
Design of Cyclic Peptides Targeting Protein–Protein Interactions Using AlphaFold



Abstract
:1. Introduction
- Due to the absence of termini, cyclic peptides are more resistant to digestive enzymes like peptidases and exoproteases.
- The constraint of the cyclic structure facilitates more stable folding without relying on secondary structures.
2. Results
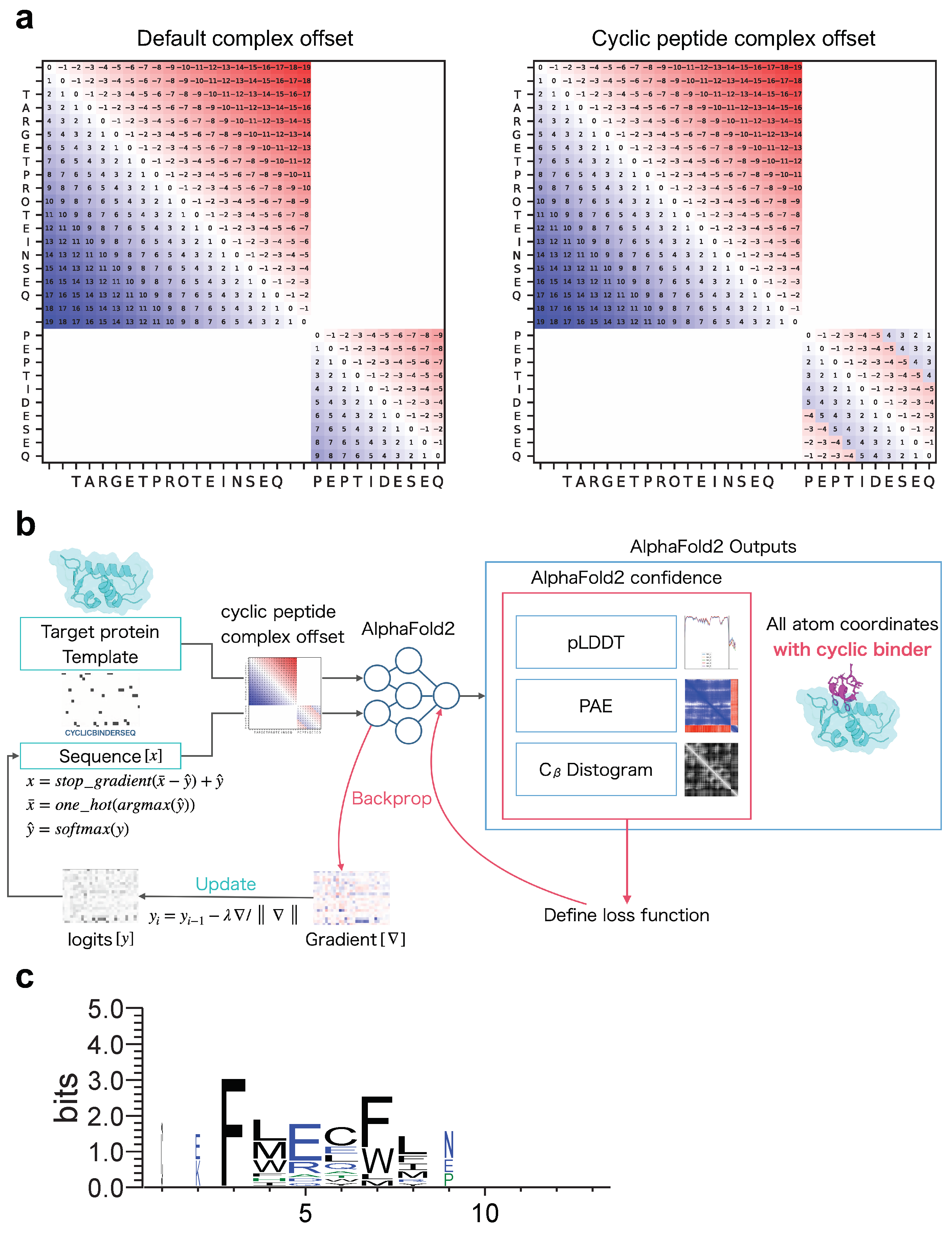
2.1. Structure Prediction of the Protein and Cyclic Peptide Complex
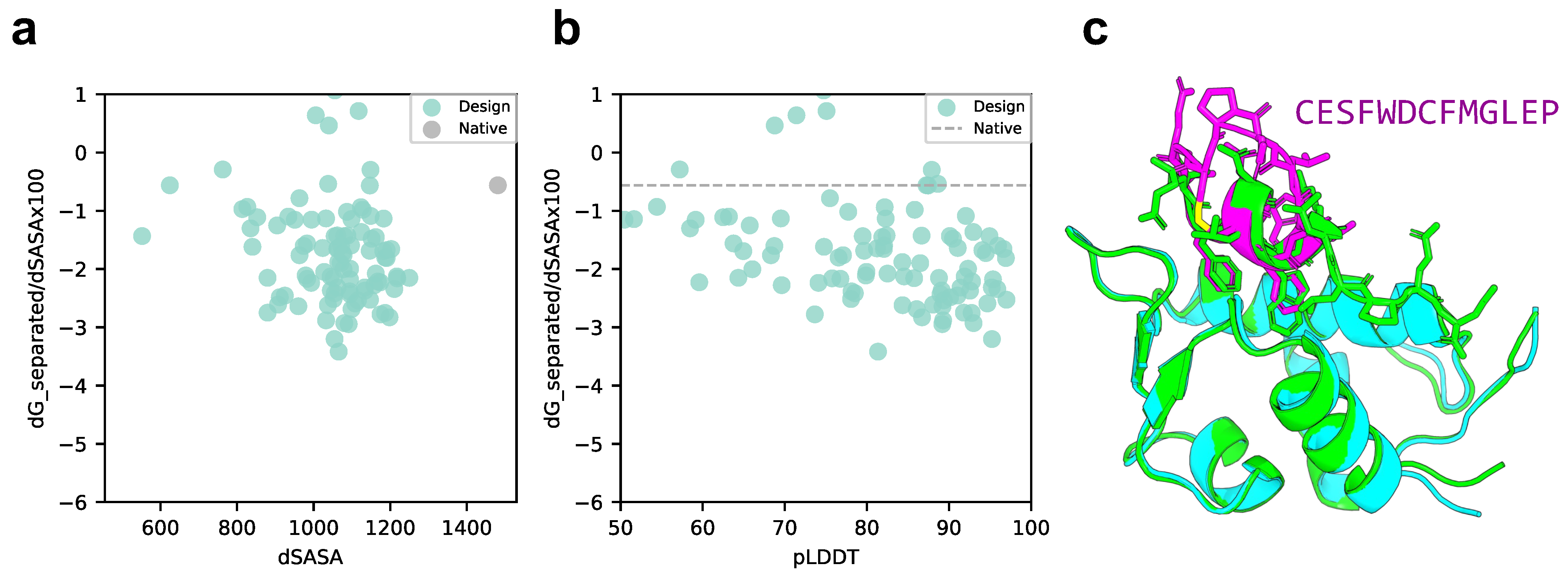
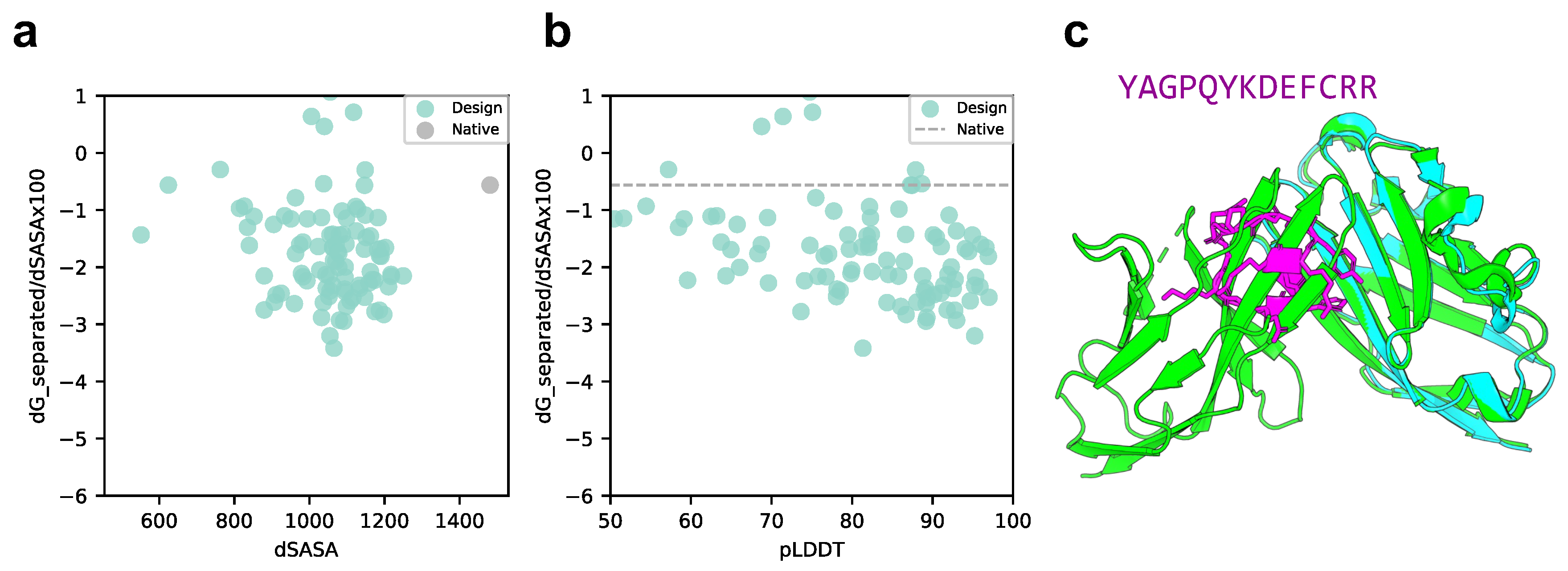
2.2. Cyclic Peptide Binder Hallucination Targeting the Protein-Peptide Complex
3. Discussion
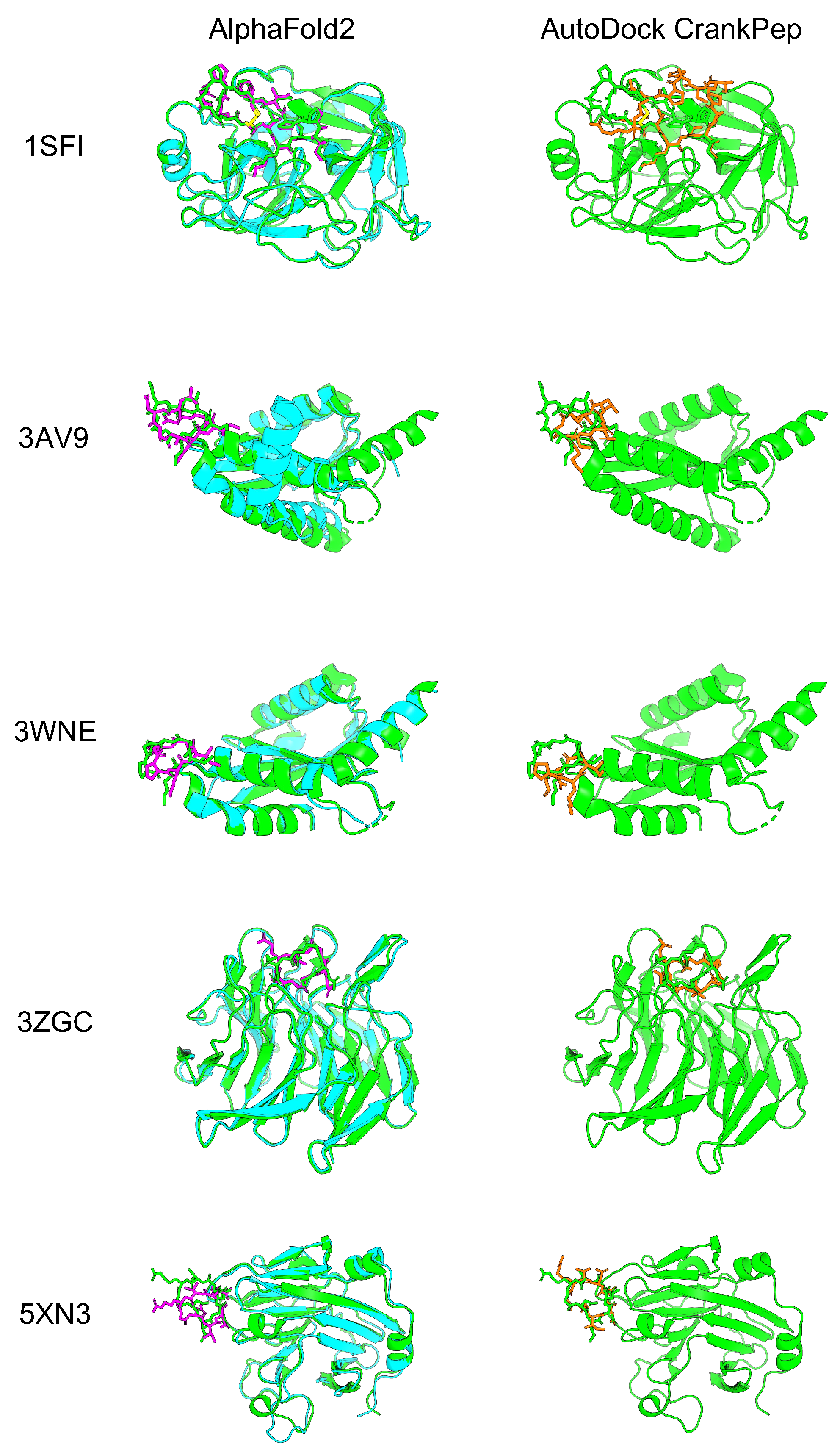
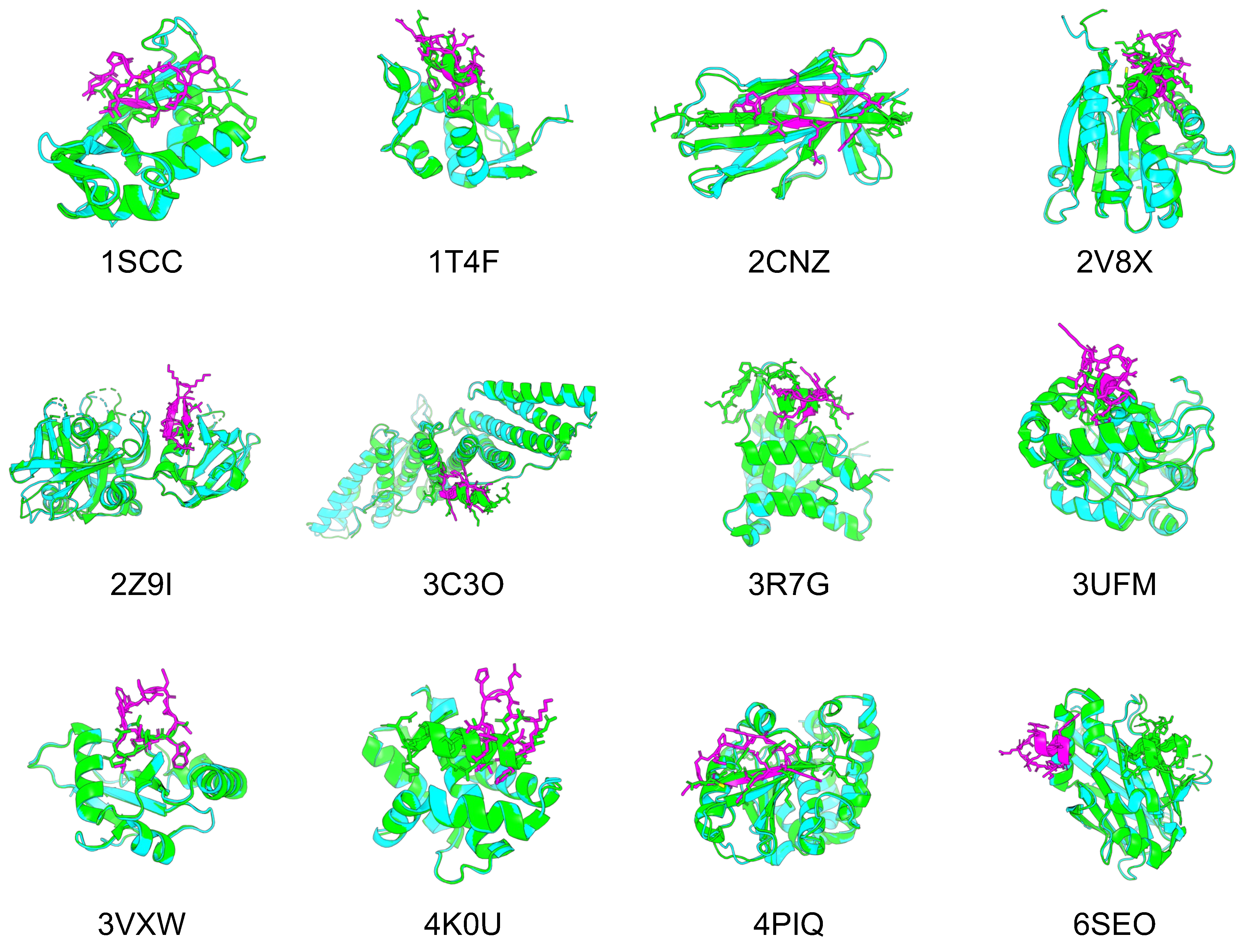
3.1. Application of Cyclic Peptide Binder Hallucinations to Other Target Protein-Peptide Complexes
3.2. Cyclic Peptide Binder Hallucination Targeting Protein and Protein Complex
3.3. Limitations and Challenges
4. Materials and Methods
4.1. ColabFold Settings
- To predict target protein–peptide complexes, we first used the target protein and cyclic peptide sequences connected by “:” and input to colab_batch as “TARGETPROTEINSEQ:CYCLICPEPTIDESEQ”.
- Using the chain brake residue_index, an offset matrix was created using the default method of AlphaFold-Multimer.
- Using the offset matrix for the default complex created in step 3 and the offset matrix for the cyclic peptide created in step 4, we replaced cyclic offsets only in the default offset matrix that corresponds to the cyclic peptide (Figure 1a). This enables the prediction of complexes of protein and cyclic peptide, where target proteins are linearized and peptides are cyclized.
4.2. AfDesign Settings
4.3. Calculation of Protein-Peptide Local Docking for Cyclic Peptide
4.4. Visualization of Sequence Logos
- The PDB target protein was aligned with target proteins in the complex with cyclic peptides predicted by AF2.
- A window with each native peptide and cyclic peptide shifted by one residue was created with a width of 6 residues (from the first residue to the sixth residue, from the second residue to the seventh residue and so on. The native peptide windows will have a length of , and the cyclic peptide will have the same number of windows as its length).
- The RMSD of the C of each window in 1 was calculated using rms_cur. Using 1YCR as an example, RMSDs were calculated per design.
- To compare with native “linear” peptides, using the window index with the lowest RMSD of the C among them, the design sequence was aligned based on the index numbers of the native peptide sequence and the cyclic peptide. The lowest RMSD was named RMSD_best in this study.
- After alignment, the non-6 letters were filled in with ‘-’. Sequence logos were created via WebLogo using the thresholding alignment sequence in RMSD_best as input (Figure 1c). The sequence logo represents each column of alignment as a stack of letters, with the height of each letter proportional to the observed frequency of the corresponding amino acid, and the overall height of each stack proportional to the degree of sequence conservation (measured in bits) at that location. The maximum sequence conservation per site is bits for amino acids. The width of the letters is proportional to ungaps of each column (the more ‘-’ there are in each column instead of amino acid represents, the thinner the letters).
4.5. Rosetta Interface Analyzer
4.6. Calculation of Solubility and Lipophilicity
4.7. Interatomic Interactions between PD-L1 and the Designed Cyclic Peptide
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PPI | protein–protein interaction |
| HTVS | High-throughput virtual screening |
| DL | deep learning |
| AF2 | AlphaFold2 |
| RMSD | root mean square deviation |
| MSA | multiple sequence alignment |
| PDB | Protein Data Bank |
| ADCP | AutoDock CrankPep |
| PAE | predicted aligned error |
| pLDDT | predicted local distance difference test |
| AF2_v3 | alphafold2_multimer_v3 model |
| AF2_v2 | alphafold2_multimer_v2 model |
| AF2_ptm | alphafold2_ptm model |
| SASA | solvent accessible surface area |
References
- Bonetta, L. Interactome under Construction. Nature 2010, 468, 851–852. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.-J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Toogood, P.L. Inhibition of Protein-Protein Association by Small Molecules: Approaches and Progress. J. Med. Chem. 2002, 45, 1543–1558. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-Molecule Inhibitors of Protein–Protein Interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]
- Dev, K.K. Making Protein Interactions Druggable: Targeting PDZ Domains. Nat. Rev. Drug Discov. 2004, 3, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wang, W.; Fang, G. Targeting Protein-Protein Interaction by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting Protein–Protein Interactions as an Anticancer Strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef]
- Shin, W.-H.; Kumazawa, K.; Imai, K.; Hirokawa, T.; Kihara, D. Current Challenges and Opportunities in Designing Protein–Protein Interaction Targeted Drugs. Adv. Appl. Bioinform. Chem. 2020, 13, 11–25. [Google Scholar] [CrossRef]
- Kosugi, T.; Ohue, M. Quantitative Estimate Index for Early-Stage Screening of Compounds Targeting Protein-Protein Interactions. Int. J. Mol. Sci. 2021, 22, 10925. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in Peptide Drug Discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Tsomaia, N. Peptide Therapeutics: Targeting the Undruggable Space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef]
- Whitty, A.; Zhong, M.; Viarengo, L.; Beglov, D.; Hall, D.R.; Vajda, S. Quantifying the Chameleonic Properties of Macrocycles and Other High-Molecular-Weight Drugs. Drug Discov. Today 2016, 21, 712–717. [Google Scholar] [CrossRef]
- Lee, D.; Lee, S.; Choi, J.; Song, Y.-K.; Kim, M.J.; Shin, D.-S.; Bae, M.A.; Kim, Y.-C.; Park, C.-J.; Lee, K.-R.; et al. Interplay among Conformation, Intramolecular Hydrogen Bonds, and Chameleonicity in the Membrane Permeability and Cyclophilin A Binding of Macrocyclic Peptide Cyclosporin O Derivatives. J. Med. Chem. 2021, 64, 8272–8286. [Google Scholar] [CrossRef]
- Sugita, M.; Sugiyama, S.; Fujie, T.; Yoshikawa, Y.; Yanagisawa, K.; Ohue, M.; Akiyama, Y. Large-scale membrane permeability prediction of cyclic peptides crossing a lipid bilayer based on enhanced sampling molecular dynamics simulations. J. Chem. Inf. Model. 2021, 61, 3681–3695. [Google Scholar] [CrossRef]
- Sugita, M.; Fujie, T.; Yanagisawa, K.; Ohue, M.; Akiyama, Y. Lipid composition is critical for accurate membrane permeability prediction of cyclic peptides by molecular dynamics simulations. J. Chem. Inf. Model. 2022, 62, 4549–4560. [Google Scholar] [CrossRef]
- Wu, C.-H.; Liu, I.-J.; Lu, R.-M.; Wu, H.-C. Advancement and Applications of Peptide Phage Display Technology in Biomedical Science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef]
- Goto, Y.; Suga, H. The RaPID Platform for the Discovery of Pseudo-Natural Macrocyclic Peptides. Acc. Chem. Res. 2021, 54, 3604–3617. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Naimuddin, M.; Biyani, M.; Sasaki, T.; Machida, M.; Kubo, T.; Funatsu, T.; Husimi, Y.; Nemoto, N. CDNA Display: A Novel Screening Method for Functional Disulfide-Rich Peptides by Solid-Phase Synthesis and Stabilization of mRNA–Protein Fusions. Nucleic Acids Res. 2009, 37, e108. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; dos Santos, C.; Chen, P.-Y.; et al. Accelerated Antimicrobial Discovery via Deep Generative Models and Molecular Dynamics Simulations. Nat. Biomed. Eng. 2021, 5, 613–623. [Google Scholar] [CrossRef]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, A.; Zhang, A.; Reymond, J.-L. Populating Chemical Space with Peptides Using a Genetic Algorithm. J. Chem. Inf. Model. 2020, 60, 121–132. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Klebe, G. Virtual Ligand Screening: Strategies, Perspectives and Limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef]
- Amarasinghe, K.N.; De Maria, L.; Tyrchan, C.; Eriksson, L.A.; Sadowski, J.; Petrović, D. Virtual Screening Expands the Non-Natural Amino Acid Palette for Peptide Optimization. J. Chem. Inf. Model. 2022, 62, 2999–3007. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Tubert-Brohman, I.; Sherman, W.; Repasky, M.; Beuming, T. Improved Docking of Polypeptides with Glide. J. Chem. Inf. Model. 2013, 53, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Alogheli, H.; Olanders, G.; Schaal, W.; Brandt, P.; Karlén, A. Docking of Macrocycles: Comparing Rigid and Flexible Docking in Glide. J. Chem. Inf. Model. 2017, 57, 190–202. [Google Scholar] [CrossRef]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-Dock Web Server for the Flexible Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Pawel Ciemny, M.; Oleniecki, T.; Kuriata, A.; Badaczewska-Dawid, A.E.; Kolinski, A.; Kmiecik, S. CABS-Dock Standalone: A Toolbox for Flexible Protein–Peptide Docking. Bioinformatics 2019, 35, 4170–4172. [Google Scholar] [CrossRef]
- Porter, K.A.; Xia, B.; Beglov, D.; Bohnuud, T.; Alam, N.; Schueler-Furman, O.; Kozakov, D. ClusPro PeptiDock: Efficient Global Docking of Peptide Recognition Motifs Using FFT. Bioinformatics 2017, 33, 3299–3301. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Goldstein, O.; Xia, B.; Porter, K.A.; Kozakov, D.; Schueler-Furman, O. High-Resolution Global Peptide-Protein Docking Using Fragments-Based PIPER-FlexPepDock. PLoS Comput. Biol. 2017, 13, e1005905. [Google Scholar] [CrossRef] [PubMed]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de Novo Structure Prediction for Linear Peptides in Solution and in Complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Moll, M.; Devaurs, D.; Jackson, K.R.; Lizée, G.; Kavraki, L.E. DINC 2.0: A New Protein–Peptide Docking Webserver Using an Incremental Approach. Cancer Res. 2017, 77, e55–e57. [Google Scholar] [CrossRef]
- Charitou, V.; van Keulen, S.C.; Bonvin, A.M.J.J. Cyclization and Docking Protocol for Cyclic Peptide–Protein Modeling Using HADDOCK2.4. J. Chem. Theory Comput. 2022, 18, 4027–4040. [Google Scholar] [CrossRef]
- Zhang, Y.; Sanner, M.F. AutoDock CrankPep: Combining Folding and Docking to Predict Protein–Peptide Complexes. Bioinformatics 2019, 35, 5121–5127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sanner, M.F. Docking Flexible Cyclic Peptides with AutoDock CrankPep. J. Chem. Theory Comput. 2019, 15, 5161–5168. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, N.M.; Bandyopadhyay, A. High Throughput Virtual Screening (HTVS) of Peptide Library: Technological Advancement in Ligand Discovery. Eur. J. Med. Chem. 2022, 243, 114766. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Anishchenko, I.; Humphreys, I.R.; Cong, Q.; Baker, D.; DiMaio, F. Efficient and Accurate Prediction of Protein Structure Using RoseTTAFold2. bioRxiv 2023. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Humphreys, I.R.; Pei, J.; Baek, M.; Krishnakumar, A.; Anishchenko, I.; Ovchinnikov, S.; Zhang, J.; Ness, T.J.; Banjade, S.; Bagde, S.R.; et al. Computed Structures of Core Eukaryotic Protein Complexes. Science 2021, 374, eabm4805. [Google Scholar] [CrossRef] [PubMed]
- Gulsevin, A.; Meiler, J. Benchmarking Peptide Structure Prediction with AlphaFold2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tsaban, T.; Varga, J.K.; Avraham, O.; Ben-Aharon, Z.; Khramushin, A.; Schueler-Furman, O. Harnessing Protein Folding Neural Networks for Peptide–Protein Docking. Nat. Commun. 2022, 13, 176. [Google Scholar] [CrossRef] [PubMed]
- Roney, J.P.; Ovchinnikov, S. State-of-the-Art Estimation of Protein Model Accuracy Using AlphaFold. Phys. Rev. Lett. 2022, 129, 238101. [Google Scholar] [CrossRef]
- Anishchenko, I.; Pellock, S.J.; Chidyausiku, T.M.; Ramelot, T.A.; Ovchinnikov, S.; Hao, J.; Bafna, K.; Norn, C.; Kang, A.; Bera, A.K.; et al. De Novo Protein Design by Deep Network Hallucination. Nature 2021, 600, 547–552. [Google Scholar] [CrossRef]
- Norn, C.; Wicky, B.I.M.; Juergens, D.; Liu, S.; Kim, D.; Tischer, D.; Koepnick, B.; Anishchenko, I.; Players, F.; Baker, D.; et al. Protein sequence design using conformational landscape optimization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017228118. [Google Scholar] [CrossRef]
- Goverde, C.A.; Wolf, B.; Khakzad, H.; Rosset, S.; Correia, B.E. De Novo Protein Design by Inversion of the AlphaFold Structure Prediction Network. Protein Sci. 2023, 32, e4653. [Google Scholar] [CrossRef]
- Frank, C.; Khoshouei, A.; de Stigter, Y.; Schiewitz, D.; Feng, S.; Ovchinnikov, S.; Dietz, H. Efficient and Scalable de Novo Protein Design Using a Relaxed Sequence Space. bioRxiv 2023. [Google Scholar] [CrossRef]
- Kosugi, T.; Ohue, M. Solubility-Aware Protein Binding Peptide Design Using AlphaFold. Biomedicines 2022, 10, 1626. [Google Scholar] [CrossRef] [PubMed]
- Rettie, S.A.; Campbell, K.V.; Bera, A.K.; Kang, A.; Kozlov, S.; De La Cruz, J.; Adebomi, V.; Zhou, G.; DiMaio, F.; Ovchinnikov, S.; et al. Cyclic Peptide Structure Prediction and Design Using AlphaFold. bioRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Ovchinnikov, S. (@sokrypton). Twitter Post: Max Galettis Alerted Us to an Error in Our Cyclic Offset Implementation. Which Is Now Fixed in the Notebook. (When You Circularly Permuted the Sequences, the Solutions Were *Nearly* Identical (When Aligned), But Were Not Identical. Now with the Bugfix, They Are Identical! With the Bugfix, They Are Identical!). Available online: https://twitter.com/sokrypton/status/1670551262427840513 (accessed on 18 June 2023).
- Banhos Danneskiold-Samøe, N.; Kavi, D.; Jude, K.M.; Nissen, S.B.; Wat, L.W.; Coassolo, L.; Zhao, M.; Asae Santana-Oikawa, G.; Broido, B.B.; Garcia, K.C.; et al. Rapid and Accurate Deorphanization of Ligand-Receptor Pairs Using AlphaFold. bioRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Baek, M. (@minkbaek). Twitter Post: Adding a Big Enough Number for “residue_index” Feature is Enough to Model Hetero-Complex Using AlphaFold (Green&Cyan: Crystal Structure/Magenta: Predicted Model w/residue_index Modification). Available online: https://twitter.com/minkbaek/status/1417538291709071362 (accessed on 20 July 2021).
- Available online: https://github.com/sokrypton/ColabDesign/tree/main/af (accessed on 14 March 2022).
- Stranges, P.B.; Kuhlman, B. A Comparison of Successful and Failed Protein Interface Designs Highlights the Challenges of Designing Buried Hydrogen Bonds. Protein Sci. 2013, 22, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Peccati, F.; Alunno-Rufini, S.; Jiménez-Osés, G. Accurate Prediction of Enzyme Thermostabilization with Rosetta Using AlphaFold Ensembles. J. Chem. Inf. Model. 2023, 63, 898–909. [Google Scholar] [CrossRef]
- Bryant, A.; Elofsson, A. EvoBind: In Silico Directed Evolution of Peptide Binders with AlphaFold. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhou, X.; Huang, Y.-H.; King, G.J.; Collins, B.M.; Gao, Y.; Craik, D.J.; Wang, C.K. Rational Design of Potent Peptide Inhibitors of the PD-1:PD-L1 Interaction for Cancer Immunotherapy. J. Am. Chem. Soc. 2021, 143, 18536–18547. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein-Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of Side-Chain Amide Orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1SFI | 3AV9 | 3WNE | 3ZGC | 5XN3 | ||
|---|---|---|---|---|---|---|
| RMSD (Å) (Aligned with protein) | AF2_v2 | 0.86 | 1.57 | 1.63 | 1.85 | 4.28 |
| ADCP | 6.71 | 7.09 | 4.29 | 3.58 | 3.40 | |
| RMSD (Å) (Aligned with peptide) | AF2_v2 | 0.53 | 1.01 | 1.37 | 1.23 | 2.94 |
| ADCP | 5.01 | 3.32 | 2.49 | 2.27 | 3.28 |
| pLDDT | Rosetta Interface Analyzer Score (Design) | RMSD_best | Rosetta Interface Analyzer Score (Native) | |
|---|---|---|---|---|
| 1SSC | 88.51 | −2.51 | 2.58 | −2.23 |
| 1T4F | 86.39 | −2.49 | 0.85 | −2.40 |
| 2CNZ | 90.00 | −4.05 | 0.60 | −3.98 |
| 2V8X | 82.89 | −1.58 | 3.06 | −2.35 |
| 2Z9I | 83.79 | −0.38 | 0.52 | 2.67 |
| 3C3O | 71.23 | −1.10 | 1.40 | −0.21 |
| 3R7G | 91.53 | −1.45 | 1.34 | −1.41 |
| 3UFM | 77.05 | −2.33 | 4.53 | −2.77 |
| 3VXW | 79.40 | −1.90 | 0.65 | 4.80 |
| 4K0U | 95.24 | −2.85 | 0.51 | −2.04 |
| 4PIQ | 84.88 | −1.16 | 0.79 | −0.90 |
| 6SEO | 77.02 | −0.96 | 18.15 | 2.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosugi, T.; Ohue, M. Design of Cyclic Peptides Targeting Protein–Protein Interactions Using AlphaFold. Int. J. Mol. Sci. 2023, 24, 13257. https://doi.org/10.3390/ijms241713257
Kosugi T, Ohue M. Design of Cyclic Peptides Targeting Protein–Protein Interactions Using AlphaFold. International Journal of Molecular Sciences. 2023; 24(17):13257. https://doi.org/10.3390/ijms241713257
Chicago/Turabian StyleKosugi, Takatsugu, and Masahito Ohue. 2023. "Design of Cyclic Peptides Targeting Protein–Protein Interactions Using AlphaFold" International Journal of Molecular Sciences 24, no. 17: 13257. https://doi.org/10.3390/ijms241713257
APA StyleKosugi, T., & Ohue, M. (2023). Design of Cyclic Peptides Targeting Protein–Protein Interactions Using AlphaFold. International Journal of Molecular Sciences, 24(17), 13257. https://doi.org/10.3390/ijms241713257