

A Functional Genomics Review of Non-Small-Cell Lung Cancer in Never Smokers

 , ,
, ,  , ,
, ,  and
and
Abstract
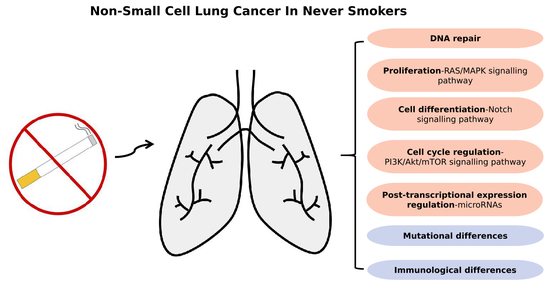
1. Introduction
2. The Genomic Profile of Lung Cancer in Never Smokers
3. Immunological Changes: Tumour Microenvironment in Never Smokers
4. Abnormalities in Growth-Stimulatory Signalling Pathways
4.1. RAS/MAPK Signalling Pathway
4.2. Mutations in Other EGFR Signalling Pathway Genes
4.2.1. Notch Signalling Pathway Genes
4.2.2. Abnormalities in Tumour Suppressor Gene Pathways: P53 and KRAS
4.2.3. PI3K–AKT–mTOR
4.2.4. microRNAs
5. Application of Molecular Biology and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, T.; Joubert, P.; Ansari-Pour, N.; Zhao, W.; Hoang, P.H.; Lokanga, R.; Moye, A.L.; Rosenbaum, J.; Gonzalez-Perez, A.; Martínez-Jiménez, F.; et al. Genomic and evolutionary classification of lung cancer in never smokers. Nat. Genet. 2021, 53, 1348–1359. [Google Scholar] [CrossRef]
- Torok, S.; Hegedus, B.; Laszlo, V.; Hoda, M.A.; Ghanim, B.; Berger, W.; Klepetko, W.; Dome, B.; Ostoros, G. Lung cancer in never smokers. Future Oncol. 2011, 7, 1195–1211. [Google Scholar] [CrossRef]
- Devarakonda, S.; Li, Y.; Martins Rodrigues, F.; Sankararaman, S.; Kadara, H.; Goparaju, C.; Lanc, L.; Pepin, K.; Waqar, S.N.; Govindan, R.; et al. Genomic Profiling of Lung Adenocarcinoma in Never-Smokers. J. Clin. Oncol. 2021, 39, 3747–3758. [Google Scholar] [CrossRef]
- Dias, M.; Linhas, R.; Campainha, S.; Conde, S.; Barroso, A. Lung cancer in never-smokers—What are the differences? Acta Oncol. 2017, 56, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Korpanty, G.J.; Kamel-Reid, S.; Pintilie, M.; Hwang, D.M.; Zer, A.; Liu, G.; Leighl, N.B.; Feld, R.; Siu, L.L.; Bedard, P.L.; et al. Lung cancer in never smokers from the Princess Margaret Cancer Centre. Oncotarget 2018, 9, 22559–22570. [Google Scholar] [CrossRef] [PubMed]
- Smolle, E.; Pichler, M. Non-Smoking-Associated Lung Cancer: A distinct Entity in Terms of Tumor Biology, Patient Characteristics and Impact of Hereditary Cancer Predisposition. Cancers 2019, 11, 204. [Google Scholar] [CrossRef]
- Hitchman, S.C.; Fong, G.T. Gender empowerment and female-to-male smoking prevalence ratios. Bull. World Health Organ. 2011, 89, 195–202. [Google Scholar] [CrossRef]
- Landi, M.T.; Synnott, N.C.; Rosenbaum, J.; Zhang, T.; Zhu, B.; Shi, J.; Zhao, W.; Kebede, M.; Sang, J.; Choi, J.; et al. Tracing Lung Cancer Risk Factors Through Mutational Signatures in Never-Smokers. Am. J. Epidemiol. 2021, 190, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Hammouz, R.Y.; Kostanek, J.K.; Dudzisz, A.; Witas, P.; Orzechowska, M.; Bednarek, A.K. Differential expression of lung adenocarcinoma transcriptome with signature of tobacco exposure. J. Appl. Genet. 2020, 61, 421–437. [Google Scholar] [CrossRef]
- Brambilla, E.; Gazdar, A. Pathogenesis of lung cancer signalling pathways: Roadmap for therapies. Eur. Respir. J. 2009, 33, 1485–1497. [Google Scholar] [CrossRef]
- Mack, P.C.; I Klein, M.; Ayers, K.L.; Zhou, X.; Guin, S.; Fink, M.; Rossi, M.; Ai-Kateb, H.; O’connell, T.; Hantash, F.M.; et al. Targeted Next-Generation Sequencing Reveals Exceptionally High Rates of Molecular Driver Mutations in Never-Smokers With Lung Adenocarcinoma. Oncologist 2022, 27, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Md, S.; Alhakamy, N.A.; Karim, S.; Gabr, G.A.; Iqubal, M.K.; Murshid, S.S.A. Signaling Pathway Inhibitors, miRNA, and Nanocarrier-Based Pharmacotherapeutics for the Treatment of Lung Cancer: A Review. Pharmaceutics 2021, 13, 2120. [Google Scholar] [CrossRef] [PubMed]
- Saab, S.; Zalzale, H.; Rahal, Z.; Khalifeh, Y.; Sinjab, A.; Kadara, H. Insights Into Lung Cancer Immune-Based Biology, Prevention, and Treatment. Front. Immunol. 2020, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J.; Wu, P.; Zhou, L.; Lu, B.; Ying, K.; Chen, E.; Lu, Y.; Liu, P. Smoker and non-smoker lung adenocarcinoma is characterized by distinct tumor immune microenvironments. OncoImmunology 2018, 7, e1494677. [Google Scholar] [CrossRef] [PubMed]
- Koba, H.; Kimura, H.; Yoneda, T.; Ogawa, N.; Tanimura, K.; Tambo, Y.; Sone, T.; Hosomichi, K.; Tajima, A.; Kasahara, K. NOTCH alteration in EGFR-mutated lung adenocarcinoma leads to histological small-cell carcinoma transformation under EGFR-TKI treatment. Transl. Lung Cancer Res. 2021, 10, 4161–4173. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Roumeliotis, T.I.; Chang, Y.H.; Chen, C.T.; Han, C.L.; Lin, M.H.; Chen, H.W.; Chang, G.C.; Chang, Y.L.; Chen, Y.J.; et al. Proteogenomics of Non-smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell 2020, 182, 226–244.e17. [Google Scholar] [CrossRef]
- Paik, P.K.; Johnson, M.L.; D’Angelo, S.P.; Sima, C.S.; Ang, D.; Dogan, S.; Miller, V.A.; Ladanyi, M.; Kris, M.G.; Riely, G.J. Driver mutations determine survival in smokers and never-smokers with stage IIIB/IV lung adenocarcinomas. Cancer 2012, 118, 5840–5847. [Google Scholar] [CrossRef]
- Mazières, J.; Rouquette, I.; Lepage, B.; Milia, J.; Brouchet, L.; Guibert, N.; Beau-Faller, M.; Validire, P.; Hofman, P.; Fouret, P. Specificities of Lung Adenocarcinoma in Women Who Have Never Smoked. J. Thorac. Oncol. 2013, 8, 923–929. [Google Scholar] [CrossRef]
- Samet, J.M.; Avila-Tang, E.; Boffetta, P.; Hannan, L.M.; Olivo-Marston, S.; Thun, M.J.; Rudin, C.M. Lung Cancer in Never Smokers: Clinical Epidemiology and Environmental Risk Factors. Clin. Cancer Res. 2009, 15, 5626–5645. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Kuśnierczyk, P. Genetic differences between smokers and never-smokers with lung cancer. Front. Immunol. 2023, 14, 1063716. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and Biological Features Associated With Epidermal Growth Factor Receptor Gene Mutations in Lung Cancers. JNCI J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.M.; Sun, K.Y.; Ruestow, P.; Cowan, D.M.; Madl, A.K. Lung cancer mutation profile of EGFR, ALK, and KRAS: Meta-analysis and comparison of never and ever smokers. Lung Cancer 2016, 102, 122–134. [Google Scholar] [CrossRef]
- Cheng, E.S.; Weber, M.F.; Steinberg, J.; Canfell, K.; Yu, X.Q. Evaluating risk factors for lung cancer among never-smoking individuals using two Australian studies. J. Cancer Res. Clin. Oncol. 2022, 148, 2827–2840. [Google Scholar] [CrossRef]
- Mountzios, G.; Planchard, D.; Besse, B.; Validire, P.; Girard, P.; Devisme, C.; Dimopoulos, M.-A.; Soria, J.-C.; Fouret, P. Mitogen-Activated Protein Kinase Activation in Lung Adenocarcinoma: A Comparative Study between Ever Smokers and Never Smokers. Clin. Cancer Res. 2008, 14, 4096–4102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hammouz, R.Y.; Orzechowska, M.; Anusewicz, D.; Bednarek, A.K. X Or Y Cancer: An Extensive Analysis of Sex Differences in Lung Adenocarcinoma. Curr. Oncol. 2023, 30, 1395–1415. [Google Scholar] [CrossRef]
- Guo, D.; Wang, M.; Shen, Z.; Zhu, J. A new immune signature for survival prediction and immune checkpoint molecules in lung adenocarcinoma. J. Transl. Med. 2020, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Yano, T.; Morodomi, Y.; Yoshida, T.; Kohno, M.; Haro, A.; Shikada, Y.; Okamoto, T.; Maruyama, R.; Maehara, Y. Serum antioxidant capacity and oxidative injury to pulmonary DNA in never-smokers with primary lung cancer. Anticancer Res. 2012, 32, 1063–1067. [Google Scholar] [PubMed]
- Sui, Q.; Liang, J.; Hu, Z.; Chen, Z.; Bi, G.; Huang, Y.; Li, M.; Zhan, C.; Lin, Z.; Wang, Q. Genetic and microenvironmental differences in non-smoking lung adenocarcinoma patients compared with smoking patients. Transl. Lung Cancer Res. 2020, 9, 1407–1421. [Google Scholar] [CrossRef]
- Wang, R.; Li, S.; Wen, W.; Zhang, J. Multi-Omics Analysis of the Effects of Smoking on Human Tumors. Front. Mol. Biosci. 2021, 8, 704910. [Google Scholar] [CrossRef]
- Shi, K.; Li, N.; Yang, M.; Li, W. Identification of Key Genes and Pathways in Female Lung Cancer Patients Who Never Smoked by a Bioinformatics Analysis. J. Cancer 2019, 10, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Zhang, L.; Zhang, Y.; Dong, G.; Yang, Y.; Xia, W.; Chen, B.; Ma, W.; Hu, J.; Jiang, F.; et al. A network-based signature to predict the survival of non-smoking lung adenocarcinoma. Cancer Manag. Res. 2018, 10, 2683–2693. [Google Scholar] [CrossRef]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer. 2020, 11, 511–518. [Google Scholar] [CrossRef]
- Jain, A.S.; Prasad, A.; Pradeep, S.; Dharmashekar, C.; Achar, R.R.; Silina, E.; Stupin, V.; Amachawadi, R.G.; Prasad, S.K.; Kollur, S.P.; et al. Everything Old Is New Again: Drug Repurposing Approach for Non-Small Cell Lung Cancer Targeting MAPK Signaling Pathway. Front. Oncol. 2021, 11, 741326. [Google Scholar] [CrossRef]
- Xu, L.; Wang, L.; Cheng, M. Identification of genes and pathways associated with sex in Non-smoking lung cancer population. Gene 2022, 831, 146566. [Google Scholar] [CrossRef]
- Planchard, D.; Camara-Clayette, V.; Dorvault, N.; Soria, J.C.; Fouret, P. p38 mitogen-activated protein kinase signaling, ERCC1 expression, and viability of lung cancer cells from never or light smoker patients: p38 MAPK and Lung Cancer. Cancer 2012, 118, 5015–5025. [Google Scholar] [CrossRef]
- Wu, H.; Meng, S.; Xu, Q.; Wang, X.; Wang, J.; Gong, R.; Song, Y.; Duan, Y.; Zhang, Y. Gene expression profiling of lung adenocarcinoma in Xuanwei, China. Eur. J. Cancer Prev. 2016, 25, 508–517. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Wozniak, A. Preclinical Rationale for PI3K/Akt/mTOR Pathway Inhibitors as Therapy for Epidermal Growth Factor Receptor Inhibitor-Resistant Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2013, 14, 322–332. [Google Scholar] [CrossRef]
- Hill, W.; Lim, E.L.; Weeden, C.E.; Lee, C.; Augustine, M.; Chen, K.; Kuan, F.-C.; Marongiu, F.; Evans, E.J.; Moore, D.A.; et al. Lung adenocarcinoma promotion by air pollutants. Nature 2023, 616, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-W.; Shie, S.-S.; Chiu, C.-T.; Wang, C.-L.; Yang, T.-Y.; Chou, S.-C.; Liu, C.-Y.; Kuo, C.-H.S.; Lin, Y.-C.; Li, L.-F.; et al. Association of smoking status with non-small cell lung cancer patients harboring uncommon epidermal growth factor receptor mutation. Front. Immunol. 2022, 13, 1011092. [Google Scholar] [CrossRef]
- Zhao, Z.-R.; Lin, Y.-B.; Ng, C.S.; Zhang, R.; Wu, X.; Ou, Q.; Chen, W.; Zhou, W.-J.; Lin, Y.-B.; Su, X.-D.; et al. Mutation Profile of Resected EGFR -Mutated Lung Adenocarcinoma by Next-Generation Sequencing. Oncol. 2019, 24, 1368–1374. [Google Scholar] [CrossRef] [PubMed]
- Macari, D.; Ibironke, O.; Jinna, S.; Stender, M.J.; Jaiyesimi, I.A. Survival differences between smokers and nonsmokers with EGFR mutated non-small cell lung cancer. J. Clin. Oncol. 2020, 38 (Suppl. 15), e21509. [Google Scholar] [CrossRef]
- Li, W.; Zhou, J.; Chen, Y.; Zhang, G.; Jiang, P.; Hong, L.; Shen, Y.; Wang, X.; Gong, X. Cigarette smoke enhances initiation and progression of lung cancer by mutating Notch1/2 and dysregulating downstream signaling molecules. Oncotarget 2017, 8, 115128–115139. [Google Scholar] [CrossRef][Green Version]
- Chen, C.-Y.; Chen, Y.-Y.; Hsieh, M.-S.; Ho, C.-C.; Chen, K.-Y.; Shih, J.-Y.; Yu, C.-J. Expression of Notch Gene and Its Impact on Survival of Patients with Resectable Non-small Cell Lung Cancer. J. Cancer. 2017, 8, 1292–1300. [Google Scholar] [CrossRef]
- Anusewicz, D.; Orzechowska, M.; Bednarek, A.K. Lung squamous cell carcinoma and lung adenocarcinoma differential gene expression regulation through pathways of Notch, Hedgehog, Wnt, and ErbB signalling. Sci. Rep. 2020, 10, 21128. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Silwal-Pandit, L.; Meza-Zepeda, L.A.; Vodak, D.; Vu, P.; Sagerup, C.; Hovig, E.; Myklebost, O.; Børresen-Dale, A.-L.; Brustugun, O.T.; et al. TP53 Mutation Spectrum in Smokers and Never Smoking Lung Cancer Patients. Front. Genet. 2016, 7, 85. [Google Scholar] [CrossRef]
- Song, P.; Zhang, F.; Li, Y.; Yang, G.; Li, W.; Ying, J.; Gao, S. Concomitant TP53 mutations with response to crizotinib treatment in patients with ALK-rearranged non-small-cell lung cancer. Cancer Med. 2019, 8, 1551–1557. [Google Scholar] [CrossRef]
- Uras, I.Z.; Moll, H.P.; Casanova, E. Targeting KRAS Mutant Non-Small-Cell Lung Cancer: Past, Present and Future. Int. J. Mol. Sci. 2020, 21, 4325. [Google Scholar] [CrossRef]
- Le Calvez, F.; Mukeria, A.; Hunt, J.D.; Kelm, O.; Hung, R.J.; Taniere, P.; Brennan, P.; Boffetta, P.; Zaridze, D.G.; Hainaut, P. TP53 and KRAS Mutation Load and Types in Lung Cancers in Relation to Tobacco Smoke: Distinct Patterns in Never, Former, and Current Smokers. Cancer Res. 2005, 65, 5076–5083. [Google Scholar] [CrossRef]
- Kosaka, T.; Yatabe, Y.; Onozato, R.; Kuwano, H.; Mitsudomi, T. Prognostic Implication of EGFR, KRAS, and TP53 Gene Mutations in a Large Cohort of Japanese Patients with Surgically Treated Lung Adenocarcinoma. J. Thorac. Oncol. 2009, 4, 22–29. [Google Scholar] [CrossRef]
- Kim, H.R.; Cho, B.C.; Shim, H.S.; Lim, S.M.; Kim, S.K.; Chang, J.; Kim, D.J.; Kim, J.H. Prediction for response duration to epidermal growth factor receptor-tyrosine kinase inhibitors in EGFR mutated never smoker lung adenocarcinoma. Lung Cancer 2014, 83, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Huang, Y.; Gu, W.; Gan, J.; Wang, W.; Zhang, S.; Wang, K.; Zhan, J.; Yang, Y.; Zhang, L.; et al. PI3K-AKT-mTOR pathway alterations in advanced NSCLC patients after progression on EGFR-TKI and clinical response to EGFR-TKI plus everolimus combination therapy. Transl. Lung Cancer Res. 2020, 9, 1258–1267. [Google Scholar] [CrossRef]
- Lee, Y.J.; Cho, B.C.; Jee, S.H.; Moon, J.W.; Kim, S.K.; Chang, J.; Chung, K.Y.; Park, I.K.; Choi, S.H.; Kim, J.H. Impact of Environmental Tobacco Smoke on the Incidence of Mutations in Epidermal Growth Factor Receptor Gene in Never-Smoker Patients With Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Dutu, T.; Michiels, S.; Fouret, P.; Penault-Llorca, F.; Validire, P.; Benhamou, S.; Taranchon, E.; Morat, L.; Grunenwald, D.; Le Chevalier, T.; et al. Differential expression of biomarkers in lung adenocarcinoma: A comparative study between smokers and never-smokers. Ann. Oncol. 2005, 16, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Seike, M.; Goto, A.; Okano, T.; Bowman, E.D.; Schetter, A.J.; Horikawa, I.; Mathe, E.A.; Jen, J.; Yang, P.; Sugimura, H.; et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Mutated Samples | Samples Tested | NS % | CS % | RS % | p-Value |
|---|---|---|---|---|---|---|
| EGFR * | 25,394 | 95,066 | 13.33 | 1.67 | 6.60 | p-value = 0.0046 |
| KRAS | 5994 | 37,187 | 6.67 | 14.17 | 16.50 | p-value = 0.095 |
| TP53 | 3246 | 9440 | 10.67 | 24.17 | 21.78 | p-value = 0.058 |
| PIK3CA | 546 | 13,654 | 1.33 | 3.33 | 2.97 | p-value = 0.800 |
| CSMD3 | 543 | 2305 | 1.33 | 7.50 | 9.57 | p-value = 0.059 |
| LRP1B * | 497 | 2492 | 1.33 | 15.83 | 15.18 | p-value = 0.0042 |
| BRAF | 493 | 24,447 | 0.00 | 5.00 | 4.62 | p-value = 0.14 |
| USH2A * | 484 | 2305 | 5.33 | 10.83 | 15.84 | p-value = 0.038 |
| RYR2 * | 444 | 2199 | 6.67 | 21.67 | 16.17 | p-value = 0.021 |
| CDKN2A | 425 | 5443 | 0.00 | 2.50 | 1.98 | p-value = 0.45 |
| MUC16 * | 402 | 2333 | 4.00 | 16.67 | 21.45 | p-value = 0.0018 |
| ZFHX4 * | 379 | 2200 | 1.33 | 15.00 | 13.86 | p-value = 0.0071 |
| SPTA1 | 308 | 2202 | 6.67 | 10.00 | 13.20 | p-value = 0.24 |
| STK11 | 298 | 5856 | 2.67 | 5.83 | 9.90 | p-value = 0.073 |
| MUC17 | 288 | 2205 | 4.00 | 12.50 | 8.91 | p-value = 0.13 |
| SYNE1 | 287 | 2204 | 0.00 | 5.83 | 3.63 | p-value = 0.072 |
| XIRP2 | 283 | 2200 | 0.00 | 10.83 | 10.23 | p-value = 0.014 |
| FLG | 282 | 2203 | 5.33 | 15.00 | 12.54 | p-value = 0.12 |
| ERBB2 | 275 | 18,935 | 4.00 | 0.00 | 0.99 | p-value = 0.054 |
| FAM135B * | 256 | 2303 | 0.00 | 5.00 | 7.92 | p-value = 0.015 |
| COL11A1 | 255 | 2302 | 1.33 | 8.33 | 9.57 | p-value = 0.063 |
| NAV3 * | 253 | 2203 | 0.00 | 12.50 | 9.24 | p-value = 0.0086 |
| RYR3 | 246 | 2199 | 1.33 | 5.00 | 6.93 | p-value = 0.16 |
| KEAP1 | 240 | 2993 | 2.67 | 6.67 | 9.57 | p-value = 0.12 |
| Gene | Pathway | Findings | References |
|---|---|---|---|
| TP53 | Notch/ASCL1 axis | Inactivation of TP53 and RB1 participated in the transformation of SCLC in female never smokers. | [15] |
| RB1 | [15] | ||
| UBA1 | Ubiquitin pathways | Mutations in UBA1 occurred before the corresponding copy number gain. | [1] |
| ARID1A | PI3K/AKT pathway | Mutations in ARID1A could promote exit from a quiescent cell state, resulting in high intra-tumoural heterogeneity. | [1] |
| CTNNB1 | WNT pathway | Never smokers had a lower TMBs and a higher frequency of mutations in CTNNB1 compared with smokers. | |
| BRCA1 | DNA repair | Germline alterations in the listed DNA repair genes were exclusively mutated in never smokers. | [3] |
| BRCA2 | |||
| FANCG | |||
| FANCM | |||
| MSH6 | |||
| POLD1 | |||
| RNF213 | WNT pathway | Somatic mutations were identified as prevalent in Taiwanese study of never smoker lung adenocarcinoma patients. | [16] |
| ATP2B3 | [16] | ||
| TET2 | [16] | ||
| EGFR-Related Mutations | |||
| KRAS | RTK–Ras pathway | EGFR and KRAS mutations were mutually exclusive (p = 0.004) | [1] |
| Mutations in KRAS were generally early events occurring prior to whole-genome doubling and most other somatic copy number alterations. | [1] | ||
| ALK | 6.0% alteration in RTK–Ras pathway. | [1] | |
| Never smoker lung adenocarcinomas had a higher proportion of ALK mutations (12%) than former/current smokers (2%) (p < 0.0001). | [17] | ||
| MET | 4.3% alteration in RTK–Ras pathway. | [1] | |
| ERBB2 | 3.9%, all indels alteration in RTK–Ras pathway. | [1] | |
| ROS1 | 2.6% alteration in RTK–Ras pathway. | [1] | |
| RET | 1.3% alteration in RTK–Ras pathway. | [1] | |
| EGFR | Higher frequency of EGFR mutations in female (31.4%) than in male patients (19.3%) (p = 0.092). | [1] | |
| Somatic mutations occurred in 85% of Taiwanese never smoker lung adenocarcinoma patients. | [16] | ||
| Statistically significant between NS and CS LUAD (p < 0.005). | [9] | ||
| Mutations in EGFR were generally early events occurring prior to whole-genome doubling and most other somatic copy number alterations. | [1] | ||
| Never smoker lung adenocarcinomas had a higher proportion of EGFR mutations 37% than former/current smokers 14% (p < 0.0001). | [17] | ||
| Erα expression was correlated with the presence of an EGFR mutation in female never smokers. | [18] | ||
| RBM10 | Mutations in RBM10 were generally early events occurring prior to whole-genome doubling and most other somatic copy number alterations. | [1] | |
| Top ranking mutations in 20% of Taiwanese never smoker lung adenocarcinoma patients. | [16] | ||
| TP53 | Top-ranking mutations in 33% of Taiwanese never smoker lung adenocarcinoma patients. | [1] | |
| Mutations in TP53 were generally early events occurring prior to whole-genome doubling and most other somatic copy number alterations. | [1] | ||
| SETD2 | Significant enrichment of SETD2 mutations in samples with oncogene fusions, particularly in TP53-proficient tumours (p = 0.06). | [1] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamouz, M.; Hammouz, R.Y.; Bajwa, M.A.; Alsayed, A.W.; Orzechowska, M.; Bednarek, A.K. A Functional Genomics Review of Non-Small-Cell Lung Cancer in Never Smokers. Int. J. Mol. Sci. 2023, 24, 13314. https://doi.org/10.3390/ijms241713314
Hamouz M, Hammouz RY, Bajwa MA, Alsayed AW, Orzechowska M, Bednarek AK. A Functional Genomics Review of Non-Small-Cell Lung Cancer in Never Smokers. International Journal of Molecular Sciences. 2023; 24(17):13314. https://doi.org/10.3390/ijms241713314
Chicago/Turabian StyleHamouz, Mohammad, Raneem Y. Hammouz, Muhammad Ahmed Bajwa, Abdelrahman Waleed Alsayed, Magdalena Orzechowska, and Andrzej K. Bednarek. 2023. "A Functional Genomics Review of Non-Small-Cell Lung Cancer in Never Smokers" International Journal of Molecular Sciences 24, no. 17: 13314. https://doi.org/10.3390/ijms241713314
APA StyleHamouz, M., Hammouz, R. Y., Bajwa, M. A., Alsayed, A. W., Orzechowska, M., & Bednarek, A. K. (2023). A Functional Genomics Review of Non-Small-Cell Lung Cancer in Never Smokers. International Journal of Molecular Sciences, 24(17), 13314. https://doi.org/10.3390/ijms241713314