
Halogen Bonding Involving Isomeric Isocyanide/Nitrile Groups

 , ,
, ,  and
and 
Abstract
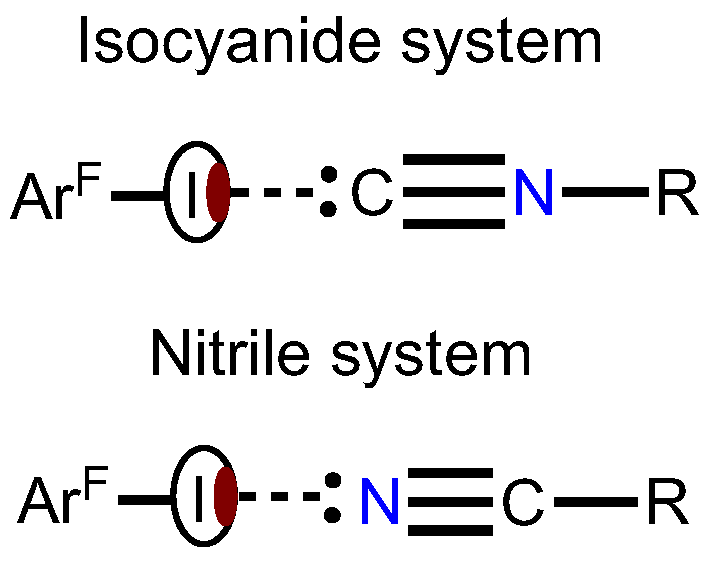
:1. Introduction
2. Results
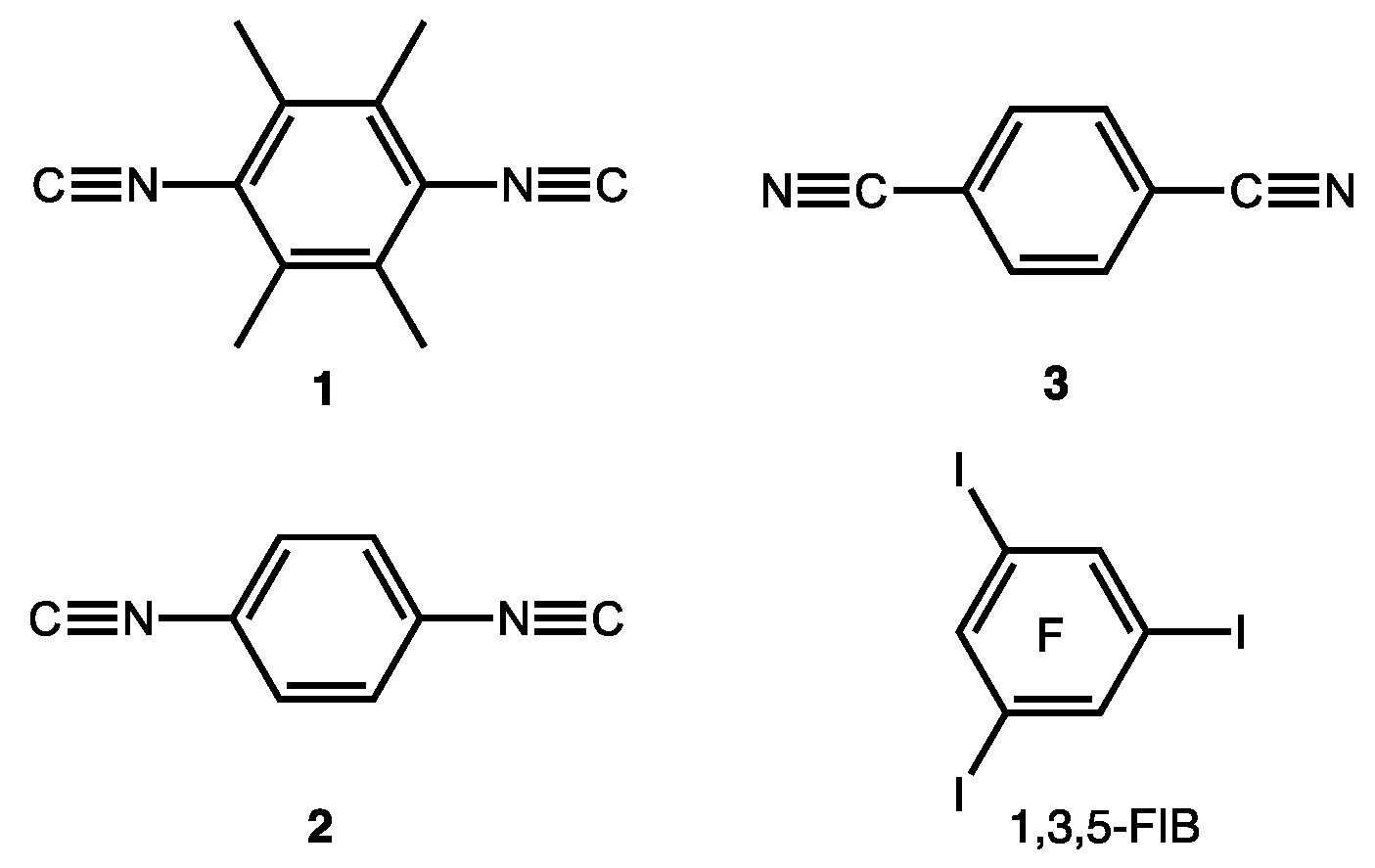
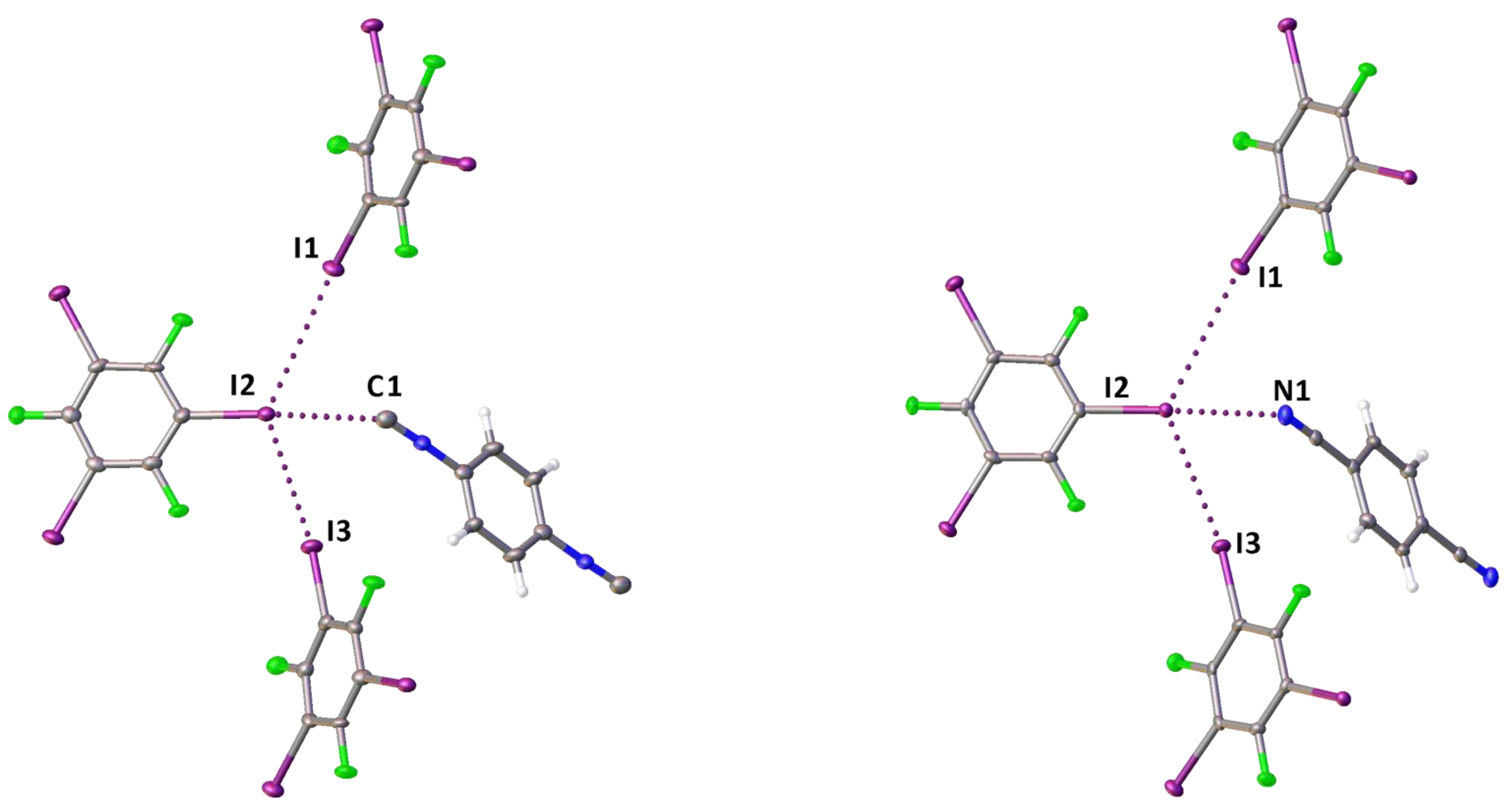
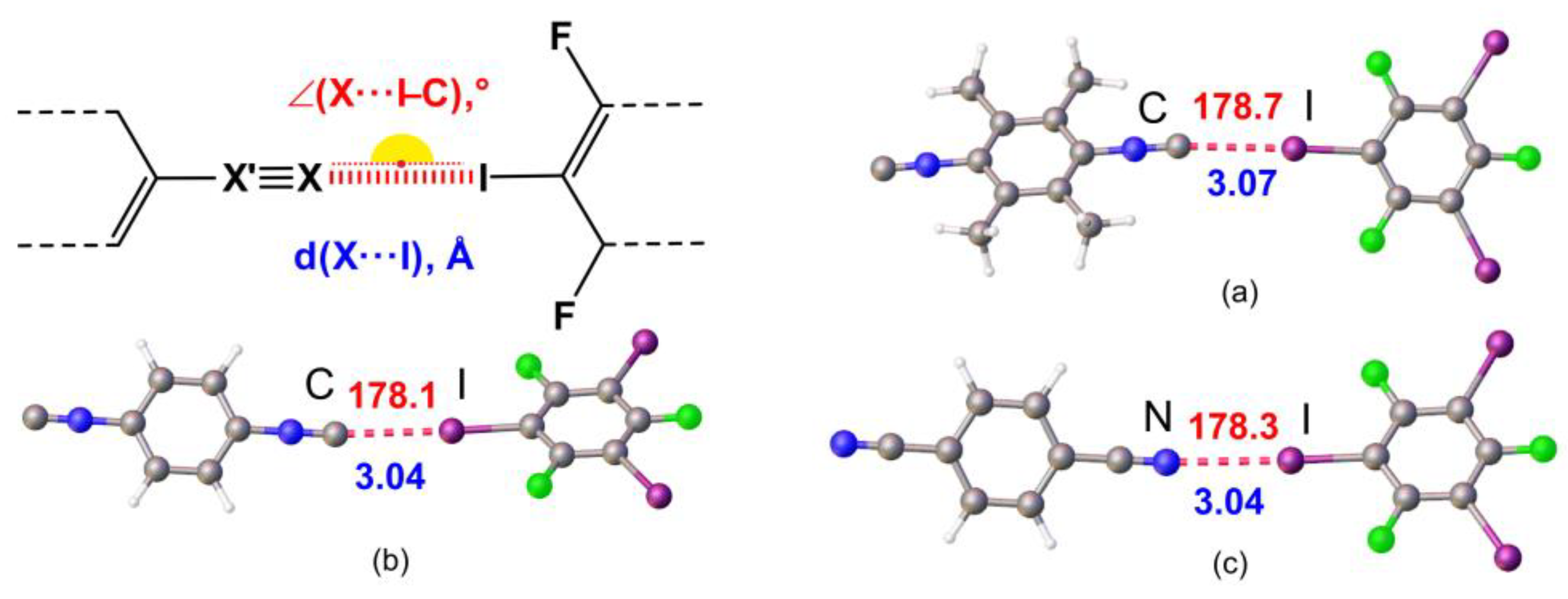
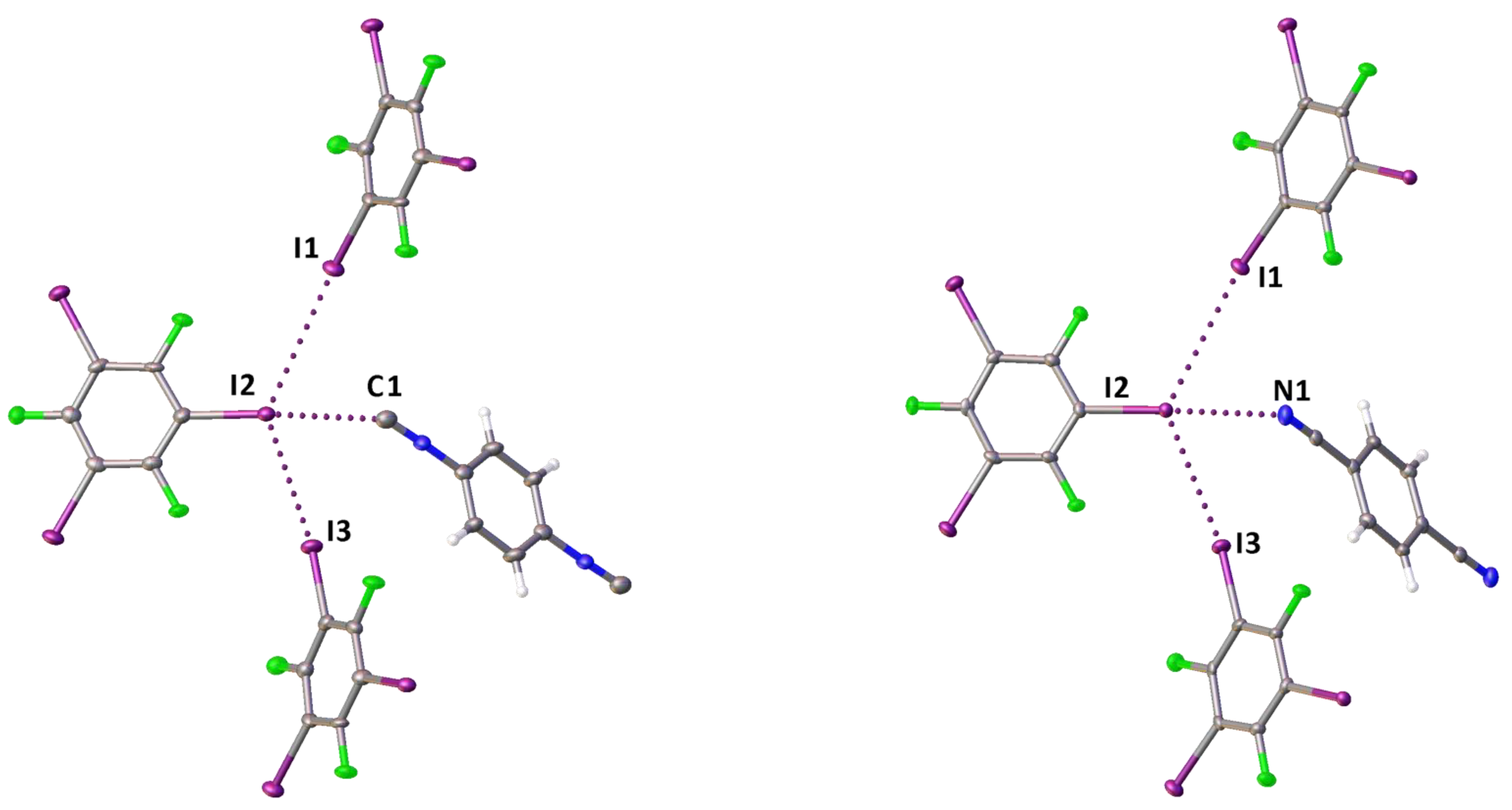
2.1. Crystal Growth and Structural Motifs of the XRD Structures
2.2. Theoretical Calculations
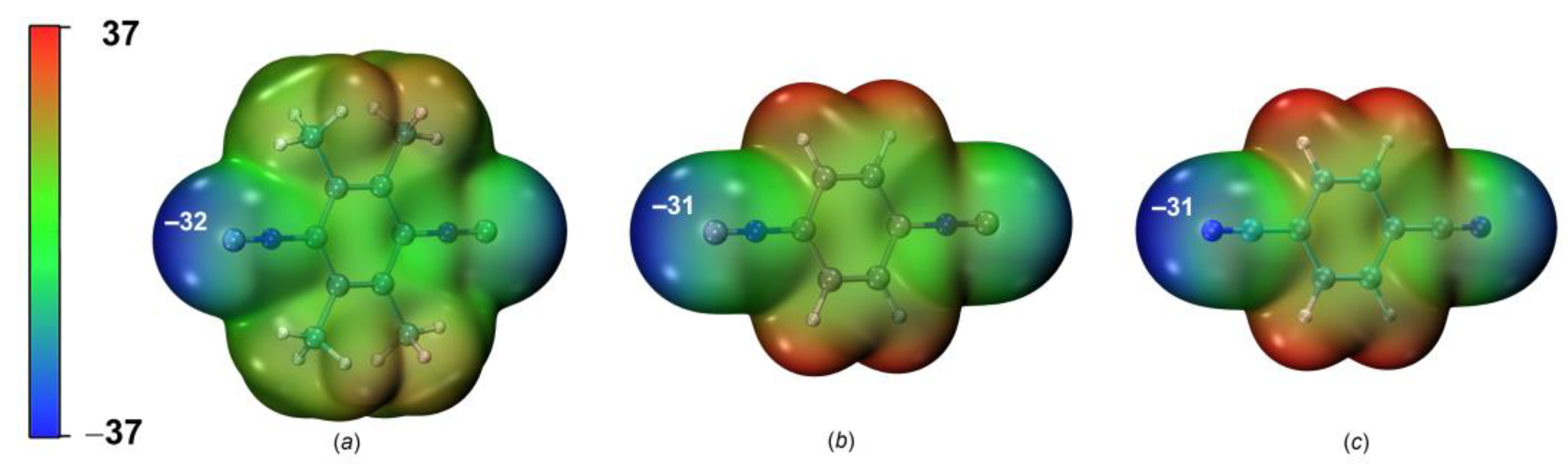
2.2.1. Nucleophilicity and Molecular Electrostatic Potential
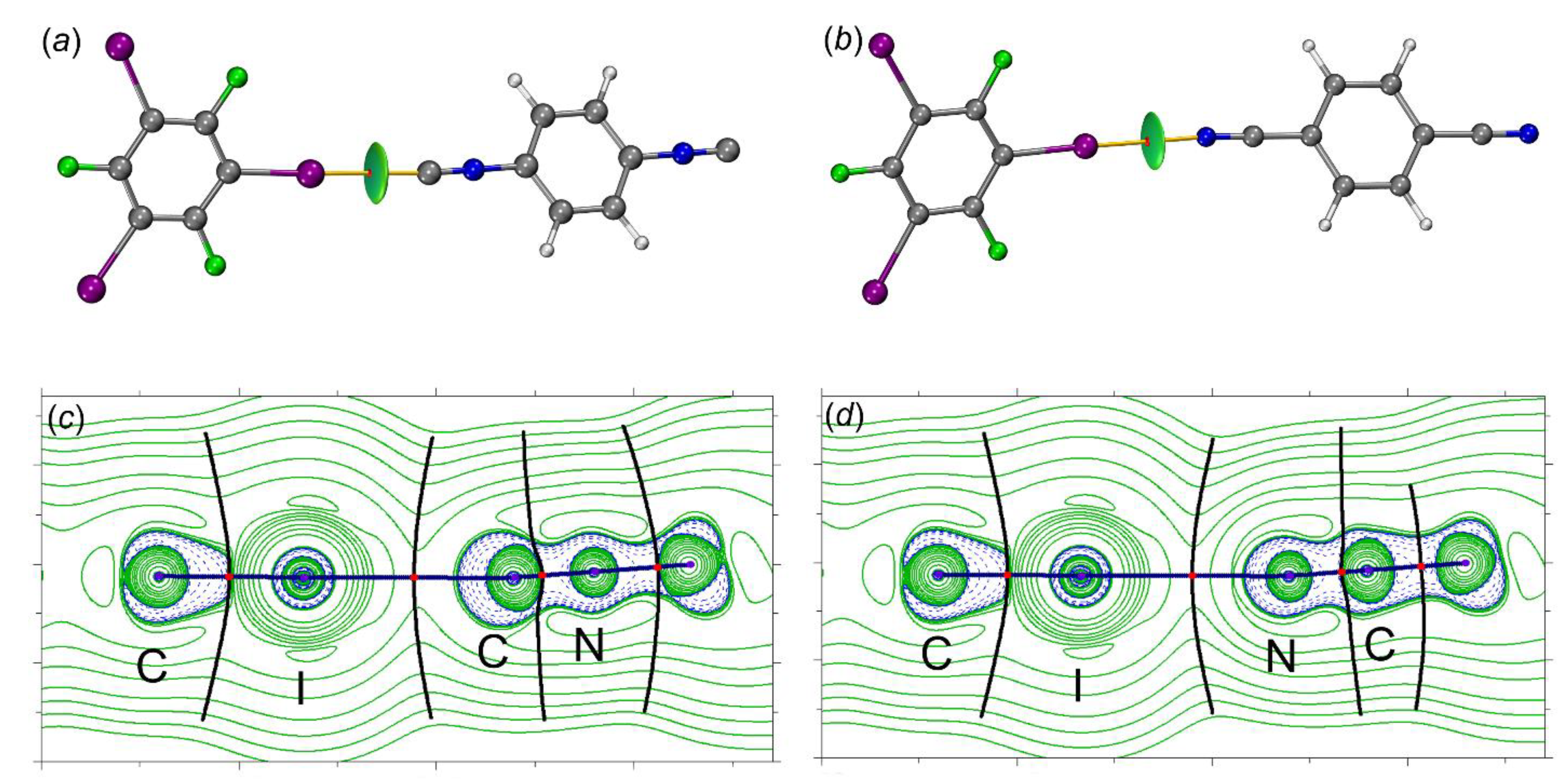
2.2.2. QTAIM-IGMH
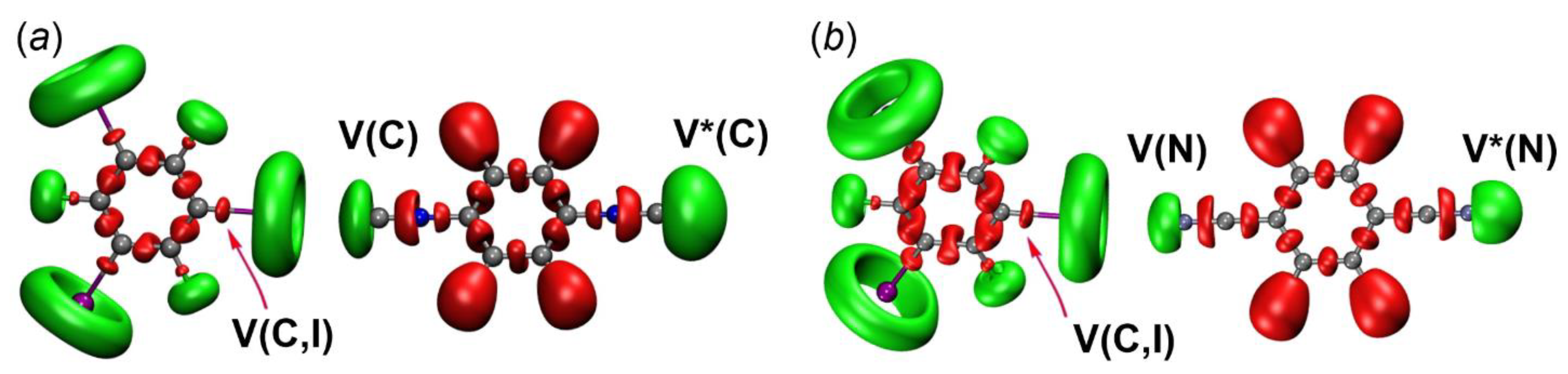
2.2.3. Electron Localization Function
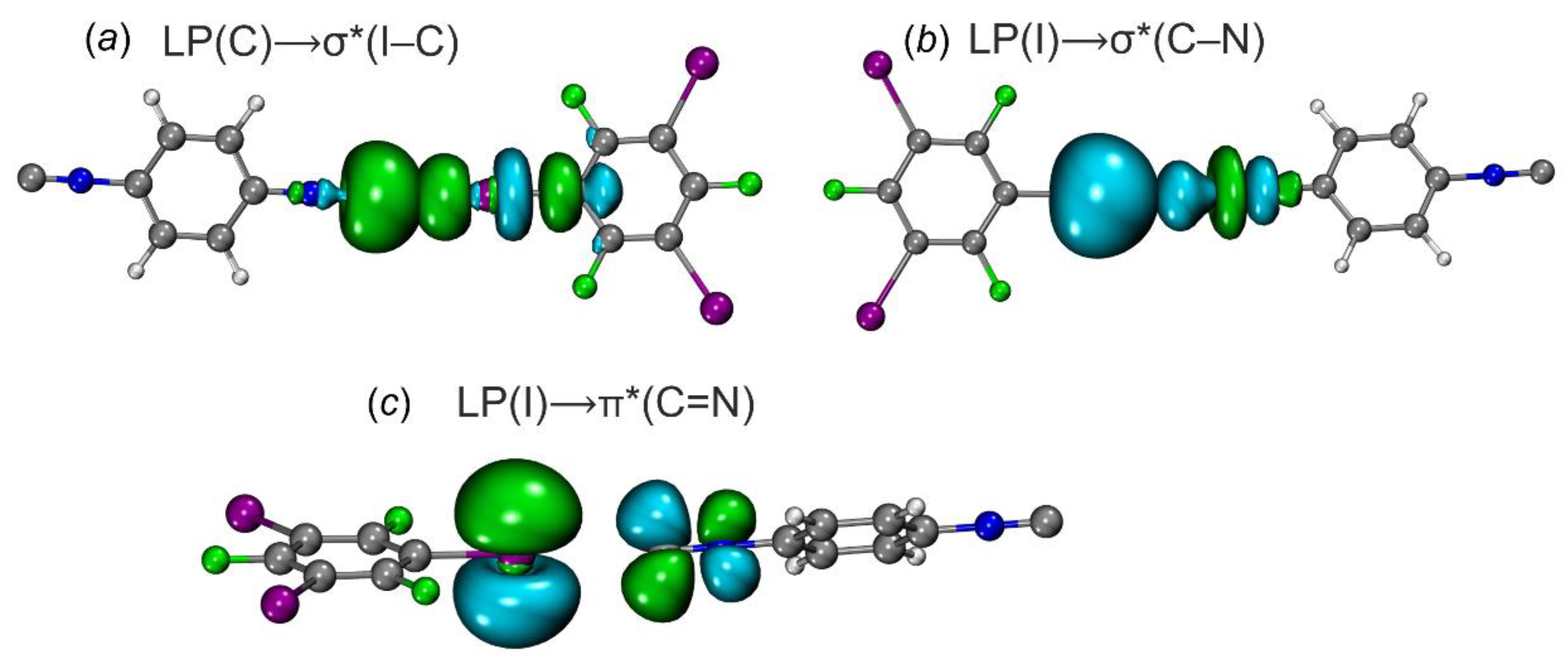
2.2.4. Natural Bond Orbital Approach
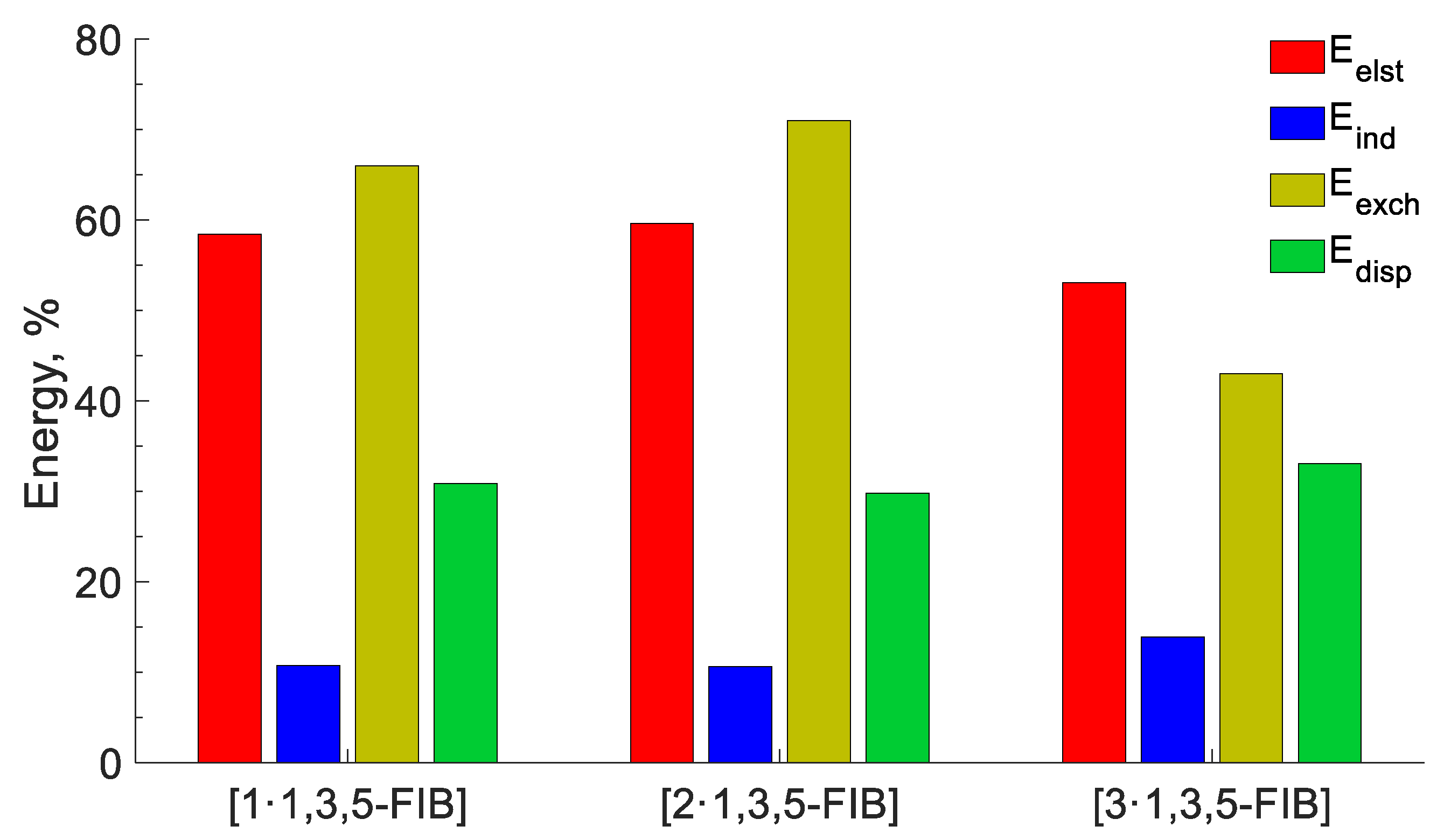
2.2.5. Energy
3. Discussion
4. Materials and Methods
4.1. Materials and Instruments
4.2. Cocrystal Growth
4.3. XRD Studies
4.4. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornaton, Y.; Djukic, J.-P. Noncovalent Interactions in Organometallic Chemistry: From Cohesion to Reactivity, a New Chapter. Acc. Chem. Res. 2021, 54, 3828–3840. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals 2021, 11, 1205. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. Chalcogen and pnictogen bonds: Insights and relevance. Curr. Sci. 2021, 120, 1848–1853. [Google Scholar] [CrossRef]
- Miller, D.K.; Chernyshov, I.Y.; Torubaev, Y.V.; Rosokha, S.V. From weak to strong interactions: Structural and electron topology analysis of the continuum from the supramolecular chalcogen bonding to covalent bonds. Phys. Chem. Chem. Phys. 2022, 24, 8251–8259. [Google Scholar] [CrossRef] [PubMed]
- Resnati, G.; Metrangolo, P. Celebrating 150 years from Mendeleev: The Periodic Table of Chemical Interactions. Coord. Chem. Rev. 2020, 420, 213409. [Google Scholar] [CrossRef]
- Brammer, L. Halogen bonding, chalcogen bonding, pnictogen bonding, tetrel bonding: Origins, current status and discussion. Faraday Discuss. 2017, 203, 485–507. [Google Scholar] [CrossRef]
- Brammer, L.; Peuronen, A.; Roseveare, T.M. Halogen bonds, chalcogen bonds, pnictogen bonds, tetrel bonds and other [sigma]-hole interactions: A snapshot of current progress. Acta Crystallogr. Sect. C 2023, 79, 204–216. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Li, B.; Zang, S.-Q.; Wang, L.-Y.; Mak, T.C.W. Halogen bonding: A powerful, emerging tool for constructing high-dimensional metal-containing supramolecular networks. Coord. Chem. Rev. 2016, 308, 1–21. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. Engl. 2018, 57, 6004–6016. [Google Scholar] [CrossRef] [PubMed]
- Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. (Eds.) Non-Covalent Interactions in the Synthesis and Design of New Compounds; Wiley Online Library: Hoboken, NJ, USA, 2016. [Google Scholar]
- Saha, B.K.; Veluthaparambath, R.V.P.; Krishna, G.V. Halogen⋯Halogen Interactions: Nature, Directionality and Applications. Chem. Asian J. 2023, 18, e202300067. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Xin, D.; Matti, T.; Matti, H. Halogen Bonding in Crystal Engineering. In Recent Advances in Crystallography; Jason, B.B., Ed.; IntechOpen: Rijeka, Croatia, 2012; Chapter 7. [Google Scholar]
- Teyssandier, J.; Mali, K.S.; De Feyter, S. Halogen Bonding in Two-Dimensional Crystal Engineering. ChemistryOpen 2020, 9, 225–241. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Bokach, N.A.; Yu. Kukushkin, V.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chem. Eur. J. 2022, 28, e202103173. [Google Scholar]
- Hein, R.; Beer, P.D. Halogen bonding and chalcogen bonding mediated sensing. Chem. Sci. 2022, 13, 7098–7125. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Suwardi, A.; Wong, C.J.E.; Loh, X.J.; Li, Z. Halogen bonding regulated functional nanomaterials. Nanoscale Adv. 2021, 3, 6342–6357. [Google Scholar] [CrossRef]
- Pancholi, J.; Beer, P.D. Halogen bonding motifs for anion recognition. Coord. Chem. Rev. 2020, 416, 213281. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; da Silva, M.F.C.G.; Pombeiro, A.J. Noncovalent Interactions in Catalysis; Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Breugst, M.; von der Heiden, D.; Schmauck, J. Novel Noncovalent Interactions in Catalysis: A Focus on Halogen, Chalcogen, and Anion-π Bonding. Synthesis 2017, 49, 3224–3236. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coord. Chem. Rev. 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen bonding in polymer science: From crystal engineering to functional supramolecular polymers and materials. Polym. Chem. 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking Back, Looking Forward at Halogen Bonding in Drug Discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef]
- Ho, P.S. Halogen bonding in medicinal chemistry: From observation to prediction. Future Med. Chem. 2017, 9, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Pavan, B.; Ferretti, V. Can pharmaceutical co-crystals provide an opportunity to modify the biological properties of drugs? Drug Discov Today 2017, 22, 1134–1138. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discov 2012, 7, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Bayse, C.A. Halogen bonding from the bonding perspective with considerations for mechanisms of thyroid hormone activation and inhibition. New J. Chem. 2018, 42, 10623–10632. [Google Scholar] [CrossRef]
- Marsan, E.S.; Bayse, C.A. A Halogen Bonding Perspective on Iodothyronine Deiodinase Activity. Molecules 2020, 25, 1328. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Novikov, A.S.; Boyarskiy, V.P.; Kukushkin, V.Y. The halogen bond with isocyano carbon reduces isocyanide odor. Nat. Commun. 2020, 11, 2921. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Popov, R.A.; Smirnov, A.S.; Eliseeva, A.A.; Novikov, A.S.; Boyarskiy, V.P.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Isocyanide and Cyanide Entities Form Isostructural Halogen Bond-Based Supramolecular Networks Featuring Five-Center Tetrafurcated Halogen···C/N Bonding. Cryst. Growth Des. 2022, 22, 6079–6087. [Google Scholar] [CrossRef]
- Smirnov, A.S.; Mikherdov, A.S.; Rozhkov, A.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Halogen Bond-Involving Supramolecular Assembly Utilizing Carbon as a Nucleophilic Partner of I⋅⋅⋅C Non-covalent Interaction. Chem. Asian J. 2023, 18, e202300037. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Kline, M.; Calabrese, J.C.; Davidson, F. Synthesis of a reverse ylide from a nucleophilic carbene. J. Am. Chem. Soc. 1991, 113, 9704–9705. [Google Scholar] [CrossRef]
- Wasilewska, A.; Gdaniec, M.; Polonski, T. CCDC 618388: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2006. [Google Scholar] [CrossRef]
- Britton, D.; Gleason, W.B. Dicyanodurene-p-tetrafluorodiiodobenzene (1/1). Acta Crystallogr. Sect. E 2002, 58, o1375–o1377. [Google Scholar] [CrossRef]
- Watari, F. Vibrational frequencies, assignments and normal-coordinate analysis for the methyl isocyanide-borane complex. Inorg. Chem. 1982, 21, 1442–1446. [Google Scholar] [CrossRef]
- Bell, A.; Walton, R.A.; Edwards, D.A.; Poulter, M.A. Cationic copper(I) isocyanide complexes, [Cu(CNR)4]+ (R = CH3, C(CH3)3 and 2,6-(CH3)2C6H3): Preparations, spectroscopic properties and reactions with neutral ligands. A comparison of the vibrational spectra of [Cu(CNCH3)4]+, [Cu(NCCH3)4]+ and [Cu(NCCD3)4]+. Inorganica Chim. Acta 1985, 104, 171–178. [Google Scholar] [CrossRef]
- Tumanov, V.V.; Tishkov, A.A.; Mayr, H. Nucleophilicity Parameters for Alkyl and Aryl Isocyanides. Angew. Chem. Int. Ed. 2007, 46, 3563–3566. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Molecular electrostatic potentials and noncovalent interactions. WIREs Comput. Mol. Sci. 2017, 7, e1326. [Google Scholar] [CrossRef]
- Bader, R.F. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Relationships between Interaction Energy and Electron Density Properties for Homo Halogen Bonds of the [(A)nY–X···X–Z(B)m] Type (X = Cl, Br, I). Molecules 2019, 24, 2733. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef]
- Zhu, S.; Asim Khan, M.; Wang, F.; Bano, Z.; Xia, M. Rapid removal of toxic metals Cu2+ and Pb2+ by amino trimethylene phosphonic acid intercalated layered double hydroxide: A combined experimental and DFT study. Chem. Eng. J. 2020, 392, 123711. [Google Scholar] [CrossRef]
- Huang, W.; Lin, R.; Zhao, X.; Li, Q.; Huang, Y.; Ye, G. How does a weak interaction change from a reactive complex to a saddle point in a reaction? Comput. Theor. Chem. 2020, 1173, 112640. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. Visualization Analysis of Weak Interactions in Chemical Systems. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Lefebvre, C.; Klein, J.; Khartabil, H.; Boisson, J.-C.; Hénon, E. IGMPlot: A program to identify, characterize, and quantify molecular interactions. J. Comput. Chem. 2023, 44, 1750–1766. [Google Scholar] [CrossRef]
- Šivickytė, O.; Costa, P.J. Intrinsic bond strength index as a halogen bond interaction energy predictor. Phys. Chem. Chem. Phys. 2023, 25, 17535–17546. [Google Scholar] [CrossRef]
- Klein, J.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. New Way for Probing Bond Strength. J. Phys. Chem. A 2020, 124, 1850–1860. [Google Scholar] [CrossRef]
- Silvi, B.; Gillespie, R.J.; Gatti, C. Electron density analysis. In Comprehensive Inorganic Chemistry II; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 9, pp. 187–226. [Google Scholar]
- Fuster, F.; Sevin, A.; Silvi, B. Topological Analysis of the Electron Localization Function (ELF) Applied to the Electrophilic Aromatic Substitution. J. Phys. Chem. A 2000, 104, 852–858. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Altun, A.; Neese, F.; Bistoni, G. Local energy decomposition analysis of hydrogen-bonded dimers within a domain-based pair natural orbital coupled cluster study. Beilstein J. Org. Chem. 2018, 14, 919–929. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Parrish, R.M.; Sherrill, C.D.; Turney, J.M.; Schaefer, H.F., 3rd. Large-scale symmetry-adapted perturbation theory computations via density fitting and Laplace transformation techniques: Investigating the fundamental forces of DNA-intercalator interactions. J. Chem. Phys. 2011, 135, 174107. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Jaeger, H.M.; Carrell, E.J.; Tschumper, G.S.; Sherrill, C.D. Accurate Interaction Energies for Problematic Dispersion-Bound Complexes: Homogeneous Dimers of NCCN, P2, and PCCP. J. Chem. Theory Comput. 2011, 7, 2842–2851. [Google Scholar] [CrossRef]
- Hayes, I.; Stone, A. An intermolecular perturbation theory for the region of moderate overlap. Mol. Phys. 1984, 53, 83–105. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation theory approach to intermolecular potential energy surfaces of van der Waals complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. WIREs Comput. Mol. Sci. 2020, 10, e1452. [Google Scholar] [CrossRef]
- Grant Hill, J.; Legon, A.C. On the directionality and non-linearity of halogen and hydrogen bonds. Phys. Chem. Chem. Phys. 2015, 17, 858–867. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-bonds to angular distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Stone, A.J.; Misquitta, A.J. Charge-transfer in Symmetry-Adapted Perturbation Theory. Chem. Phys. Lett. 2009, 473, 201–205. [Google Scholar] [CrossRef]
- NETZSCH Proteus Software v.6.1; Netzsch-Gerätebau Bayern: Selb, Germany, 2013.
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132, 154104. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, J.D.; Neese, F.; Pantazis, D.A. All-electron scalar relativistic basis sets for the elements Rb–Xe. J. Comput. Chem. 2020, 41, 1842–1849. [Google Scholar] [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Actinides. J. Chem. Theory Comput. 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO7; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Modell. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L. Psi4: An open-source ab initio electronic structure program. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Nunzi, F.; Cesario, D.; Belpassi, L.; Tarantelli, F.; Roncaratti, L.F.; Falcinelli, S.; Cappelletti, D.; Pirani, F. Insight into the halogen-bond nature of noble gas-chlorine systems by molecular beam scattering experiments, ab initio calculations and charge displacement analysis. Phys. Chem. Chem. Phys. 2019, 21, 7330–7340. [Google Scholar] [CrossRef] [PubMed]
- Belpassi, L.; Infante, I.; Tarantelli, F.; Visscher, L. The Chemical Bond between Au(I) and the Noble Gases. Comparative Study of NgAuF and NgAu+ (Ng = Ar, Kr, Xe) by Density Functional and Coupled Cluster Methods. J. Am. Chem. Soc. 2008, 130, 1048–1060. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. Intermolecular interaction characteristics of the all-carboatomic ring, cyclo[18]carbon: Focusing on molecular adsorption and stacking. Carbon 2021, 171, 514–523. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Duley, S.; Domingo, L.R. Understanding local electrophilicity/nucleophilicity activation through a single reactivity difference index. Org. Biomol. Chem. 2012, 10, 2855–2861. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cocrystal | Contact I···Nu (Nu = C, I, or F) | Interatomic Distance, Å | Nc 1 | Angle C–I···Nu, ° | Angle I···Nu–Y, ° |
|---|---|---|---|---|---|
| 1·1,3,5-FIB | I2···C1 | 3.084 (5) | 0.84 | 173.22 (16) | 153.4 (4) |
| I3···C7 | 3.035 (5) | 0.82 | 173.38 (16) | 159.5 (4) | |
| I1···F2 | 2.992 (3) | 0.87 | 171.54 (13) | 136.9 (3) | |
| 2·2(1,3,5-FIB) | I2···C1 | 2.957 (6) | 0.80 | 172.0 (2) | 138.4 (5) |
| I1···I2 | 3.9561 (5) | 1.00 | 163.51 (14) | 116.60 (15) | |
| I3···I2 | 3.8022 (4) | 0.96 | 172.52 (14) | 110.56 (14) | |
| 3·2(1,3,5-FIB) | I2···N1 | 2.935 (5) | 0.80 | 173.79 (19) | 135.9 (5) |
| I1···I2 | 3.9625 (6) | 1.00 | 163.79 (14) | 116.95 (18) | |
| I3···I2 | 3.8122 (5) | 0.96 | 171.32 (14) | 111.63 (17) |
| 1 | 2 | 3 | |
|---|---|---|---|
| NNu, eV | 2.35 | 1.80 | 1.40 |
| NNuloc (C or N), e*eV | 0.33 | 0.30 | 0.23 |
| Contact | Clusters | ρb | ∇2ρb | Vb | Gb | Hb | λ2 | ELF | IBSI | δgpair |
|---|---|---|---|---|---|---|---|---|---|---|
| I···C | [1‧1,3,5-FIB] | 0.0177 | 0.0467 | –0.0101 | 0.0109 | 0.0008 | –0.0115 | 0.09 | 0.032 | 0.027 |
| I···C | [2‧1,3,5-FIB] | 0.0186 | 0.0494 | –0.0109 | 0.0116 | 0.0007 | –0.0123 | 0.09 | 0.034 | 0.028 |
| I···N | [3‧1,3,5-FIB] | 0.0150 | 0.0505 | –0.0094 | 0.0110 | 0.0016 | –0.0103 | 0.05 | 0.025 | 0.024 |
| Clusters | V(C or N), e [VELF, Ǻ3] | V(C,I), e |
|---|---|---|
| [1‧1,3,5-FIB] | 2.62 [100]; 2.63 * [380] | 1.75; 1.67 *; 1.67 * |
| [2‧1,3,5-FIB] | 2.60 [170]; 2.61 * [610] | 1.76, 1.68 *; 1.68 * |
| [3‧1,3,5-FIB] | 3.31 [90]; 3.31 * [460] | 1.74, 1.67 *; 1.67 * |
| Clusters | Transition | E(2) | Δocc |
|---|---|---|---|
| [1‧1,3,5-FIB] | LP(C)⟶σ*(I–C) | 9.6 | 44 |
| LP(I)⟶σ*/π*(C≡N) | 2.1 | 15 | |
| [2‧1,3,5-FIB] | LP(C)⟶σ*(I–C) | 10.2 | 46 |
| LP(I)⟶σ*/π*(C≡N) | 2.3 | 16 | |
| [3‧1,3,5-FIB] | LP(N)⟶σ*(I–C) | 5.2 | 16 |
| LP(I)⟶σ*/π*(N≡C) | 1.8 | 12 |
| Cluster | Eelst | Eind (Ect) | Eexch | Edisp | EintSAPT * | EintSM | EbSM |
|---|---|---|---|---|---|---|---|
| [1‧1,3,5-FIB] | –8.7 | –1.6 (0.5) | 9.8 | –4.6 | –5.1 | –4.2 | –4.1 |
| [2‧1,3,5-FIB] | –9.0 | –1.6 (0.5) | 10.6 | –4.5 | –4.6 | –3.8 | –3.8 |
| [3‧1,3,5-FIB] | –6.1 | –1.6 (0.1) | 6.4 | –3.8 | –5.0 | –3.7 | –3.7 |
| Methods (Descriptors) | HaB I···Ciso vs. I···Nnitr | Comments |
|---|---|---|
| Global Nucleophilicity (NNu) | NNiso > NNnitr | Diisocyanides are more nucleophilic than the dinitrile |
| Local Nucleophilicity (NNuloc) | NNuloc(iso) > NNuloc(nitr) | |
| MEP (Vs,min) | Vs,miniso~Vs,minnitr | Electrostatic potentials are nearly the same for both systems |
| QTAIM (ρb) | ρbiso > ρbnitr | The electron density values at the BCP of the HaB are slightly higher for the diisocyanide system; this indicates a stronger binding of the diisocyanides to 1,3,5-FIB |
| IBSI | IBSIiso > IBSInitr | The index IBSI indicates a stronger binding of the diisocyanides to 1,3,5-FIB |
| ELF (V(C,I)) | V(C,I)iso > V(C,I)nitr | Charge transfer effect is higher for the isocyanide systems |
| NBO (Δocc (σ*(I–C)) | Δocc iso > Δocc nitr | |
| DLPNO-CCSD(T) (EbSM) | Ebiso > Ebnitr | Binding energy between the coformers is larger for the diisocyanide structures |
| SAPT (Eexch) | Eexchiso > Eexchnitr | Repulsive Pauli energy for the I···C/N HaBs is higher for the isocyanides. This is in agreement with increased orbital interactions for the diisocyanide systems |
| SAPT (Eelst) | ΔEelstiso~ΔEelstnitrl | SAPT results indicate the dominance of Coulomb interactions in the HaB of both systems; they contribute approximately 60% to the sum of negative energy components. The dispersion contribution is no more than 30% |
| SAPT (Edisp) | ΔEdispiso~ΔEdispnitrl |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnov, A.S.; Katlenok, E.A.; Mikherdov, A.S.; Kryukova, M.A.; Bokach, N.A.; Kukushkin, V.Y. Halogen Bonding Involving Isomeric Isocyanide/Nitrile Groups. Int. J. Mol. Sci. 2023, 24, 13324. https://doi.org/10.3390/ijms241713324
Smirnov AS, Katlenok EA, Mikherdov AS, Kryukova MA, Bokach NA, Kukushkin VY. Halogen Bonding Involving Isomeric Isocyanide/Nitrile Groups. International Journal of Molecular Sciences. 2023; 24(17):13324. https://doi.org/10.3390/ijms241713324
Chicago/Turabian StyleSmirnov, Andrey S., Eugene A. Katlenok, Alexander S. Mikherdov, Mariya A. Kryukova, Nadezhda A. Bokach, and Vadim Yu. Kukushkin. 2023. "Halogen Bonding Involving Isomeric Isocyanide/Nitrile Groups" International Journal of Molecular Sciences 24, no. 17: 13324. https://doi.org/10.3390/ijms241713324
APA StyleSmirnov, A. S., Katlenok, E. A., Mikherdov, A. S., Kryukova, M. A., Bokach, N. A., & Kukushkin, V. Y. (2023). Halogen Bonding Involving Isomeric Isocyanide/Nitrile Groups. International Journal of Molecular Sciences, 24(17), 13324. https://doi.org/10.3390/ijms241713324