
A Digital PCR Method Based on Highly Specific Taq for Detecting Gene Editing and Mutations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
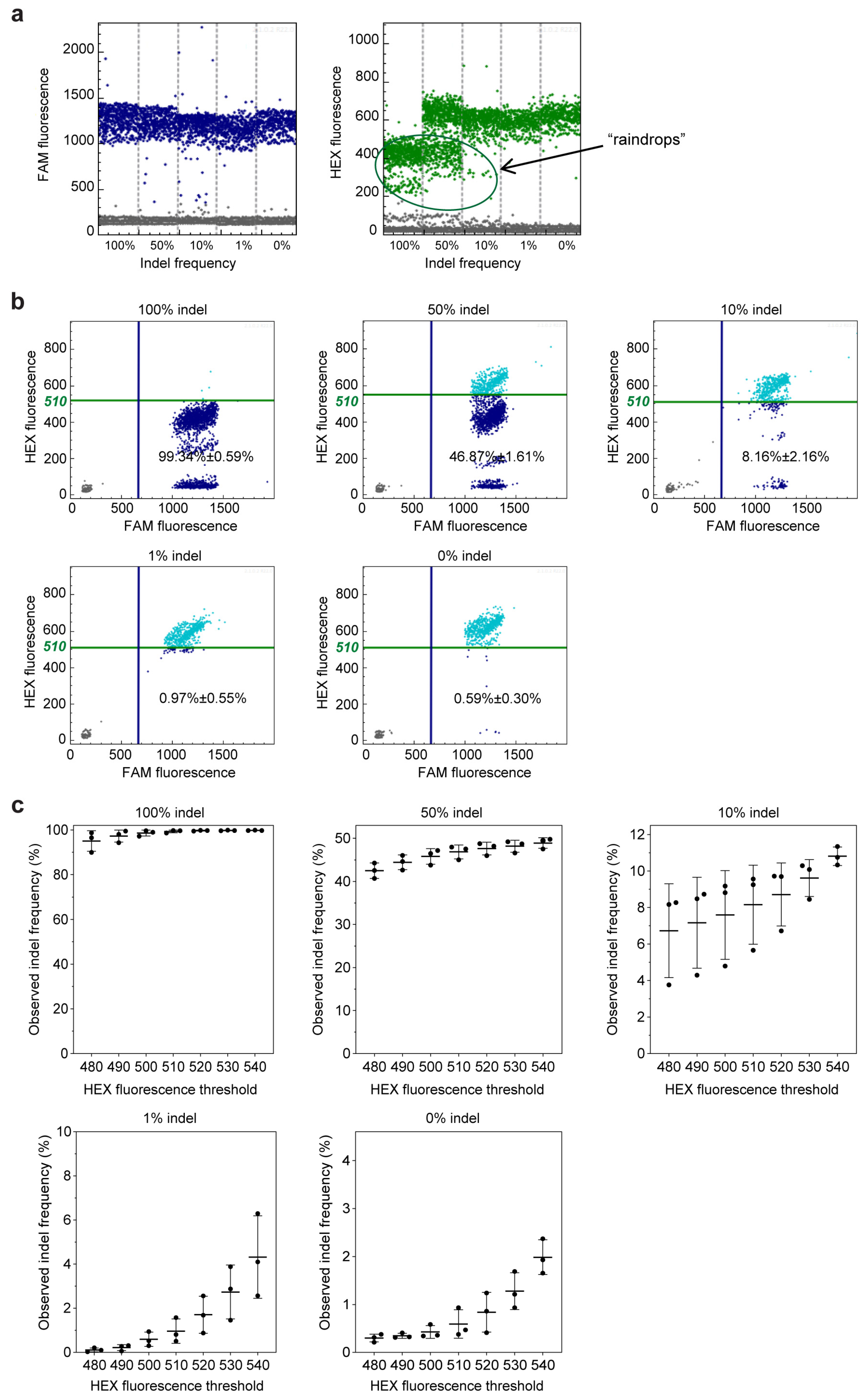
2.1. Ability of GEF-dPCR to Assess Indels
2.2. get-dPCR Distinguishes Indels Clearly
2.3. get-dPCR Using Taq388 Quantify Indel Frequency Accurately
2.4. Gene Mutation Detection Using get-dPCR
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids
4.3. Genomic DNA
4.4. Primers and Probes
4.5. Conditions for dPCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.L.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.B.; Jiang, W.Y.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef]
- Skryabin, B.V.; Kummerfeld, D.M.; Gubar, L.; Seeger, B.; Kaiser, H.; Stegemann, A.; Roth, J.; Meuth, S.G.; Pavenstadt, H.; Sherwood, J.; et al. Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9-mediated genome editing events. Sci. Adv. 2020, 6, eaax2941. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, J.; Chen, B.; Mo, S.; Lian, L.; Luo, Y.; Ding, D.; Ding, Y.; Cao, Q.; Li, Y.; et al. A donor-DNA-free CRISPR/Cas-based approach to gene knock-up in rice. Nat. Plants 2021, 7, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gallagher, J.; Arevalo, E.D.; Chen, R.; Skopelitis, T.; Wu, Q.; Bartlett, M.; Jackson, D. Enhancing grain-yield-related traits by CRISPR-Cas9 promoter editing of maize CLE genes. Nat. Plants 2021, 7, 287–294. [Google Scholar] [CrossRef]
- Zhu, J.; Ananthaswamy, N.; Jain, S.; Batra, H.; Tang, W.C.; Lewry, D.A.; Richards, M.L.; David, S.A.; Kilgore, P.B.; Sha, J.; et al. A universal bacteriophage T4 nanoparticle platform to design multiplex SARS-CoV-2 vaccine candidates by CRISPR engineering. Sci. Adv. 2021, 7, eabh1547. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.E.; Diamond, S.; Cress, B.F.; Crits-Christoph, A.; Lou, Y.C.; Borges, A.L.; Shivram, H.; He, C.; Xu, M.; Zhou, Z.; et al. Species- and site-specific genome editing in complex bacterial communities. Nat. Microbiol. 2022, 7, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Germini, D.; Tsfasman, T.; Zakharova, V.V.; Sjakste, N.; Lipinski, M.; Vassetzky, Y. A Comparison of Techniques to Evaluate the Effectiveness of Genome Editing. Trends Biotechnol. 2018, 36, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Vouillot, L.; Thelie, A.; Pollet, N. Comparison of T7E1 and Surveyor Mismatch Cleavage Assays to Detect Mutations Triggered by Engineered Nucleases. G3 Genes Genomes Genet. 2015, 5, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Fuccio, A.; Iorio, M.; Amato, F.; Elce, A.; Ingino, R.; Filocamo, M.; Castaldo, G.; Salvatore, F.; Tomaiuolo, R. A Novel DHPLC-Based Procedure for the Analysis of COL1A1 and COL1A2 Mutations in Osteogenesis Imperfecta. J. Mol. Diagn. 2011, 13, 648–656. [Google Scholar] [CrossRef]
- Gasser, R.B.; Hu, M.; Chilton, N.B.; Campbell, B.E.; Jex, A.J.; Otranto, D.; Cafarchia, C.; Beveridge, I.; Zhu, X. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat. Protoc. 2006, 1, 3121–3128. [Google Scholar] [CrossRef] [PubMed]
- Kennerson, L.; Warburton, T.; Nelis, E.; Brewer, M.; Polly, P.; De Jonghe, P.; Timmerman, V.; Nicholson, G.A. Mutation scanning the GJB1 gene with high-resolution melting analysis: Implications for mutation scanning of genes for Charcot-Marie-Tooth disease. Clin. Chem. 2007, 53, 349–352. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Kluesner, M.G.; Nedveck, D.A.; Lahr, W.S.; Garbe, J.R.; Abrahante, J.E.; Webber, B.R.; Moriarity, B.S. EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J. 2018, 1, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Guell, M.; Yang, L.H.; Church, G.M. Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 2014, 30, 2968–2970. [Google Scholar] [CrossRef] [PubMed]
- Boel, A.; Steyaert, W.; De Rocker, N.; Menten, B.; Callewaert, B.; De Paepe, A.; Coucke, P.; Willaert, A. BATCH-GE: Batch analysis of Next-Generation Sequencing data for genome editing assessment. Sci. Rep. 2016, 6, 30330. [Google Scholar] [CrossRef] [PubMed]
- Pinello, L.; Canver, M.C.; Hoban, M.D.; Orkin, S.H.; Kohn, D.B.; Bauer, D.E.; Yuan, G.C. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016, 34, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Haeussler, M.; Bauer, D.E.; Orkin, S.H.; Sanjana, N.E.; Shalem, O.; Yuan, G.C.; Zhang, F.; Concordet, J.P.; Pinello, L. Integrated design, execution, and analysis of arrayed and pooled CRISPR genome-editing experiments. Nat. Protoc. 2018, 13, 946–986. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lim, K.; Kim, J.S.; Bae, S. Cas-analyzer: An online tool for assessing genome editing results using NGS data. Bioinformatics 2017, 33, 286–288. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Zhong, Z.H.; Ren, Q.R.; Hassan, F.; Zhang, Y.; Zhang, T. CRISPRMatch: An Automatic Calculation and Visualization Tool for High-throughput CRISPR Genome-editing Data Analysis. Int. J. Biol. Sci. 2018, 14, 858–862. [Google Scholar] [CrossRef]
- Xie, K.; Yang, Y. RNA-guided genome editing in plants using a CRISPR-Cas system. Mol. Plant 2013, 6, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Mock, U.; Hauber, I.; Fehse, B. Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat. Protoc. 2016, 11, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ren, N.; Yang, L.; Liu, J.; Huang, Q. A qPCR method for genome editing efficiency determination and single-cell clone screening in human cells. Sci. Rep. 2019, 9, 18877. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, Y.G.; Yao, S.H.; Wei, Y.Q. A PCR Based Protocol for Detecting Indel Mutations Induced by TALENs and CRISPR/Cas9 in Zebrafish. PLoS ONE 2014, 9, e98282. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.F.; Wang, C.; Huang, J.; Wang, K.J. A simple and efficient method for CRISPR/Cas9-induced mutant screening. J. Genet. Genom. 2017, 44, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Yuno, M.; Kitaura, F.; Fujii, H. A refined two-step oligoribonucleotide interference-PCR method for precise discrimination of nucleotide differences. Sci. Rep. 2018, 8, 17195. [Google Scholar] [CrossRef] [PubMed]
- Germini, D.; Saada, Y.B.; Tsfasman, T.; Osina, K.; Robin, C.; Lomov, N.; Rubtsov, M.; Sjakste, N.; Lipinski, M.; Vassetzky, Y. A One-Step PCR-Based Assay to Evaluate the Efficiency and Precision of Genomic DNA-Editing Tools. Mol. Ther. Methods Clin. Dev. 2017, 5, 43–50. [Google Scholar] [CrossRef]
- Baker, M. Digital PCR hits its stride. Nat. Methods 2012, 9, 541–544. [Google Scholar] [CrossRef]
- Tan, L.L.; Loganathan, N.; Agarwalla, S.; Yang, C.; Yuan, W.Y.; Zeng, J.; Wu, R.G.; Wang, W.; Duraiswamy, S. Current commercial dPCR platforms: Technology and market review. Crit. Rev. Biotechnol. 2023, 43, 433–464. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.; Dasgupta, K.; Xing, Y.P.; Hernandez, B.T.; Shao, M.; Rohozinski, D.; Kovak, E.; Lin, J.; de Oliveira, M.L.P.; Stover, E.; et al. Accurate measurement of transgene copy number in crop plants using droplet digital PCR. Plant J. 2017, 90, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Mizukami, Y.; Ono, Y.; Sato, H.; Hayashi, A.; Kawabata, H.; Koizumi, K.; Masuda, S.; Teshima, S.; Takahashi, K.; et al. Digital PCR-based plasma cell-free DNA mutation analysis for early-stage pancreatic tumor diagnosis and surveillance. J. Gastroenterol. 2020, 55, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Provencio, M.; Serna-Blasco, R.; Franco, F.; Calvo, V.; Royuela, A.; Auglyte, M.; Sanchez-Hernandez, A.; de Julian Campayo, M.; Garcia-Giron, C.; Domine, M.; et al. Analysis of circulating tumour DNA to identify patients with epidermal growth factor receptor-positive non-small cell lung cancer who might benefit from sequential tyrosine kinase inhibitor treatment. Eur. J. Cancer 2021, 149, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Parvin, R.; Fan, Q.; Ye, F. Emerging digital PCR technology in precision medicine. Biosens. Bioelectron. 2022, 211, 114344. [Google Scholar] [CrossRef]
- Nyaruaba, R.; Mwaliko, C.; Dobnik, D.; Neuzil, P.; Amoth, P.; Mwau, M.; Yu, J.P.; Yang, H.; Wei, H.P. Digital PCR Applications in the SARS-CoV-2/COVID-19 Era: A Roadmap for Future Outbreaks. Clin. Microbiol. Rev. 2022, 35, e0016821. [Google Scholar] [CrossRef]
- Falabella, M.; Sun, L.; Barr, J.; Pena, A.Z.; Kershaw, E.E.; Gingras, S.; Goncharova, E.A.; Kaufman, B.A. Single-Step qPCR and dPCR Detection of Diverse CRISPR-Cas9 Gene Editing Events In Vivo. G3 Genes Genomes Genet. 2017, 7, 3533–3542. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Y.; Ugozzoli, L.; Pal, B.K.; Wallace, R.B. Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle cell anemia. Proc. Natl. Acad. Sci. USA 1989, 86, 2757–2760. [Google Scholar] [CrossRef]
- Du, P.; Li, B.; Liu, X.; Yang, L.; Ren, N.; Li, Y.; Huang, Q. Enhanced Taq Variant Enables Efficient Genome Editing Testing and Mutation Detection. CRISPR J. 2022, 5, 131–145. [Google Scholar] [CrossRef]
- Kundu, S.; Ali, M.A.; Handin, N.; Conway, L.P.; Rendo, V.; Artursson, P.; He, L.; Globisch, D.; Sjoblom, T. Common and mutation specific phenotypes of KRAS and BRAF mutations in colorectal cancer cells revealed by integrative -omics analysis. J. Exp. Clin. Cancer Res. 2021, 40, 225. [Google Scholar] [CrossRef]
- Jacob, L.S.; Vanharanta, S.; Obenauf, A.C.; Pirun, M.; Viale, A.; Socci, N.D.; Massague, J. Metastatic Competence Can Emerge with Selection of Preexisting Oncogenic Alleles without a Need of New Mutations. Cancer Res. 2015, 75, 3713–3719. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Huang, Q.; Whitington, T.; Gao, P.; Lindberg, J.F.; Yang, Y.; Sun, J.; Vaisanen, M.R.; Szulkin, R.; Annala, M.; Yan, J.; et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat. Genet. 2014, 46, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.C.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Liu, J.; Huang, Q. A Digital PCR Method Based on Highly Specific Taq for Detecting Gene Editing and Mutations. Int. J. Mol. Sci. 2023, 24, 13405. https://doi.org/10.3390/ijms241713405
Li B, Liu J, Huang Q. A Digital PCR Method Based on Highly Specific Taq for Detecting Gene Editing and Mutations. International Journal of Molecular Sciences. 2023; 24(17):13405. https://doi.org/10.3390/ijms241713405
Chicago/Turabian StyleLi, Bo, Junhao Liu, and Qilai Huang. 2023. "A Digital PCR Method Based on Highly Specific Taq for Detecting Gene Editing and Mutations" International Journal of Molecular Sciences 24, no. 17: 13405. https://doi.org/10.3390/ijms241713405
APA StyleLi, B., Liu, J., & Huang, Q. (2023). A Digital PCR Method Based on Highly Specific Taq for Detecting Gene Editing and Mutations. International Journal of Molecular Sciences, 24(17), 13405. https://doi.org/10.3390/ijms241713405