
Sex-Specific Effects of Prenatal Hypoxia and a Placental Antioxidant Treatment on Cardiac Mitochondrial Function in the Young Adult Offspring

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
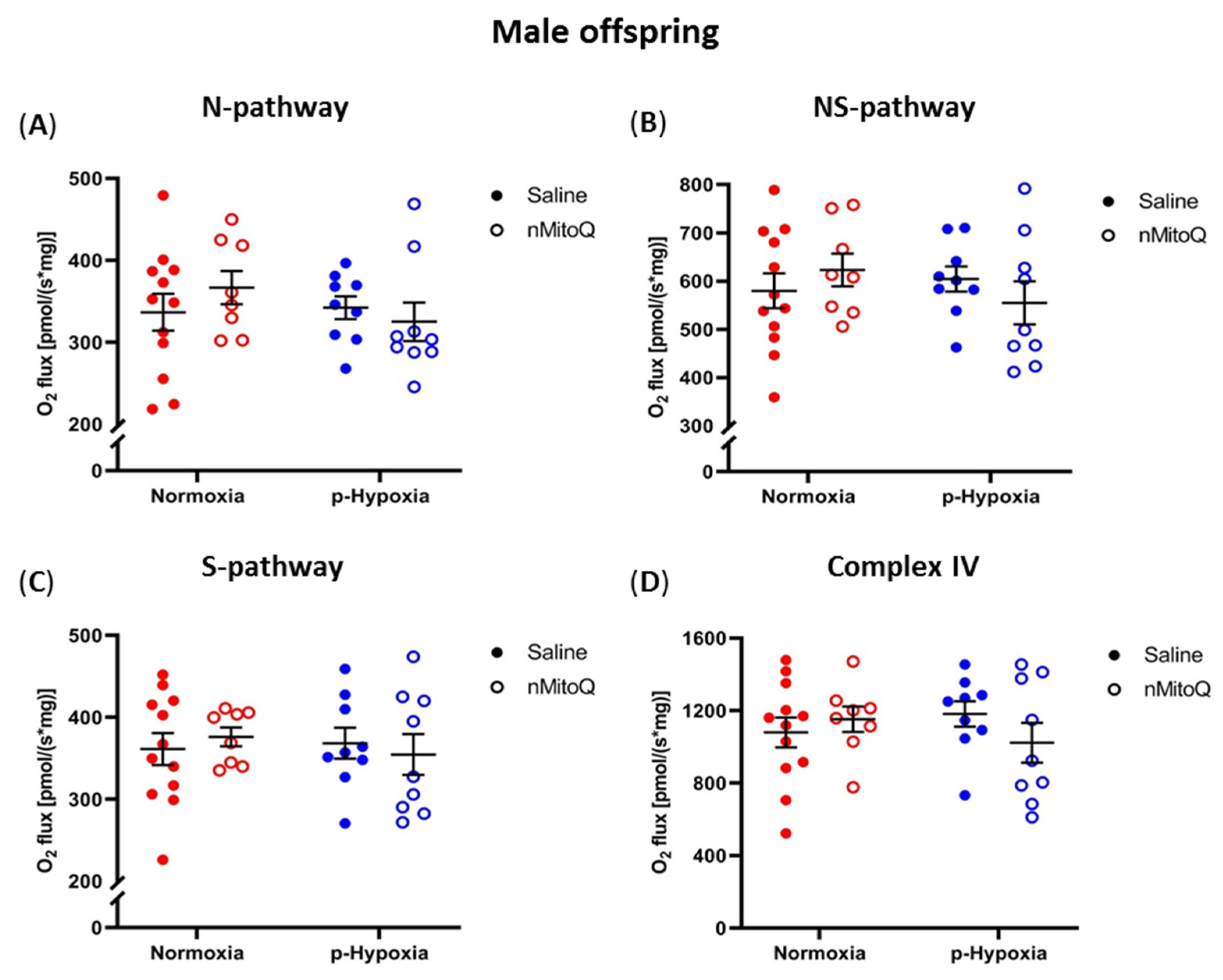
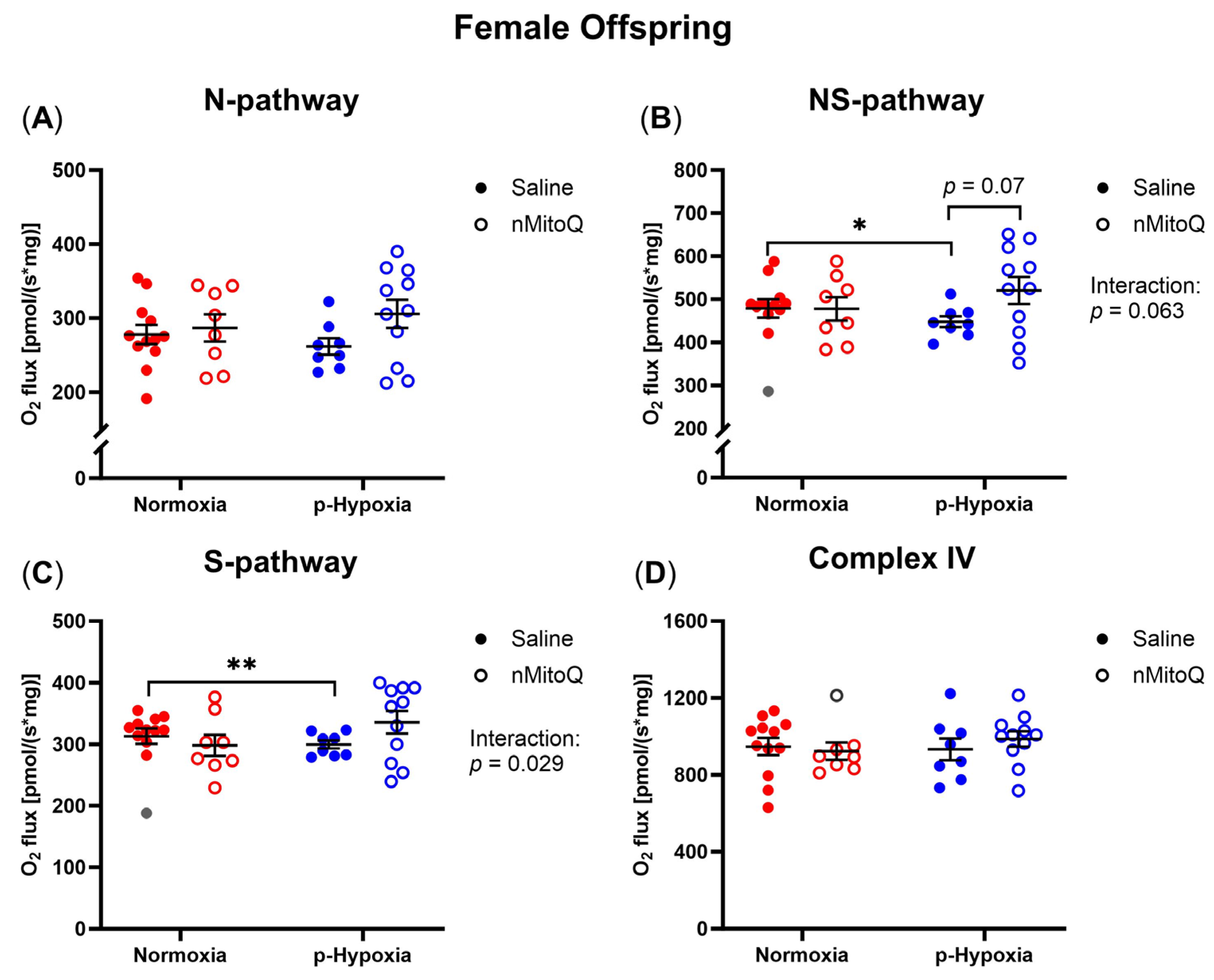
2.1. Prenatal Hypoxia Decreased Cardiac OXPHOS Capacity in Female Offspring, Which Tended to Be Improved by Maternal nMitoQ Treatment
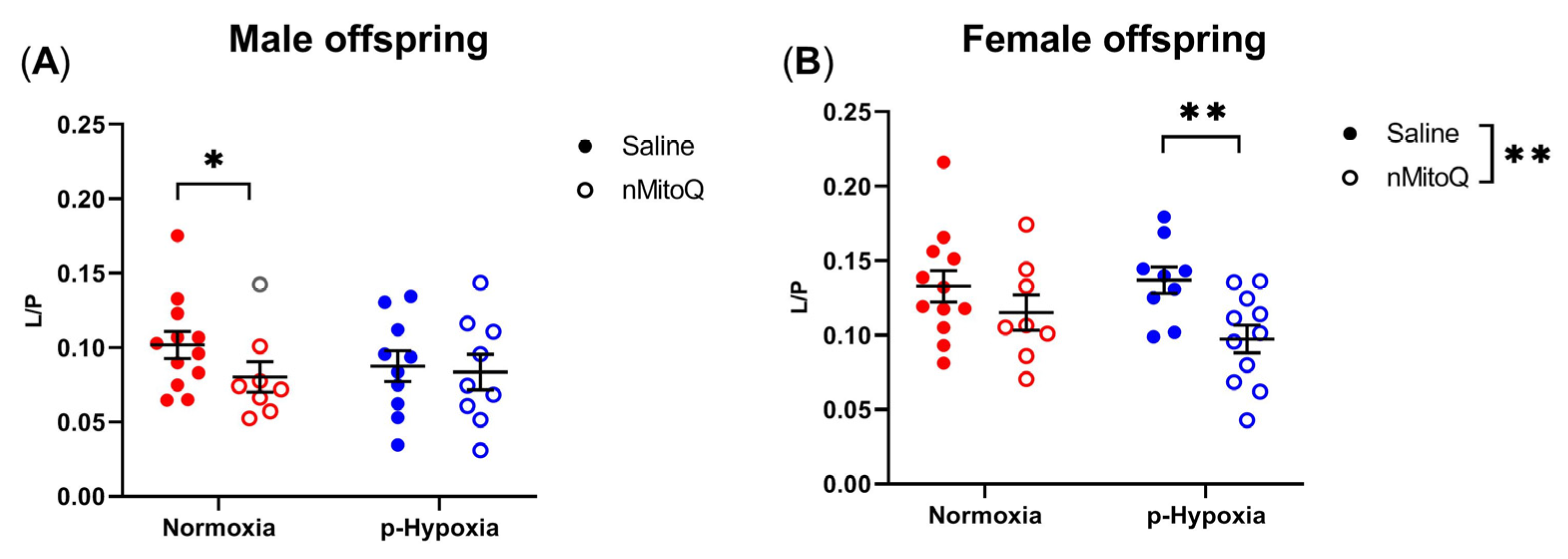
2.2. nMitoQ Treatment Increased OXPHOS Coupling Efficiency in Female Offspring Exposed to Prenatal Hypoxia
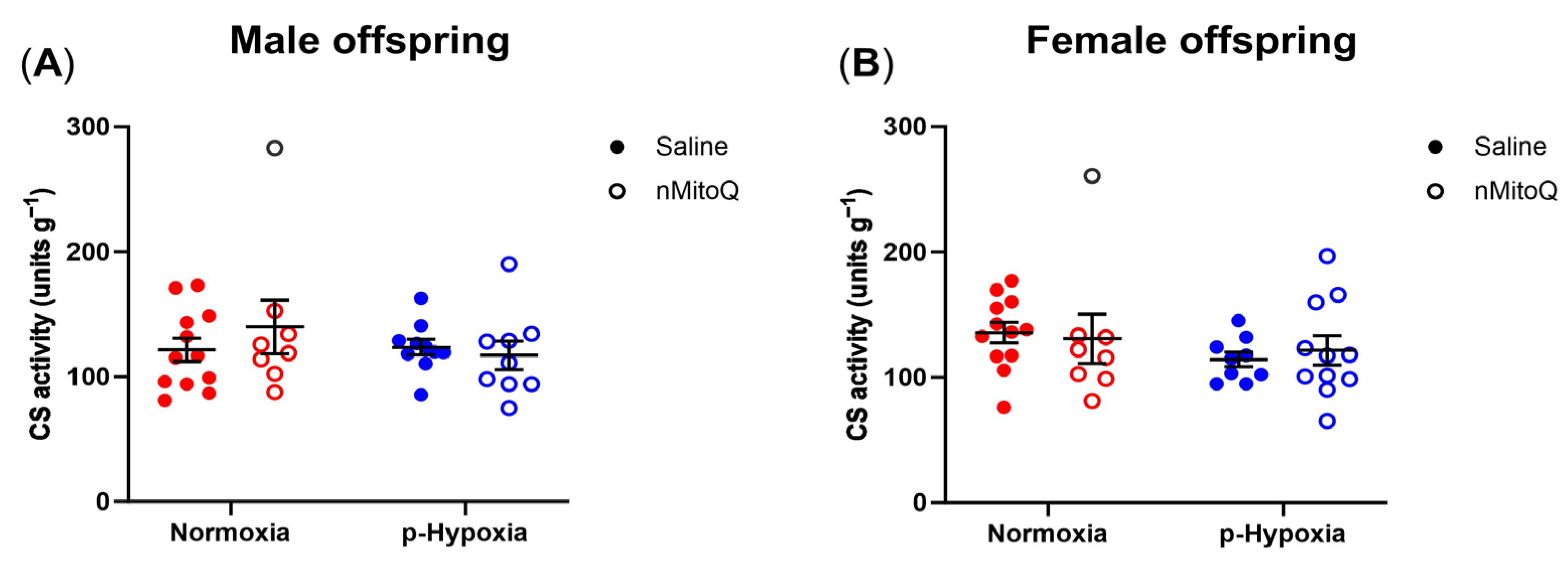
2.3. No Differences in Mitochondrial Content between the Groups
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. Prenatal Hypoxia Rat Model and Experimental Design
4.3. High-Resolution Respirometry
4.4. Mitochondrial Content
4.5. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 4 July 2023).
- Wilmot, E.G.; Edwardson, C.L.; Achana, F.A.; Davies, M.J.; Gorely, T.; Gray, L.J.; Khunti, K.; Yates, T.; Biddle, S.J. Sedentary time in adults and the association with diabetes, cardiovascular disease and death: Systematic review and meta-analysis. Diabetologia 2012, 55, 2895–2905. [Google Scholar] [CrossRef]
- Chien, S.C.; Chandramouli, C.; Lo, C.I.; Lin, C.F.; Sung, K.T.; Huang, W.H.; Lai, Y.H.; Yun, C.H.; Huang, C.; Yeh, H.I.; et al. Correction: Associations of obesity and malnutrition with cardiac remodeling and cardiovascular outcomes in Asian adults: A cohort study. PLoS Med 2021, 18, e1003784. [Google Scholar] [CrossRef]
- Rastogi, T.; Girerd, N.; Lamiral, Z.; Bresso, E.; Bozec, E.; Boivin, J.M.; Rossignol, P.; Zannad, F.; Ferreira, J.P. Impact of smoking on cardiovascular risk and premature ageing: Findings from the STANISLAS cohort. Atherosclerosis 2022, 346, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.D.; Trask, A.J. Developmental Origins of Cardiovascular Dysfunction–Doomed From Birth? Circ. J. 2016, 80, 818–820. [Google Scholar] [CrossRef]
- Arima, Y.; Fukuoka, H. Developmental origins of health and disease theory in cardiology. J. Cardiol. 2020, 76, 14–17. [Google Scholar] [CrossRef]
- Soleymanlou, N.; Jurisica, I.; Nevo, O.; Ietta, F.; Zhang, X.; Zamudio, S.; Post, M.; Caniggia, I. Molecular Evidence of Placental Hypoxia in Preeclampsia. J. Clin. Endocrinol. Metab. 2005, 90, 4299–4308. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, R. Placental insufficiency and its consequences. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 110, S99–S107. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.S.Y.; Baergen, R.N. Gross Umbilical Cord Complications are Associated with Placental Lesions of Circulatory Stasis and Fetal Hypoxia. Pediatr. Dev. Pathol. 2012, 15, 487–494. [Google Scholar] [CrossRef]
- Xu, Y.; Williams, S.J.; O’brien, D.; Davidge, S.T.; Xu, Y.; Williams, S.J.; O’brien, D.; Davidge, S.T. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J. 2006, 20, 1251–1253. [Google Scholar] [CrossRef]
- Rueda-Clausen, C.F.; Morton, J.S.; Davidge, S.T. Effects of hypoxia-induced intrauterine growth restriction on cardiopulmonary structure and function during adulthood. Cardiovasc. Res. 2009, 81, 713–722. [Google Scholar] [CrossRef]
- Rueda-Clausen, C.F.; Morton, J.S.; Lopaschuk, G.D.; Davidge, S.T. Long-term effects of intrauterine growth restriction on cardiac metabolism and susceptibility to ischaemia/reperfusion. Cardiovasc. Res. 2011, 90, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Quon, A.; Morton, J.S.; Davidge, S.T. Postnatal resveratrol supplementation improves cardiovascular function in male and female intrauterine growth restricted offspring. Physiol. Rep. 2017, 5, e13109. [Google Scholar] [CrossRef]
- Aljunaidy, M.M.; Morton, J.S.; Kirschenman, R.; Phillips, T.; Case, C.P.; Cooke, C.-L.M.; Davidge, S.T. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol. Res. 2018, 134, 332–342. [Google Scholar] [CrossRef]
- Tian, R.; Colucci, W.S.; Arany, Z.; Bachschmid, M.M.; Ballinger, S.W.; Boudina, S.; Bruce, J.E.; Busija, D.W.; Dikalov, S.; Dorn, G.W.; et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System. Circulation 2019, 140, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef]
- Bender, D.A. Oxidative phosphorylation. In Encyclopedia of Food Sciences and Nutrition; Elsevier: Amsterdam, The Netherlands, 2003; pp. 4295–4301. [Google Scholar]
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, Electron Transport Chain. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gnaiger, E. Mitochondrial pathways and respiratory control: An introduction to OXPHOS analysis. Bioenerg. Commun. 2020, 2020, 2. [Google Scholar]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Sun, Y.L.; Liu, C.Y. Structural and biochemical evidence of mitochondrial depletion in pigs with hypertrophic cardiomyopathy. Res. Vet. Sci. 2003, 74, 219–226. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Cao, Y.; Vergnes, L.; Wang, Y.-C.; Pan, C.; Chella Krishnan, K.; Moore, T.M.; Rosa-Garrido, M.; Kimball, T.H.; Zhou, Z.; Charugundla, S.; et al. Sex differences in heart mitochondria regulate diastolic dysfunction. Nat. Commun. 2022, 13, 3850. [Google Scholar] [CrossRef]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef]
- Lemieux, H.; Semsroth, S.; Antretter, H.; Höfer, D.; Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 2011, 43, 1729–1738. [Google Scholar] [CrossRef]
- Booz, G.W.; Kennedy, D.; Bowling, M.; Robinson, T.; Azubuike, D.; Fisher, B.; Brooks, K.; Chinthakuntla, P.; Hoang, N.H.; Hosler, J.P.; et al. Angiotensin II type 1 receptor agonistic autoantibody blockade improves postpartum hypertension and cardiac mitochondrial function in rat model of preeclampsia. Biol. Sex Differ. 2021, 12, 58. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes-Ferreira, L. Role of the phosphocreatine system on energetic homeostasis in skeletal and cardiac muscles. Einstein (Sao Paulo) 2014, 12, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Kane, A.D.; Lusby, C.M.; Allison, B.J.; Chua, Y.Y.; Kaandorp, J.J.; Nevin-Dolan, R.; Ashmore, T.J.; Blackmore, H.L.; Derks, J.B.; et al. Maternal Allopurinol Prevents Cardiac Dysfunction in Adult Male Offspring Programmed by Chronic Hypoxia During Pregnancy. Hypertension 2018, 72, 971–978. [Google Scholar] [CrossRef]
- Skeffington, K.L.; Beck, C.; Itani, N.; Niu, Y.; Shaw, C.J.; Giussani, D.A. Hypertension Programmed in Adult Hens by Isolated Effects of Developmental Hypoxia In Ovo. Hypertension 2020, 76, 533–544. [Google Scholar] [CrossRef]
- Xue, Q.; Zhang, L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: Role of protein kinase C epsilon. J. Pharmacol. Exp. Ther. 2009, 330, 624–632. [Google Scholar] [CrossRef]
- Li, G.; Xiao, Y.; Estrella, J.L.; Ducsay, C.A.; Gilbert, R.D.; Zhang, L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J. Soc. Gynecol. Investig. 2003, 10, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P.; Song, H.; Polster, B.M. Fetal Programming and Sexual Dimorphism of Mitochondrial Protein Expression and Activity of Hearts of Prenatally Hypoxic Guinea Pig Offspring. Oxidative Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Hellgren, K.T.; Premanandhan, H.; Quinn, C.J.; Trafford, A.W.; Galli, G.L.J. Sex-dependent effects of developmental hypoxia on cardiac mitochondria from adult murine offspring. Free Radic. Biol. Med. 2021, 162, 490–499. [Google Scholar] [CrossRef]
- Aljunaidy, M.M.; Morton, J.S.; Cooke, C.-L.M.; Davidge, S.T. Prenatal hypoxia and placental oxidative stress: Linkages to developmental origins of cardiovascular disease. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2017, 313, R395–R399. [Google Scholar] [CrossRef] [PubMed]
- Curtis, D.J.; Sood, A.; Phillips, T.J.; Leinster, V.H.L.; Nishiguchi, A.; Coyle, C.; Lacharme-Lora, L.; Beaumont, O.; Kemp, H.; Goodall, R.; et al. Secretions from placenta, after hypoxia/reoxygenation, can damage developing neurones of brain under experimental conditions. Exp. Neurol. 2014, 261, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Calcaterra, V.; Pelizzo, G.; Bramanti, P.; Mazzon, E. Prenatal Hypoxia and Placental Oxidative Stress: Insights from Animal Models to Clinical Evidences. Antioxidants 2020, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.; Phillips, T.J.; Stuart, G.C.; Rogers, M.F.; Steinkraus, B.R.; Grant, S.; Case, C.P. Preeclamptic placentae release factors that damage neurons: Implications for foetal programming of disease. Neuronal Signal. 2018, 2, NS20180139. [Google Scholar] [CrossRef]
- Ganguly, E.; Spaans, F.; Morton, J.S.; Kirschenman, R.; Aljunaidy, M.M.; Phillips, T.E.J.; Case, C.P.; Cooke, C.L.M.; Davidge, S.T. Placenta-targeted treatment in hypoxic dams improves maturation and growth of fetal cardiomyocytes in vitro via the release of placental factors. Exp. Physiol. 2020, 105, 1507–1514. [Google Scholar] [CrossRef]
- Holland, O.J.; Hickey, A.J.R.; Alvsaker, A.; Moran, S.; Hedges, C.; Chamley, L.W.; Perkins, A.V. Changes in mitochondrial respiration in the human placenta over gestation. Placenta 2017, 57, 102–112. [Google Scholar] [CrossRef]
- Mandò, C.; De Palma, C.; Stampalija, T.; Anelli, G.M.; Figus, M.; Novielli, C.; Parisi, F.; Clementi, E.; Ferrazzi, E.; Cetin, I. Placental mitochondrial content and function in intrauterine growth restriction and preeclampsia. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, E404–E413. [Google Scholar] [CrossRef]
- Ganguly, E.; Kirschenman, R.; Spaans, F.; Holody, C.D.; Phillips, T.E.J.; Case, C.P.; Cooke, C.L.M.; Murphy, M.P.; Lemieux, H.; Davidge, S.T. Nanoparticle-encapsulated antioxidant improves placental mitochondrial function in a sexually dimorphic manner in a rat model of prenatal hypoxia. FASEB J. 2021, 35, e21338. [Google Scholar] [CrossRef] [PubMed]
- Mele, J.; Muralimanoharan, S.; Maloyan, A.; Myatt, L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E419–E425. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, W.; Xu, H.; Hu, M.; Guo, X.; Jia, W.; Liu, G.; Li, J.; Cui, P.; Lager, S.; et al. Hyperandrogenism and insulin resistance-induced fetal loss: Evidence for placental mitochondrial abnormalities and elevated reactive oxygen species production in pregnant rats that mimic the clinical features of polycystic ovary syndrome. J. Physiol. 2019, 597, 3927–3950. [Google Scholar] [CrossRef]
- Bina, A.M.; Sturza, A.; Iancu, I.; Mocanu, A.G.; Bernad, E.; Chiriac, D.V.; Borza, C.; Craina, M.L.; Popa, Z.L.; Muntean, D.M.; et al. Placental oxidative stress and monoamine oxidase expression are increased in severe preeclampsia: A pilot study. Mol. Cell Biochem. 2022, 477, 2851–2861. [Google Scholar] [CrossRef]
- Phillips, T.J.; Scott, H.; Menassa, D.A.; Bignell, A.L.; Sood, A.; Morton, J.S.; Akagi, T.; Azuma, K.; Rogers, M.F.; Gilmore, C.E.; et al. Treating the placenta to prevent adverse effects of gestational hypoxia on fetal brain development. Sci. Rep. 2017, 7, 9079. [Google Scholar] [CrossRef]
- Ganguly, E.; Aljunaidy, M.M.; Kirschenman, R.; Spaans, F.; Morton, J.S.; Phillips, T.E.J.; Case, C.P.; Cooke, C.-L.M.; Davidge, S.T. Sex-Specific Effects of Nanoparticle-Encapsulated MitoQ (nMitoQ) Delivery to the Placenta in a Rat Model of Fetal Hypoxia. Front. Physiol. 2019, 10, 562. [Google Scholar] [CrossRef]
- Hula, N.; Spaans, F.; Vu, J.; Quon, A.; Kirschenman, R.; Cooke, C.-L.M.; Phillips, T.J.; Case, C.P.; Davidge, S.T. Placental treatment improves cardiac tolerance to ischemia/reperfusion insult in adult male and female offspring exposed to prenatal hypoxia. Pharmacol. Res. 2021, 165, 105461. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2005, 1706, 1–11. [Google Scholar] [CrossRef]
- Milliken, A.S.; Nadtochiy, S.M.; Brookes, P.S. Inhibiting Succinate Release Worsens Cardiac Reperfusion Injury by Enhancing Mitochondrial Reactive Oxygen Species Generation. J. Am. Heart Assoc. 2022, 11, e026135. [Google Scholar] [CrossRef] [PubMed]
- Milliken, A.S.; Kulkarni, C.A.; Brookes, P.S. Acid enhancement of ROS generation by complex-I reverse electron transport is balanced by acid inhibition of complex-II: Relevance for tissue reperfusion injury. Redox Biol. 2020, 37, 101733. [Google Scholar] [CrossRef] [PubMed]
- Bushdid, P.B.; Osinska, H.; Waclaw, R.R.; Molkentin, J.D.; Yutzey, K.E. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ. Res. 2003, 92, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P.; Chen, L.; Polster, B.M.; Pinkas, G.; Song, H. Prenatal hypoxia impairs cardiac mitochondrial and ventricular function in guinea pig offspring in a sex-related manner. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2018, 315, R1232–R1241. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol.-Heart Circ. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, P.; Holody, C.D.; Kirschenman, R.; Graton, M.E.; Spaans, F.; Phillips, T.J.; Case, C.P.; Bourque, S.L.; Lemieux, H.; Davidge, S.T. Sex-Specific Effects of Prenatal Hypoxia and a Placental Antioxidant Treatment on Cardiac Mitochondrial Function in the Young Adult Offspring. Int. J. Mol. Sci. 2023, 24, 13624. https://doi.org/10.3390/ijms241713624
Chatterjee P, Holody CD, Kirschenman R, Graton ME, Spaans F, Phillips TJ, Case CP, Bourque SL, Lemieux H, Davidge ST. Sex-Specific Effects of Prenatal Hypoxia and a Placental Antioxidant Treatment on Cardiac Mitochondrial Function in the Young Adult Offspring. International Journal of Molecular Sciences. 2023; 24(17):13624. https://doi.org/10.3390/ijms241713624
Chicago/Turabian StyleChatterjee, Paulami, Claudia D. Holody, Raven Kirschenman, Murilo E. Graton, Floor Spaans, Tom J. Phillips, C. Patrick Case, Stephane L. Bourque, Hélène Lemieux, and Sandra T. Davidge. 2023. "Sex-Specific Effects of Prenatal Hypoxia and a Placental Antioxidant Treatment on Cardiac Mitochondrial Function in the Young Adult Offspring" International Journal of Molecular Sciences 24, no. 17: 13624. https://doi.org/10.3390/ijms241713624
APA StyleChatterjee, P., Holody, C. D., Kirschenman, R., Graton, M. E., Spaans, F., Phillips, T. J., Case, C. P., Bourque, S. L., Lemieux, H., & Davidge, S. T. (2023). Sex-Specific Effects of Prenatal Hypoxia and a Placental Antioxidant Treatment on Cardiac Mitochondrial Function in the Young Adult Offspring. International Journal of Molecular Sciences, 24(17), 13624. https://doi.org/10.3390/ijms241713624