

Saturated Fat-Mediated Upregulation of IL-32 and CCL20 in Hepatocytes Contributes to Higher Expression of These Fibrosis-Driving Molecules in MASLD

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
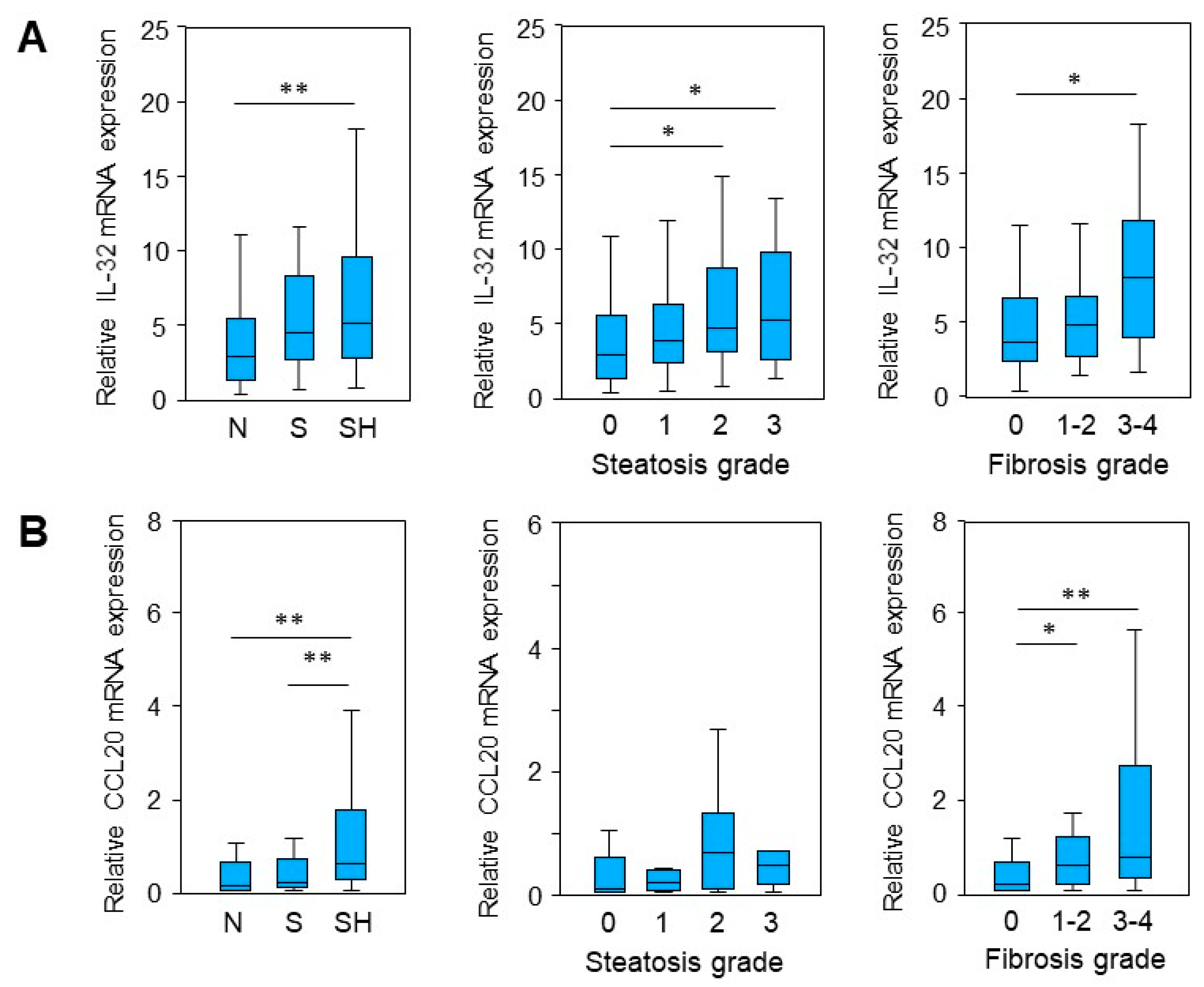
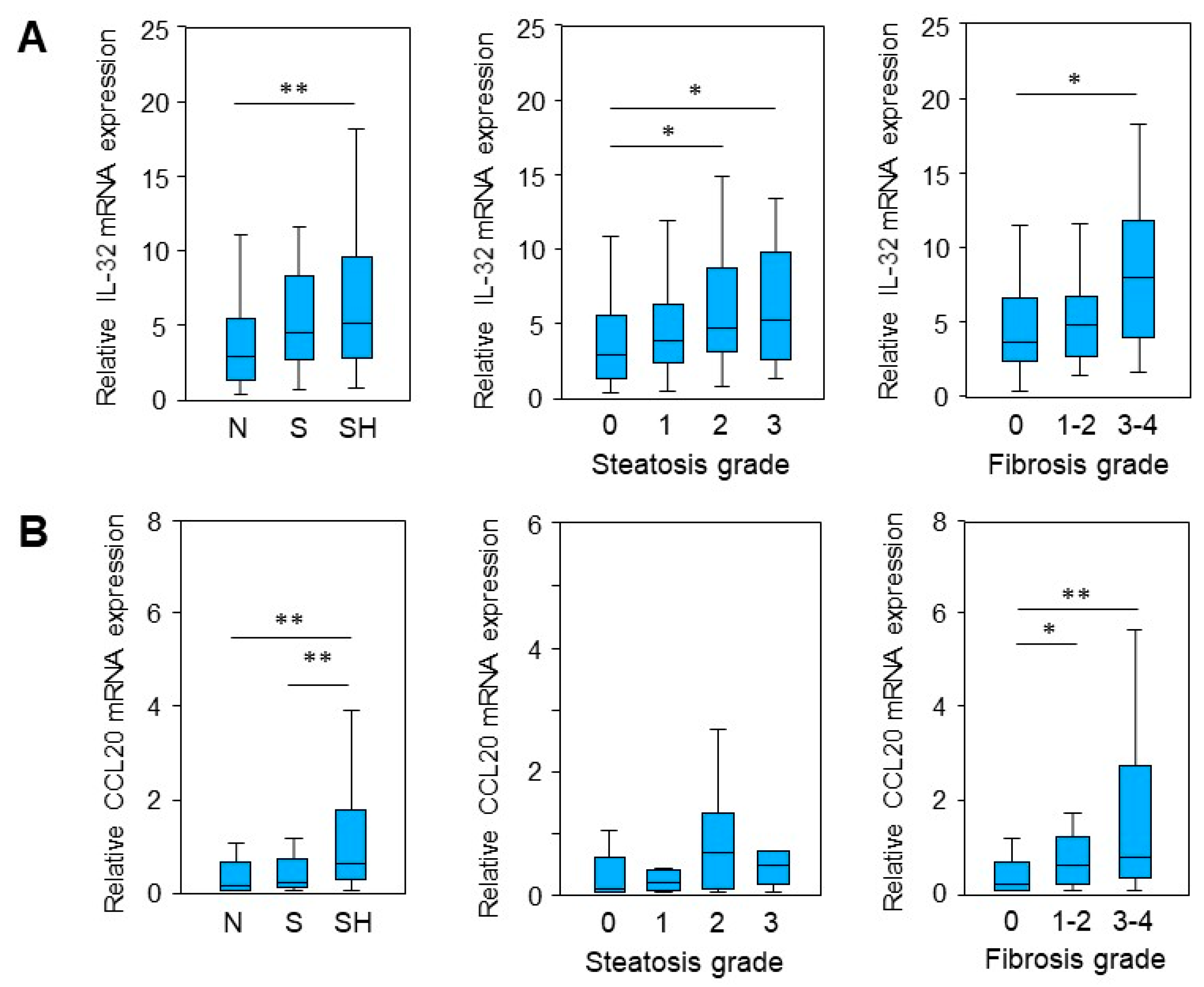
2.1. IL-32 and CCL20 mRNA Expression Is Enhanced in MASLD
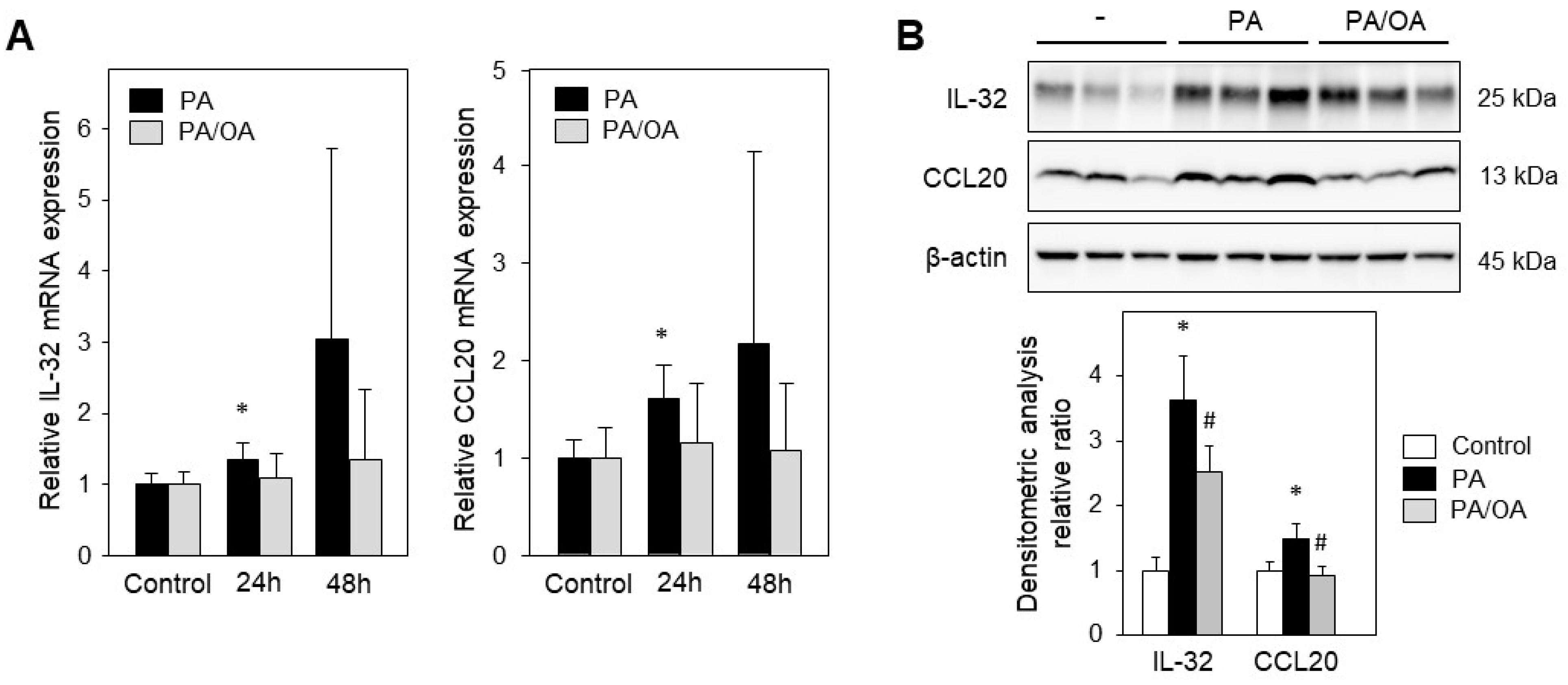
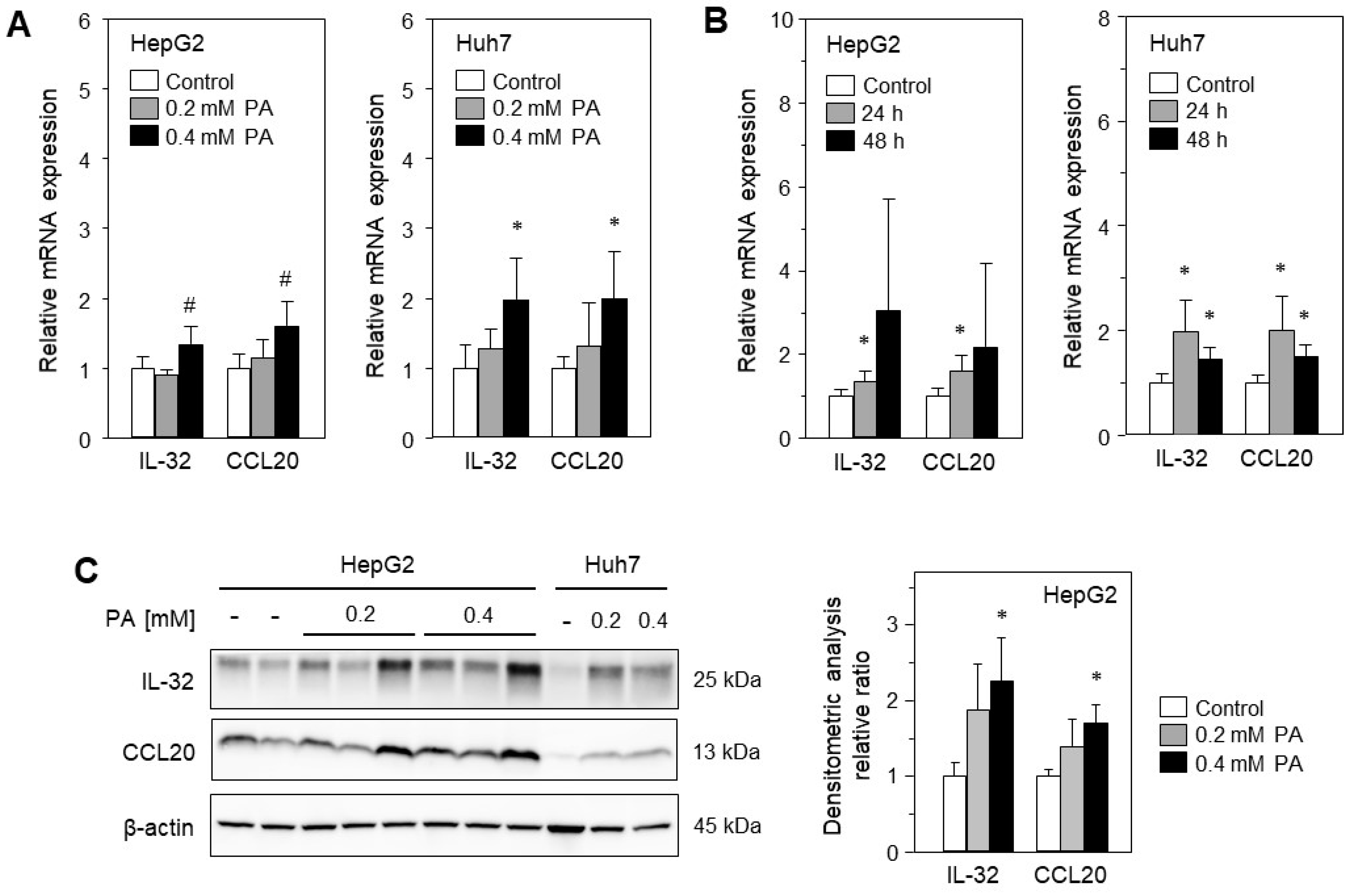
2.2. Treatment with Palmitic Acid Leads to Fat Deposition and Induction of IL-32 and CCL20 Expression
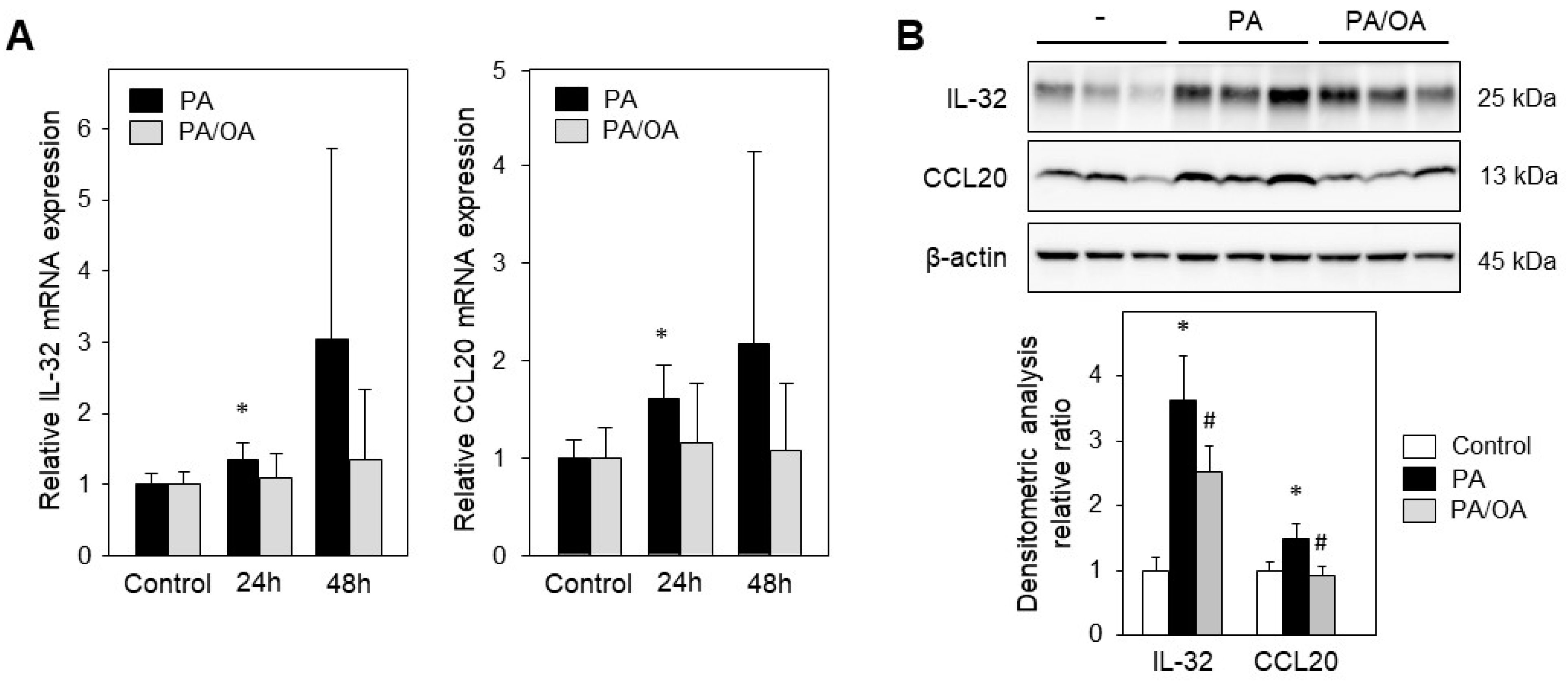
2.3. Monounsaturated Oleic Acid Compared to Palmitic Acid Does Not Induce IL-32 and CCL20 Expression
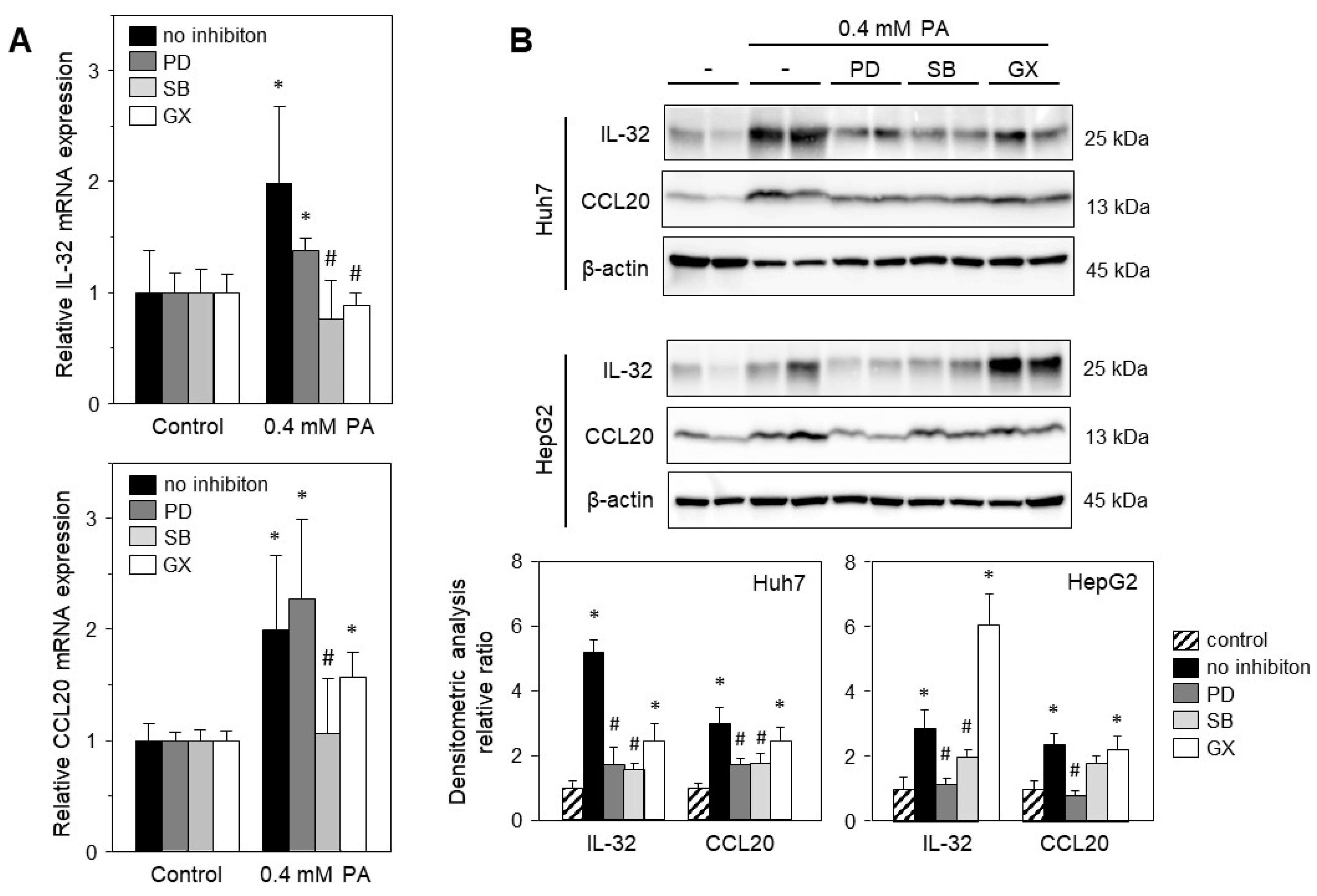
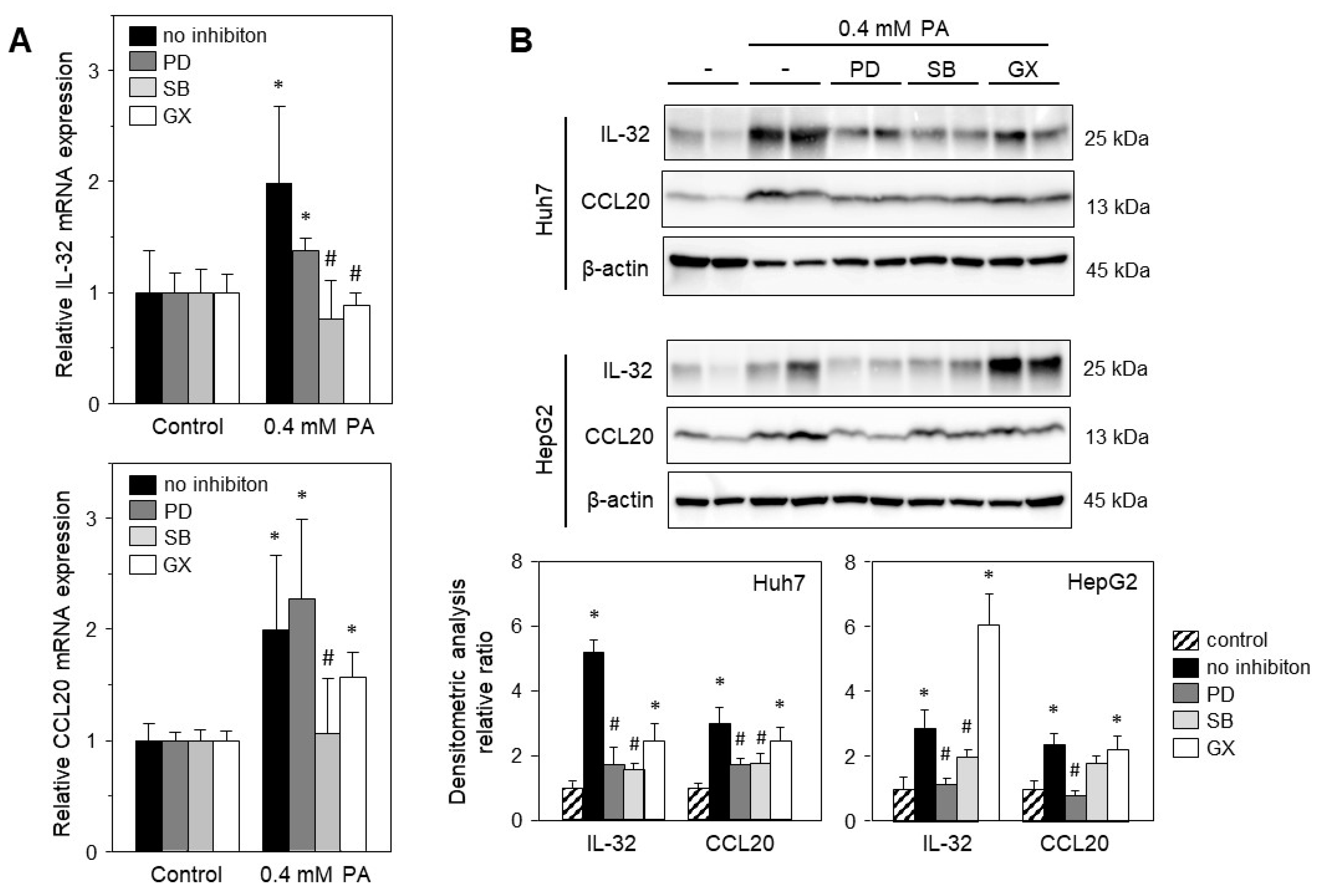
2.4. Palmitic Acid-Induced Gene Expression Is Mediated via Stress-Induced Pathways
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malaguarnera, M.; Di Rosa, M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 2009, 87, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in NAFLD—Revisited after a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell. Mol. Life Sci. 2016, 73, 1969–1987. [Google Scholar] [CrossRef]
- Lee, Y.A.; Friedman, S.L. Inflammatory and fibrotic mechanisms in NAFLD—Implications for new treatment strategies. J. Intern. Med. 2022, 291, 11–31. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Baselli, G.A.; Dongiovanni, P.; Rametta, R.; Meroni, M.; Pelusi, S.; Maggioni, M.; Badiali, S.; Pingitore, P.; Maurotti, S.; Montalcini, T.; et al. Liver transcriptomics highlights interleukin-32 as novel NAFLD-related cytokine and candidate biomarker. Gut 2020, 69, 1855–1866. [Google Scholar] [CrossRef]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C. PNPLA3—A Potential Therapeutic Target for Personalized Treatment of Chronic Liver Disease. Front. Med. 2019, 6, 304. [Google Scholar] [CrossRef]
- Kaur, S.; Rawal, P.; Siddiqui, H.; Rohilla, S.; Sharma, S.; Tripathi, D.M.; Baweja, S.; Hassan, M.; Vlaic, S.; Guthke, R.; et al. Increased Expression of RUNX1 in Liver Correlates with NASH Activity Score in Patients with Non-Alcoholic Steatohepatitis (NASH). Cells 2019, 8, 1277. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.T.; Son, D.J.; Lee, C.K.; Yoon, D.Y.; Lee, D.H.; Park, M.H. Interleukin 32, inflammation and cancer. Pharmacol. Therapeut. 2017, 174, 127–137. [Google Scholar]
- Scheiermann, P.; Bachmann, M.; Härdle, L.; Pleli, T.; Piiper, A.; Zwissler, B.; Pfeilschifter, J.; Mühl, H. Application of IL-36 receptor antagonist weakens CCL20 expression and impairs recovery in the late phase of murine acetaminophen-induced liver injury. Sci. Rep. 2015, 5, srep08521. [Google Scholar] [CrossRef]
- Starmann, J.; Fälth, M.; Spindelböck, W.; Lanz, K.-L.; Lackner, C.; Zatloukal, K.; Trauner, M.; Sültmann, H. Gene Expression Profiling Unravels Cancer-Related Hepatic Molecular Signatures in Steatohepatitis but Not in Steatosis. PLoS ONE 2012, 7, e46584. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’connor, J.-E. A human hepatocellular in vitro model to investigate steatosis. Chem. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Weiss, T.S.; Lupke, M.; Ibrahim, S.; Buechler, C.; Lorenz, J.; Ruemmele, P.; Hofmann, U.; Melter, M.; Dayoub, R. Attenuated lipotoxicity and apoptosis is linked to exogenous and endogenous augmenter of liver regeneration by different pathways. PLoS ONE 2017, 12, e0184282. [Google Scholar] [CrossRef]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Akazawa, Y.; Cazanave, S.; Mott, J.L.; Elmi, N.; Bronk, S.F.; Kohno, S.; Charlton, M.R.; Gores, G.J. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J. Hepatol. 2010, 52, 586–593. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Kohli, R.; Gores, G.J. Mechanisms of Lipotoxicity in NAFLD and Clinical Implications. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Mantzaris, M.D.; Tsianos, E.V.; Galaris, D. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS J. 2011, 278, 519–530. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, M.; Liu, X.H.; Chen, X.M.; Yuan, Y.J.; Li, L.; Liu, J.P.; Lu, Y.R.; Cheng, J.Q.; Chen, Y.N. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis. Nutr. Metab. 2020, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Assmann, A.; Möhlig, M.; Osterhoff, M.; Pfeiffer, A.F.; Spranger, J. Fatty acids differentially modify the expression of urokinase type plasminogen activator receptor in monocytes. Biochem. Biophys. Res. Commun. 2008, 376, 196–199. [Google Scholar] [CrossRef]
- Aass, K.R.; Kastnes, M.H.; Standal, T. Molecular interactions and functions of IL-32. J. Leukoc. Biol. 2021, 109, 143–159. [Google Scholar] [CrossRef]
- Gasiuniene, E.; Lavinskiene, S.; Sakalauskas, R.; Sitkauskiene, B. Levels of IL-32 in Serum, Induced Sputum Supernatant, and Bronchial Lavage Fluid of Patients with Chronic Obstructive Pulmonary Disease. COPD: J. Chronic Obstr. Pulm. Dis. 2016, 13, 569–575. [Google Scholar] [CrossRef]
- Fadaei, R.; Bagheri, N.; Heidarian, E.; Nouri, A.; Hesari, Z.; Moradi, N.; Ahmadi, A.; Ahmadi, R. Serum levels of IL-32 in patients with type 2 diabetes mellitus and its relationship with TNF-α and IL-6. Cytokine 2020, 125, 154832. [Google Scholar] [CrossRef]
- Rasool, S.T.; Tang, H.; Wu, J.; Li, W.; Mukhtar, M.M.; Zhang, J.; Mu, Y.; Xing, H.X.; Wu, J.; Zhu, Y. Increased level of IL-32 during human immunodeficiency virus infection suppresses HIV replication. Immunol. Lett. 2008, 117, 161–167. [Google Scholar] [CrossRef]
- Xu, Q.; Pan, X.; Shu, X.; Cao, H.; Li, X.; Zhang, K.; Lu, J.; Zou, Y.; Li, X.; Liu, H.; et al. Increased interleukin-32 expression in chronic hepatitis B virus-infected liver. J. Infect. 2012, 65, 336–342. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, S.; Jeong, A.L.; Han, S.; Ka, H.I.; Lim, J.S.; Lee, M.S.; Yoon, D.Y.; Lee, J.H.; Yang, Y. Hypoxia-induced IL-32 beta increases glycolysis in breast cancer cells. Cancer Lett. 2015, 356, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Keeley, T.P.; Giuntoli, B.; White, M.D.; Lavilla Puerta, M.; Perata, P.; Hopkinson, R.J.; Flashman, E.; Licausi, F.; Ratcliffe, P.J. Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 2019, 365, 65–69. [Google Scholar] [CrossRef]
- Dali-Youcef, N.; Vix, M.; Costantino, F.; El-Saghire, H.; Lhermitte, B.; Callari, C.; D’agostino, J.; Perretta, S.; Paveliu, S.; Gualtierotti, M.; et al. Interleukin-32 Contributes to Human Nonalcoholic Fatty Liver Disease and Insulin Resistance. Hepatol. Commun. 2019, 3, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Wang, X.L.; Vikash, V.; Ye, Q.; Wu, D.D.; Liu, Y.L.; Dong, W.G. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar]
- Moschen, A.R.; Fritz, T.; Clouston, A.D.; Rebhan, I.; Bauhofer, O.; Barrie, H.D.; Powell, E.E.; Kim, S.; Dinarello, C.A.; Bartenschlager, R.; et al. Interleukin-32: A new proinflammatory cytokine involved in hepatitis C virus-related liver inflammation and fibrosis. Hepatology 2011, 53, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Park, E.S.; Lee, A.R.; Park, S.; Park, Y.K.; Ahn, S.H.; Kang, H.S.; Won, J.H.; Ha, Y.N.; Jae, B.; et al. Intracellular interleukin-32 gamma mediates antiviral activity of cytokines against hepatitis B virus. Nat. Commun. 2018, 9, 3284. [Google Scholar] [CrossRef]
- Damen, M.S.; dos Santos, J.C.; Hermsen, R.; van der Vliet, J.A.; Netea, M.G.; Riksen, N.P.; Dinarello, C.A.; Joosten, L.A.; Heinhuis, B. Interleukin-32 upregulates the expression of ABCA1 and ABCG1 resulting in reduced intracellular lipid concentrations in primary human hepatocytes. Atherosclerosis 2018, 271, 193–202. [Google Scholar] [CrossRef]
- Lee, D.H.; Hong, J.E.; Yun, H.M.; Hwang, C.J.; Park, J.H.; Han, S.B.; Yoon, D.Y.; Song, M.J.; Hong, J.T. Interleukin-32 beta Ameliorates Metabolic Disorder and Liver Damage in Mice Fed High-Fat Diet. Obesity 2015, 23, 615–622. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Valentí, V.; Moncada, R.; Landecho, M.F.; Silva, C.; Salvador, J.; Frühbeck, G. Increased Interleukin-32 Levels in Obesity Promote Adipose Tissue Inflammation and Extracellular Matrix Remodeling: Effect of Weight Loss. Diabetes 2016, 65, 3636–3648. [Google Scholar] [CrossRef]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef]
- Affò, S.; Morales-Ibanez, O.; Rodrigo-Torres, D.; Altamirano, J.; Blaya, D.; Dapito, D.H.; Millán, C.; Coll, M.; Caviglia, J.M.; Arroyo, V.; et al. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut 2014, 63, 1782–1792. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wei, T.; Cui, X.; Wei, R.; Hong, T. Identification of key genes and pathways in mild and severe nonalcoholic fatty liver disease by integrative analysis. Chronic Dis. Transl. Med. 2021, 7, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xia, J.; Wang, X.; Xu, F. Transcriptional regulation of CCL20 expression. Microbes Infect. 2014, 16, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Della Fazia, M.A.; Servillo, G. Foie gras and liver regeneration: A fat dilemma. Cell Stress 2018, 2, 162–175. [Google Scholar] [CrossRef]
- Nimphy, J.; Ibrahim, S.; Dayoub, R.; Kubitza, M.; Melter, M.; Weiss, T.S. Interleukin-1b Attenuates Expression of Augmenter of Liver Regeneration (ALR) by Regulating HNF4α Independent of c-Jun. Int. J. Mol. Sci. 2023, 24, 8107. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilcher, K.; Dayoub, R.; Kubitza, M.; Riepl, J.; Klein, K.; Buechler, C.; Melter, M.; Weiss, T.S. Saturated Fat-Mediated Upregulation of IL-32 and CCL20 in Hepatocytes Contributes to Higher Expression of These Fibrosis-Driving Molecules in MASLD. Int. J. Mol. Sci. 2023, 24, 13222. https://doi.org/10.3390/ijms241713222
Schilcher K, Dayoub R, Kubitza M, Riepl J, Klein K, Buechler C, Melter M, Weiss TS. Saturated Fat-Mediated Upregulation of IL-32 and CCL20 in Hepatocytes Contributes to Higher Expression of These Fibrosis-Driving Molecules in MASLD. International Journal of Molecular Sciences. 2023; 24(17):13222. https://doi.org/10.3390/ijms241713222
Chicago/Turabian StyleSchilcher, Katharina, Rania Dayoub, Marion Kubitza, Jakob Riepl, Kathrin Klein, Christa Buechler, Michael Melter, and Thomas S. Weiss. 2023. "Saturated Fat-Mediated Upregulation of IL-32 and CCL20 in Hepatocytes Contributes to Higher Expression of These Fibrosis-Driving Molecules in MASLD" International Journal of Molecular Sciences 24, no. 17: 13222. https://doi.org/10.3390/ijms241713222
APA StyleSchilcher, K., Dayoub, R., Kubitza, M., Riepl, J., Klein, K., Buechler, C., Melter, M., & Weiss, T. S. (2023). Saturated Fat-Mediated Upregulation of IL-32 and CCL20 in Hepatocytes Contributes to Higher Expression of These Fibrosis-Driving Molecules in MASLD. International Journal of Molecular Sciences, 24(17), 13222. https://doi.org/10.3390/ijms241713222