Lopez, B.S. The secret life of Bcl-2: Apoptosis-independent inhibition of DNA repair by Bcl-2 family members. Mutat. Res. /Rev. Mutat. Res. 2012, 751, 247–257. [Google Scholar] [CrossRef]
- Kelekar, A.; Thompson, C.B. Bcl-2-family proteins: The role of the BH3 domain in apoptosis. Trends Cell Biol. 1998, 8, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Shiina, H.; Urakami, S.; Kikuno, N.; Yoneda, T.; Shigeno, K.; Igawa, M. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6116–6124. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Hess, J.L.; Sentman, C.L.; Korsmeyer, S.J. Peripheral T-Cell Lymphoma in lckpr-bcl-2 Transgenic Mice. Blood 1995, 86, 1255–1260. [Google Scholar] [CrossRef]
- Youn, C.K.; Cho, H.J.; Kim, S.H.; Kim, H.B.; Kim, M.H.; Chang, I.Y.; Lee, J.S.; Chung, M.H.; Hahm, K.S.; You, H.J. Bcl-2 expression suppresses mismatch repair activity through inhibition of E2F transcriptional activity. Nat. Cell Biol. 2005, 7, 137–147. [Google Scholar] [CrossRef]
- Kuo, M.L.; Shiah, S.G.; Wang, C.J.; Chuang, S.E. Suppression of apoptosis by Bcl-2 to enhance benzene metabolites- induced oxidative DNA damage and mutagenesis: A possible mechanism of carcinogenesis. Mol. Pharmacol. 1999, 55, 894–901. [Google Scholar]
- Wang, Q.; Gao, F.; May, W.S.; Zhang, Y.; Flagg, T.; Deng, X. Bcl2 negatively regulates DNA double-strand-break repair through a nonhomologous end-joining pathway. Mol. Cell 2008, 29, 488–498. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Meek, K. Repair of DNA double strand breaks by non-homologous end joining. Biochimie 2003, 85, 1161–1173. [Google Scholar] [CrossRef]
- Feldmann, E.; Schmiemann, V.; Goedecke, W.; Reichenberger, S.; Pfeiffer, P. DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: Implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res. 2000, 28, 2585–2596. [Google Scholar] [CrossRef]
- Laulier, C.; Barascu, A.; Guirouilh-Barbat, J.; Pennarun, G.; Le Chalony, C.; Chevalier, F.; Palierne, G.; Bertrand, P.; Verbavatz, J.M.; Lopez, B.S. Bcl-2 inhibits nuclear homologous recombination by localizing BRCA1 to the endomembranes. Cancer Res. 2011, 71, 3590–3602. [Google Scholar] [CrossRef]
- Liu, Y.; Naumovski, L.; Hanawalt, P. Nucleotide Excision Repair Capacity Is Attenuated in Human Promyelocytic HL60 Cells That Overexpress BCL21. Cancer Res. 1997, 57, 1650–1653. [Google Scholar] [PubMed]
- Jin, Z.; May, W.S.; Gao, F.; Flagg, T.; Deng, X. Bcl2 Suppresses DNA Repair by Enhancing c-Myc Transcriptional Activity. J. Biol. Chem. 2006, 281, 14446–14456. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gao, F.; Zhang, Y.; Wei, K.; Liu, Y.; Deng, X. Bcl2 Inhibits Abasic Site Repair by Down-regulating APE1 Endonuclease Activity. J. Biol. Chem. 2008, 283, 9925–9932. [Google Scholar] [CrossRef]
- Dutta, C.; Day, T.; Kopp, N.; van Bodegom, D.; Davids, M.S.; Ryan, J.; Bird, L.; Kommajosyula, N.; Weigert, O.; Yoda, A.; et al. BCL2 Suppresses PARP1 Function and Nonapoptotic Cell Death. Cancer Res. 2012, 72, 4193–4203. [Google Scholar] [CrossRef] [PubMed]
- Huambachano, O.; Herrera, F.; Rancourt, A.; Satoh, M.S. Double-stranded DNA binding domain of poly(ADP-ribose) polymerase-1 and molecular insight into the regulation of its activity. J. Biol. Chem. 2011, 286, 7149–7160. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Krietsch, J.; Rouleau, M.; Pic, É.; Ethier, C.; Dawson, T.M.; Dawson, V.L.; Masson, J.Y.; Poirier, G.G.; Gagné, J.P. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Asp. Med. 2013, 34, 1066–1087. [Google Scholar] [CrossRef]
- Hou, Y.; Gao, F.; Wang, Q.; Zhao, J.; Flagg, T.; Zhang, Y.; Deng, X. Bcl2 impedes DNA mismatch repair by directly regulating the hMSH2-hMSH6 heterodimeric complex. J. Biol. Chem. 2007, 282, 9279–9287. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. New York Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Paggi, D.; Hannibal, L.; Castro, M.A.; Oviedo-Rouco, S.; Demicheli, V.; Tortora, V.; Tomasina, F.; Radi, R.; Murgida, D.H. Multifunctional Cytochrome c: Learning New Tricks from an Old Dog. Chem. Rev. 2017, 117, 13382–13460. [Google Scholar] [CrossRef] [PubMed]
- Balk, J.; Leaver, C.J.; McCabe, P.F. Translocation of cytochrome c from the mitochondria to the cytosol occurs during heat-induced programmed cell death in cucumber plants. FEBS Lett. 1999, 463, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Arama, E.; Bader, M.; Srivastava, M.; Bergmann, A.; Steller, H. The two Drosophila cytochrome C proteins can function in both respiration and caspase activation. Embo J. 2006, 25, 232–243. [Google Scholar] [CrossRef]
- Giannattasio, S.; Atlante, A.; Antonacci, L.; Guaragnella, N.; Lattanzio, P.; Passarella, S.; Marra, E. Cytochrome c is released from coupled mitochondria of yeast en route to acetic acid-induced programmed cell death and can work as an electron donor and a ROS scavenger. FEBS Lett. 2008, 582, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Elena-Real, C.A.; Díaz-Quintana, A.; González-Arzola, K.; Velázquez-Campoy, A.; Orzáez, M.; López-Rivas, A.; Gil-Caballero, S.; De la Rosa, M.Á.; Díaz-Moreno, I. Cytochrome c speeds up caspase cascade activation by blocking 14-3-3ε-dependent Apaf-1 inhibition. Cell Death Dis. 2018, 9, 365–376. [Google Scholar] [CrossRef]
- Elena-Real, C.A.; González-Arzola, K.; Pérez-Mejías, G.; Díaz-Quintana, A.; Velázquez-Campoy, A.; Desvoyes, B.; Gutiérrez, C.; De la Rosa, M.A.; Díaz-Moreno, I. Proposed mechanism for regulation of H(2) O(2) -induced programmed cell death in plants by binding of cytochrome c to 14-3-3 proteins. Plant J. Cell Mol. Biol. 2021, 106, 74–85. [Google Scholar] [CrossRef]
- Ruiz-Vela, A.; González de Buitrago, G.; Martínez-A, C. Nuclear Apaf-1 and cytochrome c redistribution following stress-induced apoptosis. FEBS Lett. 2002, 517, 133–138. [Google Scholar] [CrossRef]
- Nur, E.K.A.; Gross, S.R.; Pan, Z.; Balklava, Z.; Ma, J.; Liu, L.F. Nuclear translocation of cytochrome c during apoptosis. J. Biol. Chem. 2004, 279, 24911–24914. [Google Scholar] [CrossRef]
- Godoy, L.C.; Muñoz-Pinedo, C.; Castro, L.; Cardaci, S.; Schonhoff, C.M.; King, M.; Tórtora, V.; Marín, M.; Miao, Q.; Jiang, J.F.; et al. Disruption of the M80-Fe ligation stimulates the translocation of cytochrome c to the cytoplasm and nucleus in nonapoptotic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2653–2658. [Google Scholar] [CrossRef]
- González-Arzola, K.; Díaz-Moreno, I.; Cano-González, A.; Díaz-Quintana, A.; Velázquez-Campoy, A.; Moreno-Beltrán, B.; López-Rivas, A.; De la Rosa, M.A. Structural basis for inhibition of the histone chaperone activity of SET/TAF-Iβ by cytochrome c. Proc. Natl. Acad. Sci. USA 2015, 112, 9908–9913. [Google Scholar] [CrossRef] [PubMed]
- Nolin, F.; Michel, J.; Wortham, L.; Tchelidze, P.; Banchet, V.; Lalun, N.; Terryn, C.; Ploton, D. Stage-Specific Changes in the Water, Na+, Cl- and K+ Contents of Organelles during Apoptosis, Demonstrated by a Targeted Cryo Correlative Analytical Approach. PLoS ONE 2016, 11, e0148727. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Krelin, Y.; Shoshan-Barmatz, V. Reducing VDAC1 expression induces a non-apoptotic role for pro-apoptotic proteins in cancer cell differentiation. Biochim. Biophys. Acta 2016, 1857, 1228–1242. [Google Scholar] [CrossRef]
- Seo, S.B.; McNamara, P.; Heo, S.; Turner, A.; Lane, W.S.; Chakravarti, D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 2001, 104, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Kalousi, A.; Hoffbeck, A.S.; Selemenakis, P.N.; Pinder, J.; Savage, K.I.; Khanna, K.K.; Brino, L.; Dellaire, G.; Gorgoulis, V.G.; Soutoglou, E. The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep. 2015, 11, 149–163. [Google Scholar] [CrossRef]
- González-Arzola, K.; Díaz-Quintana, A.; Rivero-Rodríguez, F.; Velázquez-Campoy, A.; De la Rosa, M.A.; Díaz-Moreno, I. Histone chaperone activity of Arabidopsis thaliana NRP1 is blocked by cytochrome c. Nucleic Acids Res. 2017, 45, 2150–2165. [Google Scholar] [CrossRef]
- Li, M.; Guo, H.; Damuni, Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 1995, 34, 1988–1996. [Google Scholar] [CrossRef]
- Chowdhury, D.; Keogh, M.C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. Gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef]
- Casado-Combreras, M.; Rivero-Rodríguez, F.; Elena-Real, C.A.; Molodenskiy, D.; Díaz-Quintana, A.; Martinho, M.; Gerbaud, G.; González-Arzola, K.; Velázquez-Campoy, A.; Svergun, D.; et al. PP2A is activated by cytochrome c upon formation of a diffuse encounter complex with SET/TAF-Iβ. Comput. Struct. Biotechnol. J. 2022, 20, 3695–3707. [Google Scholar] [CrossRef]
- Martínez-Fábregas, J.; Díaz-Moreno, I.; González-Arzola, K.; Janocha, S.; Navarro, J.A.; Hervás, M.; Bernhardt, R.; Velázquez-Campoy, A.; Díaz-Quintana, A.; De la Rosa, M.A. Structural and Functional Analysis of Novel Human Cytochrome c Targets in Apoptosis. Mol. Cell. Proteom. 2014, 13, 1439–1456. [Google Scholar] [CrossRef]
- Scott, D.D.; Oeffinger, M. Nucleolin and nucleophosmin: Nucleolar proteins with multiple functions in DNA repair. Biochem. Cell Biol. Biochim. Et Biol. Cell. 2016, 94, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Fujimoto, H.; Sato, J.; Hayashi, I.; Burma, S.; Matsuura, S.; Chen, D.J.; Komatsu, K. Nucleolin participates in DNA double-strand break-induced damage response through MDC1-dependent pathway. PLoS ONE 2012, 7, e49245. [Google Scholar] [CrossRef] [PubMed]
- González-Arzola, K.; Díaz-Quintana, A.; Bernardo-García, N.; Martínez-Fábregas, J.; Rivero-Rodríguez, F.; Casado-Combreras, M.Á.; Elena-Real, C.A.; Velázquez-Cruz, A.; Gil-Caballero, S.; Velázquez-Campoy, A.; et al. Nucleus-translocated mitochondrial cytochrome c liberates nucleophosmin-sequestered ARF tumor suppressor by changing nucleolar liquid–liquid phase separation. Nat. Struct. Mol. Biol. 2022, 29, 1024–1036. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, J.-H.; Kim, S.; Park, E.-J.; Yun, Y.; Kwon, J. A proteomics approach for the identification of nucleophosmin and heterogeneous nuclear ribonucleoprotein C1/C2 as chromatin-binding proteins in response to DNA double-strand breaks. Biochem. J. 2005, 388, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Vadivel, K.; Bajaj, S.P.; Chun, R.F.; Hewison, M.; Adams, J.S. The heterodimeric structure of heterogeneous nuclear ribonucleoprotein C1/C2 dictates 1,25-dihydroxyvitamin D-directed transcriptional events in osteoblasts. Bone Res. 2014, 2, 14011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Schlott, B.; Görlach, M.; Grosse, F. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res 2004, 32, 1–10. [Google Scholar] [CrossRef]
- Ford, M.G.J.; Chappie, J.S. The structural biology of the dynamin-related proteins: New insights into a diverse, multitalented family. Traffic 2019, 20, 717–740. [Google Scholar] [CrossRef]
- Yamamori, T.; Ike, S.; Bo, T.; Sasagawa, T.; Sakai, Y.; Suzuki, M.; Yamamoto, K.; Nagane, M.; Yasui, H.; Inanami, O. Inhibition of the mitochondrial fission protein dynamin-related protein 1 (Drp1) impairs mitochondrial fission and mitotic catastrophe after x-irradiation. Mol. Biol. Cell 2015, 26, 4607–4617. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; Van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Lima, A.R.; Santos, L.; Correia, M.; Soares, P.; Sobrinho-Simões, M.; Melo, M.; Máximo, V. Dynamin-related protein 1 at the crossroads of cancer. Genes 2018, 9, 115. [Google Scholar] [CrossRef]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Jagasia, R.; Grote, P.; Westermann, B.; Conradt, B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature 2005, 433, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.Y.; Chen, S.L.; Hsiao, Y.T.; Huang, C.H.; Lin, T.Y.; Chiang, I.P.; Hsu, W.H.; Chow, K.C. Nuclear expression of dynamin-related protein 1 in lung adenocarcinomas. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2009, 22, 1139–1150. [Google Scholar] [CrossRef]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; Van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.-I.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Yoon, D.-S.; Lim, I.K.; Yoon, S.-H.; Chung, H.-Y.; Rojo, M.; Malka, F.; Jou, M.-J.; Martinou, J.-C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef]
- Kim, S.J.; Kwon, M.-C.; Ryu, M.J.; Chung, H.K.; Tadi, S.; Kim, Y.K.; Man Kim, J.; Lee, S.H.; Park, J.H.; Kweon, G.R.; et al. CRIF1 Is Essential for the Synthesis and Insertion of Oxidative Phosphorylation Polypeptides in the Mammalian Mitochondrial Membrane. Cell Metab. 2012, 16, 274–283. [Google Scholar] [CrossRef]
- Chen, L.; Ran, Q.; Xiang, Y.; Xiang, L.; Chen, L.; Li, F.; Wu, J.; Wu, C.; Li, Z. Co-Activation of PKC-δ by CRIF1 Modulates Oxidative Stress in Bone Marrow Multipotent Mesenchymal Stromal Cells after Irradiation by Phosphorylating NRF2 Ser40. Theranostics 2017, 7, 2634–2648. [Google Scholar] [CrossRef]
- Chung, H.K.; Yi, Y.-W.; Jung, N.-C.; Kim, D.; Suh, J.M.; Kim, H.; Park, K.C.; Song, J.H.; Kim, D.W.; Hwang, E.S.; et al. CR6-interacting Factor 1 Interacts with Gadd45 Family Proteins and Modulates the Cell Cycle. J. Biol. Chem. 2003, 278, 28079–28088. [Google Scholar] [CrossRef]
- Ran, Q.; Jin, F.; Xiang, Y.; Xiang, L.; Wang, Q.; Li, F.; Chen, L.; Zhang, Y.; Wu, C.; Zhou, L.; et al. CRIF1 as a potential target to improve the radiosensitivity of osteosarcoma. Proc. Natl. Acad. Sci. USA 2019, 116, 20511–20516. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, F.; Guo, Q.H.; Wang, Y.P.; Zhao, R. Role of succinate dehydrogenase deficiency and oncometabolites in gastrointestinal stromal tumors. World J. Gastroenterol. 2020, 26, 5074–5089. [Google Scholar] [CrossRef]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal Cyst Formation in Fh1-Deficient Mice Is Independent of the Hif/Phd Pathway: Roles for Fumarate in KEAP1 Succination and Nrf2 Signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The Proto-oncometabolite Fumarate Binds Glutathione to Amplify ROS-Dependent Signaling. Mol. Cell 2013, 51, 236–248. [Google Scholar] [CrossRef]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, G.W.; Wang, Y.; Russanova, V.R.; Hirai, T.; Qin, J.; Nakatani, Y.; Howard, B.H. Stable Histone Deacetylase Complexes Distinguished by the Presence of SANT Domain Proteins CoREST/kiaa0071 and Mta-L1. J. Biol. Chem. 2001, 276, 6817–6824. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.R.; Moraes, C.T. Nuclear-Mitochondrial Interactions. Biomolecules 2022, 12, 427. [Google Scholar] [CrossRef]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.H.; Mullokandov, M.; Wu, Y.; Voisin, V.; Gronda, M.; Hurren, R.; Wang, X.; MacLean, N.; Jeyaraju, D.V.; Jitkova, Y.; et al. Mitochondrial carrier homolog 2 is necessary for AML survival. Blood 2020, 136, 81–92. [Google Scholar] [CrossRef]
- Li, W.; Long, Q.; Wu, H.; Zhou, Y.; Duan, L.; Yuan, H.; Ding, Y.; Huang, Y.; Wu, Y.; Huang, J.; et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 2022, 13, 7414. [Google Scholar] [CrossRef]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef]
- Pastukh, V.; Shokolenko, I.; Wang, B.; Wilson, G.; Alexeyev, M. Human mitochondrial transcription factor A possesses multiple subcellular targeting signals. FEBS J. 2007, 274, 6488–6499. [Google Scholar] [CrossRef]
- Lionaki, E.; Gkikas, I.; Tavernarakis, N. Differential Protein Distribution between the Nucleus and Mitochondria: Implications in Aging. Front. Genet. 2016, 7, 162. [Google Scholar] [CrossRef]
- Han, B.; Izumi, H.; Yasuniwa, Y.; Akiyama, M.; Yamaguchi, T.; Fujimoto, N.; Matsumoto, T.; Wu, B.; Tanimoto, A.; Sasaguri, Y.; et al. Human mitochondrial transcription factor A functions in both nuclei and mitochondria and regulates cancer cell growth. Biochem. Biophys. Res. Commun. 2011, 408, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kang, Y.C.; Park, W.H.; Jeong, J.H.; Pak, Y.K. Negative transcriptional regulation of mitochondrial transcription factor A (TFAM) by nuclear TFAM. Biochem. Biophys. Res. Commun. 2014, 450, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Rea, S. CLK-1/Coq7p is a DMQ mono-oxygenase and a new member of the di-iron carboxylate protein family. FEBS Lett. 2001, 509, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Lee, S.J.; Apfel, U.P.; Lippard, S.J. Aging-associated enzyme human clock-1: Substrate-mediated reduction of the diiron center for 5-demethoxyubiquinone hydroxylation. Biochemistry 2013, 52, 2236–2244. [Google Scholar] [CrossRef][Green Version]
- Ploumakis, A.; Coleman, M.L. OH, the places you’ll go! hydroxylation, gene expression, and cancer. Mol. Cell 2015, 58, 729–741. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Arzola, K.; Díaz-Quintana, A. Mitochondrial Factors in the Cell Nucleus. Int. J. Mol. Sci. 2023, 24, 13656. https://doi.org/10.3390/ijms241713656
González-Arzola K, Díaz-Quintana A. Mitochondrial Factors in the Cell Nucleus. International Journal of Molecular Sciences. 2023; 24(17):13656. https://doi.org/10.3390/ijms241713656
Chicago/Turabian StyleGonzález-Arzola, Katiuska, and Antonio Díaz-Quintana. 2023. "Mitochondrial Factors in the Cell Nucleus" International Journal of Molecular Sciences 24, no. 17: 13656. https://doi.org/10.3390/ijms241713656
APA StyleGonzález-Arzola, K., & Díaz-Quintana, A. (2023). Mitochondrial Factors in the Cell Nucleus. International Journal of Molecular Sciences, 24(17), 13656. https://doi.org/10.3390/ijms241713656