

Transferrin-Targeted Liposomes in Glioblastoma Therapy: A Review


Abstract
1. Introduction
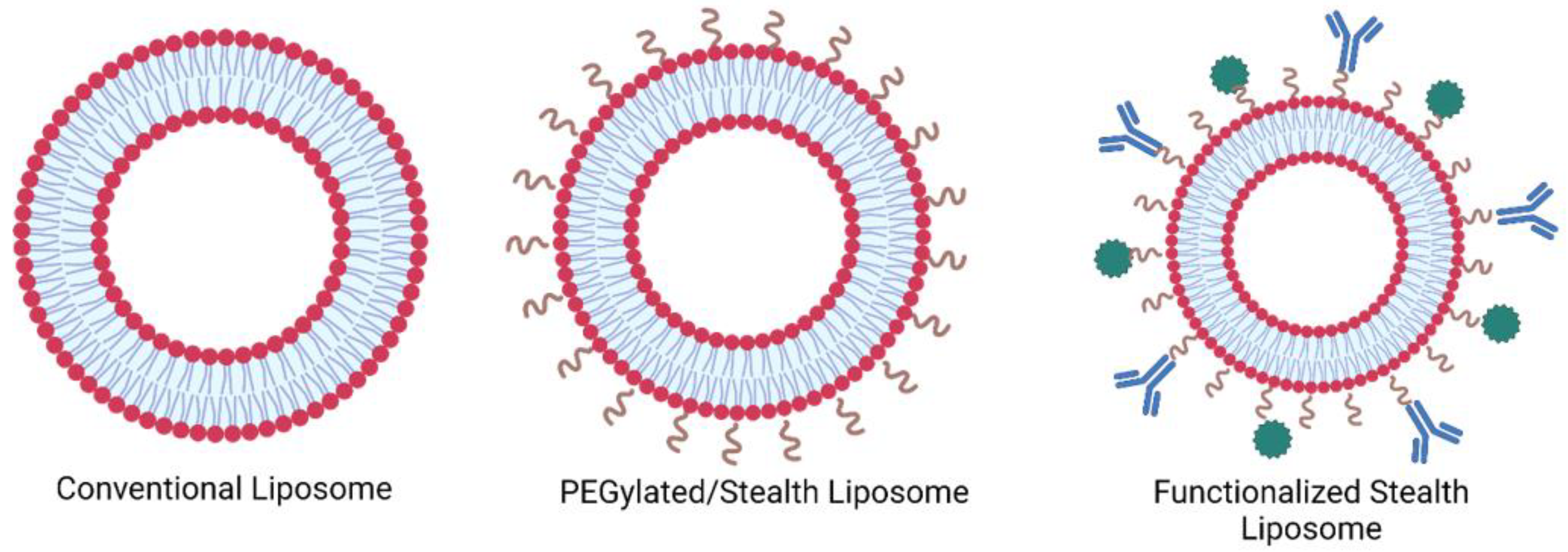
1.1. Liposomes
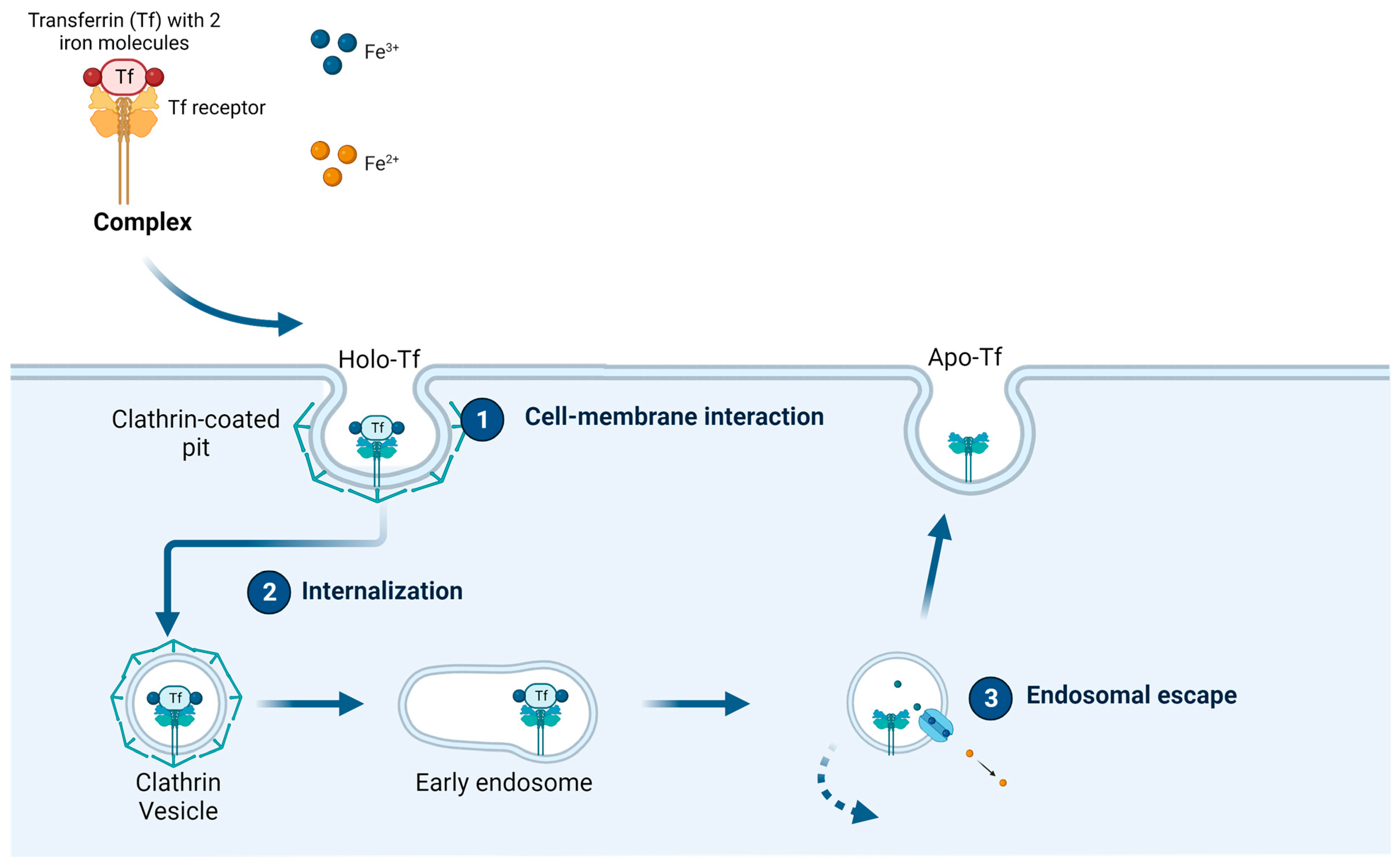
1.2. Transferrin and the Transferrin Receptor
1.3. Conjugation Chemistry of Tf onto Liposomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugation Method | Description | Schematic | Advantages | Disadvantages |
|---|---|---|---|---|
| Crosslinking of primary amines by glutaraldehyde | Dimethyl suberimidate or glutaraldehyde can be employed for imidoester or imine crosslinking, respectively. This involves reacting these substances with both the amine-functionalized liposome and the targeting ligand, which possesses an amine group as well. |  | -Preserves the affinity, specificity, and targeting -Common approach | -Homobifunctionality of the linkers -Side reactions -Loss of targeting ligand |
| Carbonyl-amine bond | Coupling the N-terminus of the peptide to preformed carboxy-terminated liposomes that contain DSPE-PEG involves amine-carboxyl coupling. |  | -Straightforward preparation | -The imine bond formed between the carbonyl group of the ligand and the amine group of the target molecule is not stable and can undergo hydrolysis, leading to bond cleavage. -pH sensitivity -The kinetics of the carbonyl amine reaction can be relatively slow, requiring longer reaction times and elevated temperatures to achieve efficient conjugation. |
| Cyanuric chloride | Nucleophilic substitution takes place under alkaline conditions. The initial two chloride substitutions can be accomplished by reacting with nucleophiles under mildly basic conditions. The first substitution on cyanuric chloride happens rapidly at 0 °C, while the second substitution takes place within 12–24 h at room temperature. The third substitution typically occurs within 12–24 h as well, but it necessitates temperatures above 60 °C. |  | -The safety of cyanuric chloride can be attributed to its pH-regulated reactivity. | -Cyanuric chloride has low reactivity under neutral physiological pH |
| Amide bond | An amide bond is formed through the reaction of para-nitrophenylcarbonyl with primary amine |  | -Undesirable side reactions are circumvented by the hydrolysis of unreacted pNP molecules. -No activation of the carboxylic acid is required simplifying the reaction and decreasing the overall time required for the surface modification | -The pNP lipid must be maintained at acidic pH during its manipulation to prevent the hydrolysis of the pNP group |
| Disulfide bond | The conjugation of two thiol functions to form a disulfide bond |  | -The reaction can be monitored spectroscopically by the release of the chromophore 2-thiopyridone -Rapid and easy | -Disulfide bonds are relatively unstable under the reductive conditions in serum |
| Maleimide bond | Thioester bond formation by the maleimide-thiol addition reaction. This process involves the rapid reaction of thiolated ligands with maleimide-modified PEGylated phospholipids such as maleimide-polyethyleneglycol |  | -Most frequently used approach to conjugate targeting ligands onto PEGylated lipids. | -Hydrolytic instability -Sensitivity to reducing agents -Variable reaction kinetics |
| EDC/NHS | The carboxyl group of a targeting ligand is activated by 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) and rapidly reacts with phospholipid-PEG-NH2 via nucleophilic substitution reaction to form a stable amide bond. |  | -High specificity -Mild reaction conditions -Compatible with a wide range of moieties | -Generally, EDC/NHS coupling reaction occurs faster in a weak acid solution. |
2. Transferrin-Liposomes in Cancer Therapy
3. Tf-Targeted Liposomes for Glioblastoma Therapy
4. Challenges and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nature Education, Cell Division and Cancer. Available online: https://www.nature.com/scitable/topicpage/cell-division-and-cancer-14046590/ (accessed on 1 June 2023).
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef]
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427. [Google Scholar] [CrossRef]
- Shapira, A.; Livney, Y.D.; Broxterman, H.J.; Assaraf, Y.G. Nanomedicine for targeted cancer therapy: Towards the overcoming of drug resistance. Drug Resist. Updates 2011, 14, 150–163. [Google Scholar] [CrossRef]
- Andresen, T.L.; Jensen, S.S.; Jørgensen, K. Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release. Prog. Lipid Res. 2005, 44, 68–97. [Google Scholar] [CrossRef]
- Mitra, A.K.; Agrahari, V.; Mandal, A.; Cholkar, K.; Natarajan, C.; Shah, S.; Joseph, M.; Trinh, H.M.; Vaishya, R.; Yang, X.; et al. Novel delivery approaches for cancer therapeutics. J. Control. Release 2015, 219, 248–268. [Google Scholar] [CrossRef] [PubMed]
- Zangabad, P.S.; Mirkiani, S.; Shahsavari, S.; Masoudi, B.; Masroor, M.; Hamed, H.; Jafari, Z.; Taghipour, Y.D.; Hashemi, H.; Karimi, M.; et al. Stimulus-Responsive Liposomes as Smart Nanoplatforms for Drug Delivery Applications. Available online: https://www.degruyter.com/document/doi/10.1515/ntrev-2017-0154/html (accessed on 23 June 2023).
- AlSawaftah, N.; Pitt, W.G.; Husseini, G.A. Dual-Targeting and Stimuli-Triggered Liposomal Drug Delivery in Cancer Treatment. ACS Pharmacol. Transl. Sci. 2021, 4, 1028–1049. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.; Zappavigna, S.; Sannolo, N.; Porto, S.; Caraglia, M. Advantages and risks of nanotechnologies in cancer patients and occupationally exposed workers. Expert Opin. Drug Deliv. 2014, 11, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Siddique, S.; Chow, J.C.L. Application of nanomaterials in biomedical imaging and cancer therapy. Nanomaterials 2020, 10, 1700. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Ju, D.-T.; Chang, C.-F.; Reddy, P.M.; Velmurugan, B.K. A review on the effects of current chemotherapy drugs and natural agents in treating non–small cell lung cancer. BioMedicine 2017, 7, 23. [Google Scholar] [CrossRef]
- Zhang, Y.; He, J. Tumor vasculature-targeting nanomedicines. Acta Biomater. 2021, 134, 1–12. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W., Jr. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2014, 10, 81–98. [Google Scholar] [CrossRef]
- Sawant, R.R.; Torchilin, V.P. Challenges in Development of Targeted Liposomal Therapeutics. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22415612 (accessed on 23 June 2023).
- Çağdaş, M.; Sezer, A.D.; Bucak, S. Liposomes as Potential Drug Carrier Systems for Drug Delivery. In Application of Nanotechnology in Drug Delivery; InTech: London, UK, 2014. [Google Scholar]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Liposome application: Problems and prospects. Curr. Opin. Colloid Interface Sci. 2001, 6, 66–77. [Google Scholar] [CrossRef]
- Pandey, H.; Rani, R.; Agarwal, V. Liposome and their applications in cancer therapy. Braz. Arch. Biol. Technol. 2016, 59, 16150477. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Olusanya, T.O.B.B.; Ahmad, R.R.H.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef]
- Clemons, T.D.; Singh, R.; Sorolla, A.; Chaudhari, N.; Hubbard, A.; Iyer, K.S. Distinction Between Active and Passive Targeting of Nanoparticles Dictate Their Overall Therapeutic Efficacy. Langmuir 2018, 34, 15343–15349. [Google Scholar] [CrossRef]
- Awad, N.S.; Paul, V.; Alsawaftah, N.M.; ter Haar, G.; Allen, T.M.; Pitt, W.G.; Husseini, G.A. Ultrasound-Responsive Nanocarriers in Cancer Treatment: A Review. ACS Pharmacol. Transl. Sci. 2021, 4, 589–612. [Google Scholar] [CrossRef]
- Bahutair, W.N.; Abuwatfa, W.H.; Husseini, G.A. Ultrasound Triggering of Liposomal Nanodrugs for Cancer Therapy: A Review. Nanomaterials 2022, 12, 3051. [Google Scholar] [CrossRef]
- AlSawaftah, N.M.; Abusamra, R.H.; Husseini, G.A. Carbohydrate-Functionalized Liposomes In Cancer Therapy—A Minireview Review. Curr. Cancer Ther. Rev. 2021, 17, 4–20. [Google Scholar] [CrossRef]
- van Vlerken, L.E.; Vyas, T.K.; Amiji, M.M. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 2007, 24, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Muro, S. Challenges in design and characterization of ligand-targeted drug delivery systems. J. Control. Release 2012, 164, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhao, Y.; Xie, C.; Chen, C.; Lin, D.; Wang, S.; Cui, X.; Guo, Z.; Zhou, J. Dual-active targeting liposomes drug delivery system for bone metastatic breast cancer: Synthesis and biological evaluation. Chem. Phys. Lipids 2019, 223, 104785. [Google Scholar] [CrossRef]
- Koneru, T.; McCord, E.; Pawar, S.; Tatiparti, K.; Sau, S.; Iyer, A.K. Transferrin: Biology and Use in Receptor-Targeted Nanotherapy of Gliomas. ACS Omega 2021, 6, 8727–8733. [Google Scholar] [CrossRef]
- Mojarad-Jabali, S.; Mahdinloo, S.; Farshbaf, M.; Sarfraz, M.; Fatahi, Y.; Atyabi, F.; Valizadeh, H. Transferrin receptor-mediated liposomal drug delivery: Recent trends in targeted therapy of cancer. Expert Opin. Drug Deliv. 2022, 19, 685–705. [Google Scholar] [CrossRef]
- Ramalho, M.J.; Loureiro, J.A.; Coelho, M.A.N.; Pereira, M.C. Transferrin receptor-targeted nanocarriers: Overcoming barriers to treat glioblastoma. Pharmaceutics 2022, 14, 279. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xiao, Y.; Di, Q.; Ma, W.; Ma, X.; Wang, Q.; Chen, W. Transferrin receptor-targeted peg-pla polymeric micelles for chemotherapy against glioblastoma multiforme. Int. J. Nanomed. 2020, 15, 6673–6688. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zak, O.; Aisen, P.; Harrison, S.C.; Walz, T. Structure of the human transferrin receptor–transferrin complex. Cell 2004, 116, 565–576. [Google Scholar] [CrossRef]
- AlSawaftah, N.M.; Awad, N.S.; Paul, V.; Kawak, P.S.; Al-Sayah, M.H.; Husseini, G.A. Transferrin-modified liposomes triggered with ultrasound to treat HeLa cells. Sci. Rep. 2021, 11, 11589. [Google Scholar] [CrossRef]
- Keenan, J.; Pearson, D.; O’Driscoll, L.; Gammell, P.; Clynes, M. Evaluation of recombinant human transferrin (DeltaFerrin(TM)) as an iron chelator in serum-free media for mammalian cell culture. Cytotechnology 2006, 51, 29–37. [Google Scholar] [CrossRef]
- Qian, Z.M.; Li, H.; Sun, H.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef] [PubMed]
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 2012, 1820, 264–281. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, H.; Pandey, M.; Chin, P.X.; Phang, Y.L.; Cheah, J.Y.; Ooi, S.C.; Mak, K.-K.; Pichika, M.R.; Kesharwani, P.; Hussain, Z.; et al. Transferrin receptors-targeting nanocarriers for efficient targeted delivery and transcytosis of drugs into the brain tumors: A review of recent advancements and emerging trends. Drug Deliv. Transl. Res. 2018, 8, 1545–1563. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Terpstra, T.R. The thermodynamic and binding properties of the transferrins as studied by isothermal titration calorimetry. Biochim. Biophys. Acta 2012, 1820, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qian, Z.M. Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 2002, 22, 225–250. [Google Scholar] [CrossRef]
- Ferguson, J.P.; Huber, S.D.; Willy, N.M.; Aygün, E.; Goker, S.; Atabey, T.; Kural, C. Mechanoregulation of clathrin-mediated endocytosis. J. Cell Sci. 2017, 130, 3631–3636. [Google Scholar] [CrossRef]
- Godlee, C.; Kaksonen, M. Review series: From uncertain beginnings: Initiation mechanisms of clathrin-mediated endocytosis. J. Cell Biol. 2013, 203, 717–725. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Nobs, L.; Buchegger, F.; Gurny, R.; Allémann, E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J. Pharm. Sci. 2004, 93, 1980–1992. [Google Scholar] [CrossRef]
- Noble, C.O.; Kirpotin, D.B.; Hayes, M.E.; Mamot, C.; Hong, K.; Park, J.W.; Benz, C.C.; Marks, J.D.; Drummond, D.C. Development of ligand-targeted liposomes for cancer therapy. Expert Opin. Ther. Targets 2004, 8, 335–353. [Google Scholar] [CrossRef]
- Nsairat, H.; Khater, D.; Sayed, U.; Odeh, F.; Al Bawab, A.; Alshaer, W. Liposomes: Structure, composition, types, and clinical applications. Heliyon 2022, 8, e09394. [Google Scholar] [CrossRef]
- Manjappa, A.S.; Chaudhari, K.R.; Venkataraju, M.P.; Dantuluri, P.; Nanda, B.; Sidda, C.; Sawant, K.K.; Ramachandra Murthy, R.S. Antibody derivatization and conjugation strategies: Application in preparation of stealth immunoliposome to target chemotherapeutics to tumor. J. Control. Release 2011, 150, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Marqués-Gallego, P.; de Kroon, A.I.P.M. Ligation strategies for targeting liposomal nanocarriers. BioMed Res. Int. 2014, 2014, 129458. [Google Scholar] [CrossRef] [PubMed]
- Frisch, B.; Hassane, F.S.; Schuber, F. Conjugation of Ligands to the Surface of Preformed Liposomes by Click Chemistry BT—Liposomes: Methods and Protocols, Volume 1: Pharmaceutical Nanocarriers; Weissig, V., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 267–277. [Google Scholar]
- Salkho, N.M.; Paul, V.; Kawak, P.; Vitor, R.F.; Martins, A.M.; Al Sayah, M.; Husseini, G.A. Ultrasonically controlled estrone-modified liposomes for estrogen-positive breast cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 2), 462–472. [Google Scholar] [CrossRef] [PubMed]
- Liposome: Encapsula’s Scientific Blog: Immunoliposomes. Available online: http://www.liposomes.org/2011/09/immunoliposomes.html (accessed on 27 June 2023).
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Evasion of Apoptosis as a Cellular Stress Response in Cancer. Int. J. Cell Biol. 2010, 2010, 370835. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zhang, P.W.; Chen, L.; Huang, T.; Zhang, N.; Kong, X.Y.; Cai, Y.D. Classifying ten types of major cancers based on reverse phase protein array profiles. PLoS ONE 2015, 10, e0123147. [Google Scholar] [CrossRef]
- Chifman, J.; Laubenbacher, R.; Torti, S.V. A systems biology approach to iron metabolism. Adv. Exp. Med. Biol. 2014, 844, 201–225. [Google Scholar]
- Abbate, V.; Hider, R. Iron in biology. Metallomics 2017, 9, 1467–1469. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Li, L.; Hou, S.; Yuan, Z.; Li, C.; Zhang, W.; Zheng, L.; Li, X. The Role of Iron in Cancer Progression. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2021.778492 (accessed on 27 June 2023).
- Indraccolo, S.; Walenta, S.; Mueller-Klieser, W. Uncovering metabolic effects of anti-angiogenic therapy in tumors by induced metabolic bioluminescence imaging. Methods Mol. Biol. 2016, 1464, 175–184. [Google Scholar] [PubMed]
- Awad, N.S.; Salkho, N.M.; Abuwatfa, W.H.; Paul, V.; AlSawaftah, N.M.; Husseini, G.A. Tumor vasculature vs tumor cell targeting: Understanding the latest trends in using functional nanoparticles for cancer treatment. OpenNano 2023, 11, 100136. [Google Scholar] [CrossRef]
- Maeshima, Y. Angiogenesis and Cancer. In Apoptosis, Cell Signaling, and Human Diseases: Molecular Mechanisms; Srivastava, R., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 35–61. [Google Scholar]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]
- Salnikow, K. Role of iron in cancer. Semin. Cancer Biol. 2021, 76, 189–194. [Google Scholar] [CrossRef]
- Liu, S.; Cao, X.; Wang, D.; Zhu, H. Iron metabolism: State of the art in hypoxic cancer cell biology. Arch. Biochem. Biophys. 2022, 723, 109199. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Urbańska, K.; Sokołowska, J.; Szmidt, M.; Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol./Współczesna Onkol. 2014, 18, 307–312. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar]
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino, M.d.C.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef]
- Liu, A.; Hou, C.; Chen, H.; Zong, X.; Zong, P. Genetics and Epigenetics of Glioblastoma: Applications and Overall Incidence of IDH1 Mutation. Front. Oncol. 2016, 6, 16. [Google Scholar] [CrossRef]
- Joshi, S.K.; Lucic, N.; Zuniga, R. Molecular pathogenesis of glioblastoma multiforme: Nuances, obstacles, and implications for treatment. World J. Neurol. 2015, 5, 88–101. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef]
- Batash, R.; Asna, N.; Schaffer, P.; Francis, N.; Schaffer, M. Glioblastoma multiforme, diagnosis and treatment; recent literature review. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar] [CrossRef] [PubMed]
- Kanderi, T.; Gupta, V. Glioblastoma Multiforme. [Updated 2022 Sep 12]. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, January 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK558954/ (accessed on 27 June 2023).
- Luiz, M.T.; Viegas, J.S.R.; Abriata, J.P.; Tofani, L.B.; Vaidergorn, M.d.M.; Emery, F.d.S.; Chorilli, M.; Marchetti, J.M. Docetaxel-loaded folate-modified TPGS-transfersomes for glioblastoma multiforme treatment. Mater. Sci. Eng. C 2021, 124, 112033. [Google Scholar] [CrossRef] [PubMed]
- Cha, G.D.; Kang, T.; Baik, S.; Kim, D.; Choi, S.H.; Hyeon, T.; Kim, D.-H. Advances in drug delivery technology for the treatment of glioblastoma multiforme. J. Control. Release 2020, 328, 350–367. [Google Scholar] [CrossRef]
- Ozdemir-Kaynak, E.; Qutub, A.A.; Yesil-Celiktas, O. Advances in glioblastoma multiforme treatment: New models for nanoparticle therapy. Front. Physiol. 2018, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Juhairiyah, F.; de Lange, E.C.M. Understanding Drug Delivery to the Brain Using Liposome-Based Strategies: Studies that Provide Mechanistic Insights Are Essential. AAPS J. 2021, 23, 114. [Google Scholar] [CrossRef] [PubMed]
- Voth, B.; Nagasawa, D.T.; Pelargos, P.E.; Chung, L.K.; Ung, N.; Gopen, Q.; Tenn, S.; Kamei, D.T.; Yang, I. Transferrin receptors and glioblastoma multiforme: Current findings and potential for treatment. J. Clin. Neurosci. 2015, 22, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Vu Han, T.-L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef]
- Li, J.; Zeng, H.; You, Y.; Wang, R.; Tan, T.; Wang, W.; Yin, L.; Zeng, Z.; Zeng, Y.; Xie, T. Active targeting of orthotopic glioma using biomimetic liposomes co-loaded elemene and cabazitaxel modified by transferritin. J. Nanobiotechnol. 2021, 19, 289. [Google Scholar] [CrossRef]
- Lakkadwala, S.; dos Santos Rodrigues, B.; Sun, C.; Singh, J. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J. Control. Release 2019, 307, 247–260. [Google Scholar] [CrossRef]
- Lv, Q.; Li, L.M.; Han, M.; Tang, X.J.; Yao, J.N.; Ying, X.Y.; Li, F.Z.; Gao, J.Q. Characteristics of sequential targeting of brain glioma for transferrin-modified cisplatin liposome. Int. J. Pharm. 2013, 444, 1–9. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Y.; Dong, S.; Lee, R.J.; Yang, D.; Zhang, H.; Teng, L. Cell-Penetrating Peptide and Transferrin Co-Modified Liposomes for Targeted Therapy of Glioma. Molecules 2019, 24, 3540. [Google Scholar] [CrossRef]
- Mitusova, K.; Peltek, O.O.; Karpov, T.E.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S. Overcoming the blood–brain barrier for the therapy of malignant brain tumor: Current status and prospects of drug delivery approaches. J. Nanobiotechnol. 2022, 20, 412. [Google Scholar] [CrossRef]
- Yusuf, A.; Almotairy, A.R.Z.; Henidi, H.; Alshehri, O.Y.; Aldughaim, M.S. Nanoparticles as Drug Delivery Systems: A Review of the Implication of Nanoparticles’ Physicochemical Properties on Responses in Biological Systems. Polymers 2023, 15, 1596. [Google Scholar] [CrossRef]
- Bagchi, D. Bio-Nanotechnology: A Revolution in Food, Biomedical, and Health Sciences; Wiley-Blackwell: Hoboken, NJ, USA, 2013; p. 803. [Google Scholar]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Desai, N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Butler, K.S.; Brinker, C.J.; Azzawi, M.; Conlan, S.; Dufés, C.; Owen, A.; Rannard, S.; Scott, C.; Chen, C.; et al. On the issue of transparency and reproducibility in nanomedicine. Nat. Nanotechnol. 2019, 14, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Meng, H. Consideration for the scale-up manufacture of nanotherapeutics—A critical step for technology transfer. View 2021, 2, 20200190. [Google Scholar] [CrossRef]
- Tyner, K.; Sadrieh, N. Considerations When Submitting Nanotherapeutics to FDA/CDER for Regulatory Review. Methods Mol. Biol. 2011, 697, 17–31. [Google Scholar]
- Yan, X.; Scherphof, G.L.; Kamps, J.A.A.M. Liposome Opsonization. J. Liposome Res. 2005, 15, 109–139. [Google Scholar] [CrossRef]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-targeted liposome design: Challenges and fundamental considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef]
- Koshkaryev, A.; Sawant, R.; Deshpande, M.; Torchilin, V. Immunoconjugates and long circulating systems: Origins, current state of the art and future directions. Adv. Drug Deliv. Rev. 2013, 65, 24–35. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.; Yan, M.; Ma, Y.; Zang, G.; She, Z.; Deng, Y. Repeated injection of PEGylated solid lipid nanoparticles induces accelerated blood clearance in mice and beagles. Int. J. Nanomed. 2012, 7, 2891–2900. [Google Scholar]
- Abu Lila, A.S.; Kiwada, H.; Ishida, T. The Accelerated Blood Clearance (ABC) Phenomenon: Clinical Challenge and Approaches to Manage. Available online: https://pubmed.ncbi.nlm.nih.gov/23933235/ (accessed on 27 June 2023).
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release Off. J. Control. Release Soc. 2006, 112, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Subasic, C.N.; Butcher, N.J.; Minchin, R.F.; Kaminskas, L.M. Dose-Dependent Production of Anti-PEG IgM after Intramuscular PEGylated-Hydrogenated Soy Phosphatidylcholine Liposomes, but Not Lipid Nanoparticle Formulations of DNA, Correlates with the Plasma Clearance of PEGylated Liposomal Doxorubicin in Rats. Mol Pharm. 2023, 20, 3494–3504. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles─From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef]
- Bilardo, R.; Traldi, F.; Vdovchenko, A.; Resmini, M. Influence of surface chemistry and morphology of nanoparticles on protein corona formation. Wiley interdisciplinary reviews. Nanomed. Nanobiotechnol. 2022, 14, e1788. [Google Scholar] [CrossRef]
- Singh, N.; Marets, C.; Boudon, J.; Millot, N.; Saviot, L.; Maurizi, L. In vivo protein corona on nanoparticles: Does the control of all material parameters orient the biological behavior? Nanoscale Adv. 2021, 3, 1209–1229. [Google Scholar] [CrossRef]
- Tang, Y.; Gao, J.; Wang, T.; Zhang, Q.; Wang, A.; Huang, M.; Yu, R.; Chen, H.; Gao, X. The effect of drug loading and multiple administration on the protein corona formation and brain delivery property of PEG-PLA nanoparticles. Acta Pharm. Sin. B 2022, 12, 2043–2056. [Google Scholar] [CrossRef]
- Huo, T.; Yang, Y.; Qian, M.; Jiang, H.; Du, Y.; Zhang, X.; Xie, Y.; Huang, R. Versatile hollow COF nanospheres via manipulating transferrin corona for precise glioma-targeted drug delivery. Biomaterials 2020, 260, 120305. [Google Scholar] [CrossRef]
- Morales, D.E.; Mousa, S. Intranasal delivery in glioblastoma treatment: Prospective molecular treatment modalities. Heliyon 2022, 8, e09517. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The blood–brain barrier: Structure, regulation, and drug delivery. Signal Transduct. Target. Ther. 2023, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina-Glushkovskaya, O.; Shirokov, A.; Blokhina, I.; Telnova, V.; Vodovozova, E.; Alekseeva, A.; Boldyrev, I.; Fedosov, I.; Dubrovsky, A.; Khorovodov, A.; et al. Intranasal Delivery of Liposomes to Glioblastoma by Photostimulation of the Lymphatic System. Pharmaceutics 2022, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.Y.; Fan, C.H.; Liu, H.L.; Huang, C.Y.; Hsieh, H.Y.; Yen, T.C.; Wei, K.C.; Yeh, C.K. Concurrent blood-brain barrier opening and local drug delivery using drug-carrying microbubbles and focused ultrasound for brain glioma treatment. Biomaterials 2012, 33, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Mao, K.L.; Tian, F.R.; Yang, J.J.; Chen, P.P.; Xu, J.; Fan, Z.L.; Zhao, Y.P.; Li, W.F.; Zheng, L.; et al. Brain tumor-targeted delivery and therapy by focused ultrasound introduced doxorubicin-loaded cationic liposomes. Cancer Chemother. Pharmacol. 2016, 77, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Papachristodoulou, A.; Signorell, R.D.; Werner, B.; Brambilla, D.; Luciani, P.; Cavusoglu, M.; Grandjean, J.; Silginer, M.; Rudin, M.; Martin, E.; et al. Chemotherapy sensitization of glioblastoma by focused ultrasound-mediated delivery of therapeutic liposomes. J. Control. Release 2019, 295, 130–139. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Guo, J.; Huang, Q. Liposomes for Tumor Targeted Therapy: A Review. Int. J. Mol. Sci. 2023, 24, 2643. [Google Scholar] [CrossRef]
- van der Koog, L.; Gandek, T.B.; Nagelkerke, A. Liposomes and extracellular vesicles as drug delivery systems: A comparison of composition, pharmacokinetics, and functionalization. Adv. Healthc. Mater. 2022, 11, 2100639. [Google Scholar] [CrossRef]
- Li, X.M.; Ding, L.Y.; Xu, Y.; Wang, Y.; Ping, Q.N. Targeted delivery of doxorubicin using stealth liposomes modified with transferrin. Int. J. Pharm. 2009, 373, 116–123. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Zhang, B.; Yan, X.; Fan, K. Transferrin receptor 1 targeted nanomedicine for brain tumor therapy. Biomater. Sci. 2023, 11, 3394–3413. [Google Scholar] [CrossRef]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef]
- Sachdeva, S.; Joo, H.; Tsai, J.; Jasti, B.; Li, X. A Rational Approach for Creating Peptides Mimicking Antibody Binding. Sci. Rep. 2019, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano 2013, 7, 2935–2947. [Google Scholar] [CrossRef] [PubMed]





| Trade Name | Year Approved | Active Ingredient | Use |
|---|---|---|---|
| Doxil/Caelyx | 1995 | Doxorubicin | Antineoplastic |
| Daunoxome | 1996 | Daunorubicin | Antineoplastic |
| DepoCyte | 1999 | Cytarabine | Lymphomatous meningitis |
| Myocet | 2001 | Doxorubicin | Metastatic breast cancer |
| Mepact | 2009 | Mifamurtide | Osteosarcoma |
| Marqibo® (Onco TCS) | 2012 | Vincristine | Acute lymphoblastic leukemia |
| Gene Name | Abbreviation | Function of Encoded Protein |
|---|---|---|
| Epidermal growth factor receptor | EGFR | Regulating cell proliferation and survival |
| Isocitrate dehydrogenase 1 | IDH1 | Production of NADPH |
| Neurofibromin 1 | NF1 | Regulating cell proliferation and survival |
| Phosphoinositide-3-kinase catalytic alpha | PIK3CA | Regulating cell proliferation and survival |
| Phosphoinositide-3-kinase regulatory 1 | PIK3R1 | Regulating cell proliferation and survival |
| Phosphatase and tensin homolog | PTEN | Regulating cell proliferation and survival |
| Protein tyrosine phosphatase receptor type D | PTPRD | Regulating cell proliferation and survival |
| Retinoblastoma 1 | RB1 | Regulating cell cycle |
| Tumor protein p53 | TP53 | Apoptosis |
| V-erb-b2-erythroblastic leukemia viral oncogene homolog 2 | ERBB2 | Regulating cell proliferation and survival |
| Payload | Tf conjugation Method | Glioma Cell Line Used | Animal Model | Main Findings | Ref. |
|---|---|---|---|---|---|
| Cisplatin | Coupling the terminal carboxy group of DSPE-PEG2000-COOH | bEnd3/C6 co-culture | - | -Tf-modified liposomes improved transport across BBB using clathrin-mediated endocytosis -Tf-modified liposomes showed higher toxicity to glioma cells in vitro | [88] |
| Temozolomide, bromodomain inhibitor JQ1 | Post insertion | U87MG, GL261 | NCR nude mice | -Tf-modified liposomes improved delivery through receptor-mediated transcytosis -Mice in Tf-modified liposomes treatment group showed increased markers for DNA damage and apoptosis -1.5-to-2-fold decrease in tumor burden in mice | [84] |
| Resveratrol | Post insertion | U-87 MG | Female athymic NCr-nu/nu nude mice | -There was a 2.3-fold higher association between Tf-modified liposomes and glioma cells compared to normal cells expressing TfRs -The percent cell viability for Tf-RES-liposomes was 54.2 ± 3.8% while that for RES-liposomes was 61.9 ± 3.9% -Mice in the Tf-modified liposomes had the longest survival rate of 28 days. | [85] |
| Elemene, Cabazitaxel | - | U251, C6, RG2 | Female nude mice | -In vitro, studies showed a 5.83-fold higher intake rate of Tf-modified liposomes -Tf-modified liposomes increased the mean survival time from 28 days (for non-targeted liposomes) to 30 days | [86] |
| Doxorubicin, erlotinib | Tf was coupled to the terminal end of DSPE-PEG(2000)-NHS using nucleophilic substitution | U87, bEnd.3 | Male/female nude mice | -The transport of the drugs by Tf-Pen-modified liposomes was improved by 15% in vitro -Tf-Pen liposomes achieved tumor regression in mice brains by ~90% -Tf-Pen liposomes achieved the highest median survival time of 36 days | [87] |
| Doxorubicin | Post insertion | U87, GL261 | Balb/c nude mice | -The MTT assay showed that the antitumor activity of Tf-liposomes was higher than that of non-targeted liposomes and free DOX -There was not much difference in the survival time of mice treated with Tf-liposomes and non-targeted liposomes (25 and 24 days, respectively) | [89] |
| Name of Institution | Liposomal Payload | Number of Patients | Description | Status | Identifier (Clinicaltrials.gov accessed on 10 July 2023) |
|---|---|---|---|---|---|
| University of Regensburg | Doxorubicin, Temozolomide | 63 | -Pegylated liposomal doxorubicin is combined with temozolomide and radiotherapy -In phase I: pegylated liposomal doxorubicin increased in increments of 5 mg/m2 (starting from 5 to 20 mg/m2) -In phase II: the pegylated liposomal doxorubicin targeted dose of 20 mg/m2 is administered up to a cumulative dose of 550 mg/m2 or until tumor progression | Completed | NCT00944801 |
| Medical University of South Carolina | cytarabine (DepoCyt), Temozolomide | 12 | -Phase I and phase II intraventricular liposomal cytarabine (DepoCyt) in combination with oral temozolomide | Terminated | NCT01044966 |
| University of California, San Francisco | CPT-11 | 34 | -Phase I dose escalation scheme with a starting dose of 120 mg/m2 or 60 mg/m2 through IV | Completed | NCT00734682 |
| SignPath Pharma, Inc. | Curcumin | 30 | -Phase I and phase II dose escalating scheme of liposomal curcumin concurrent with temozolomide and radiation therapy as well as adjuvant temozolomide after radiation therapy | Recruiting | NCT05768919 |
| Plus Therapeutics | 186Rhenium | 30 | -Phase I to determine the maximum tolerated dose of nanoliposomal 186Rhenium -Phase II to determine overall survival | Recruiting | NCT01906385 |
| Emory University | Verteporfin | 24 | -Phase I and phase II dose escalation study to study the side effects and determine the optimal dose of visudyne (liposomal verteporfin) | Recruiting | NCT04590664 |
| University of Florida | RNA | 28 | -Phase I study to determine manufacturing feasibility, safety, and the maximum tolerated dose -The trial consists of three parts, surgery, radiation, and immunotherapy -Liposomal RNA vaccine will be administered monthly for a total of 15 vaccines after radiation | Recruiting | NCT04573140 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawak, P.; Sawaftah, N.M.A.; Pitt, W.G.; Husseini, G.A. Transferrin-Targeted Liposomes in Glioblastoma Therapy: A Review. Int. J. Mol. Sci. 2023, 24, 13262. https://doi.org/10.3390/ijms241713262
Kawak P, Sawaftah NMA, Pitt WG, Husseini GA. Transferrin-Targeted Liposomes in Glioblastoma Therapy: A Review. International Journal of Molecular Sciences. 2023; 24(17):13262. https://doi.org/10.3390/ijms241713262
Chicago/Turabian StyleKawak, Paul, Nour M. Al Sawaftah, William G. Pitt, and Ghaleb A. Husseini. 2023. "Transferrin-Targeted Liposomes in Glioblastoma Therapy: A Review" International Journal of Molecular Sciences 24, no. 17: 13262. https://doi.org/10.3390/ijms241713262
APA StyleKawak, P., Sawaftah, N. M. A., Pitt, W. G., & Husseini, G. A. (2023). Transferrin-Targeted Liposomes in Glioblastoma Therapy: A Review. International Journal of Molecular Sciences, 24(17), 13262. https://doi.org/10.3390/ijms241713262