
Structural Insights into Neonicotinoids and N-Unsubstituted Metabolites on Human nAChRs by Molecular Docking, Dynamics Simulations, and Calcium Imaging
 , , and
, , and
Abstract
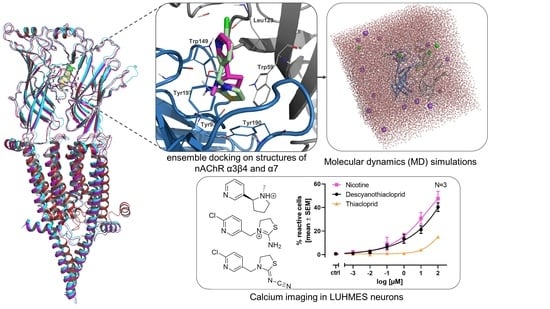
:
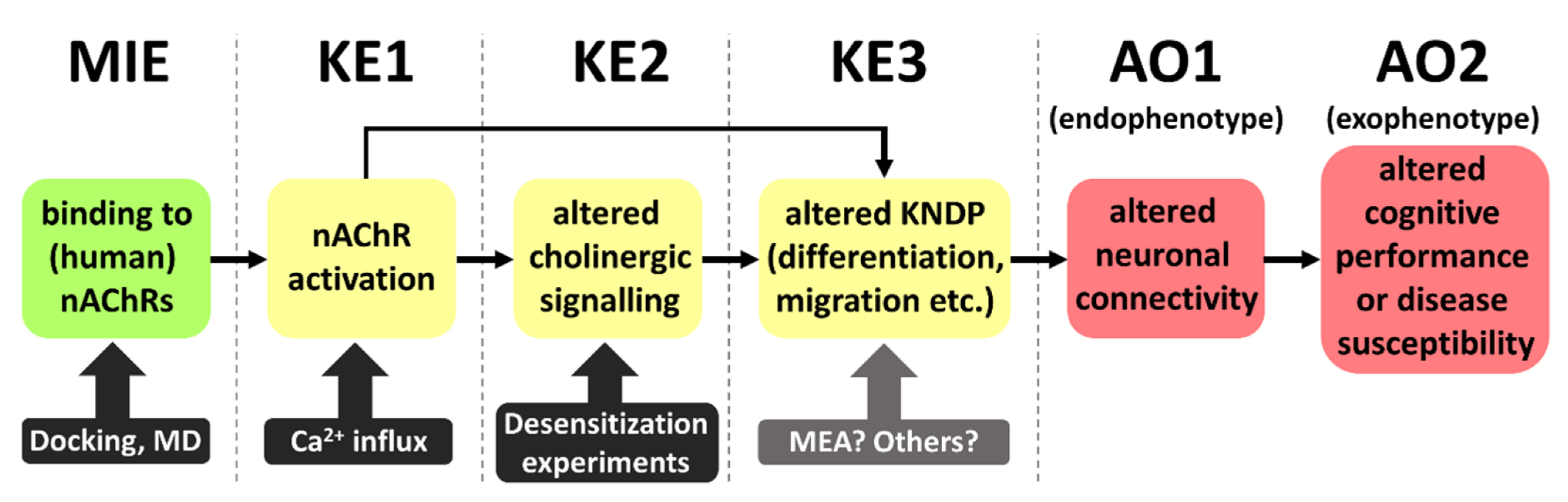
1. Introduction
2. Results and Discussion
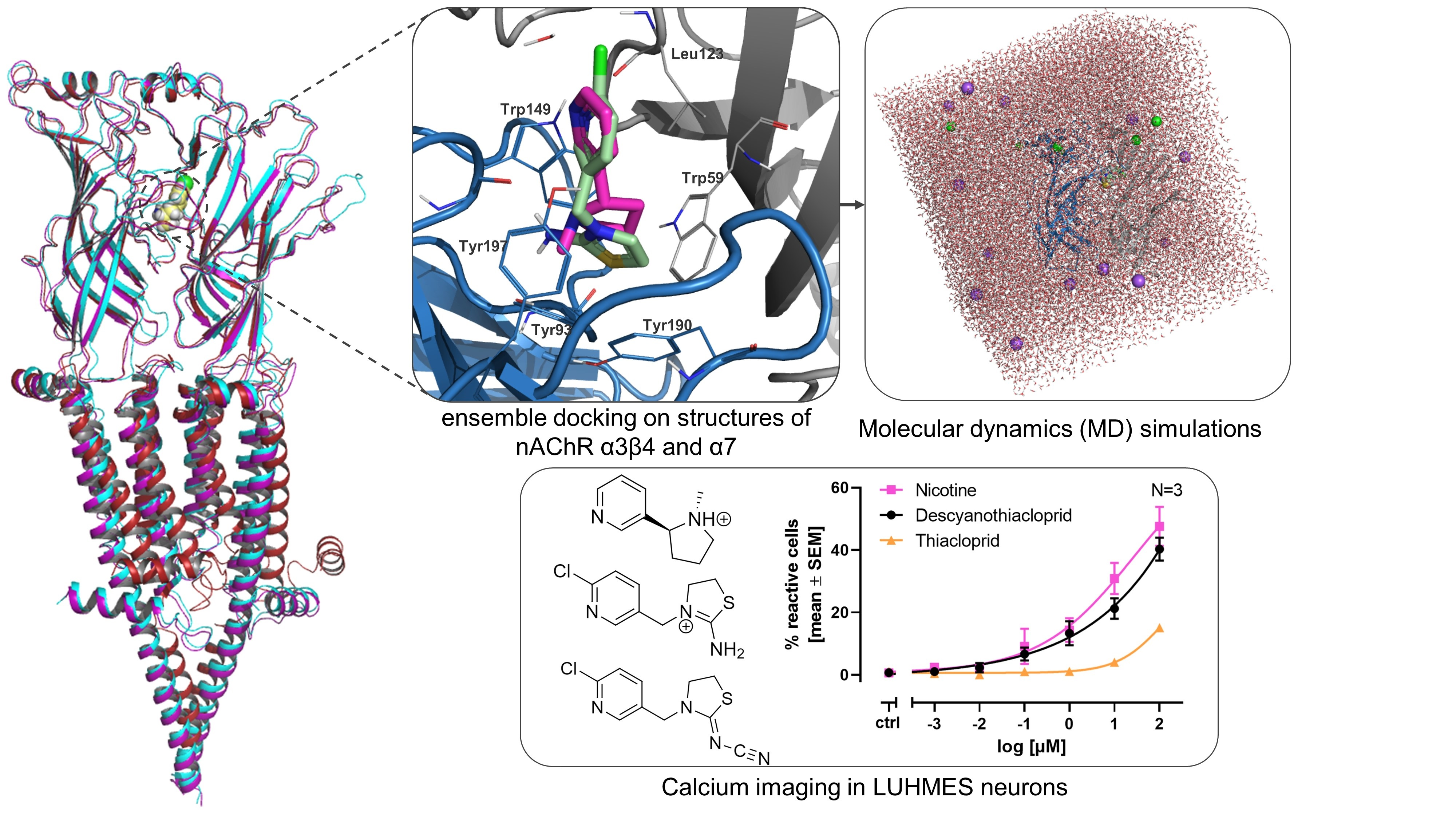
2.1. Ensemble Docking Analysis
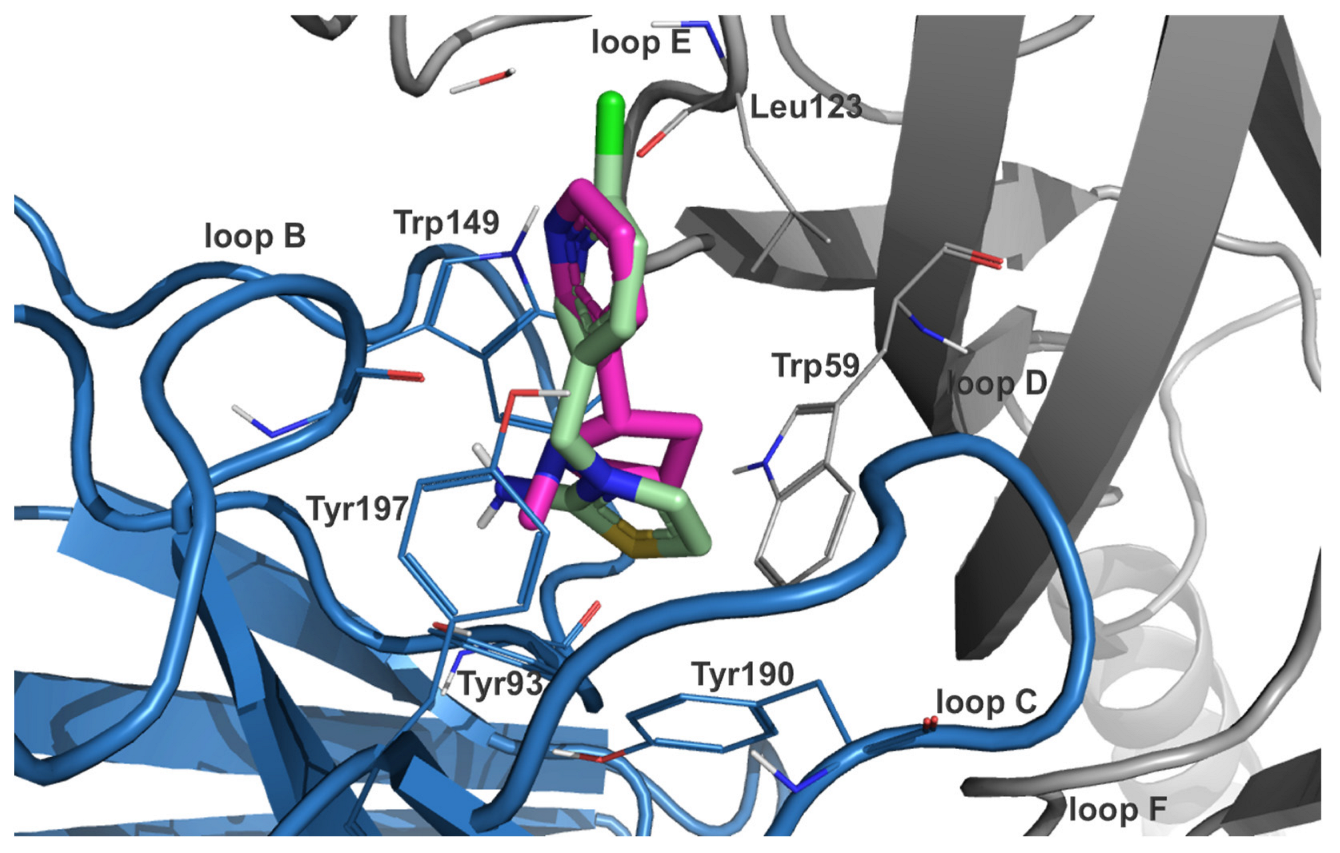
2.2. Ensemble Docking and Representative Binding Poses in nAChRs α7 and α3β4
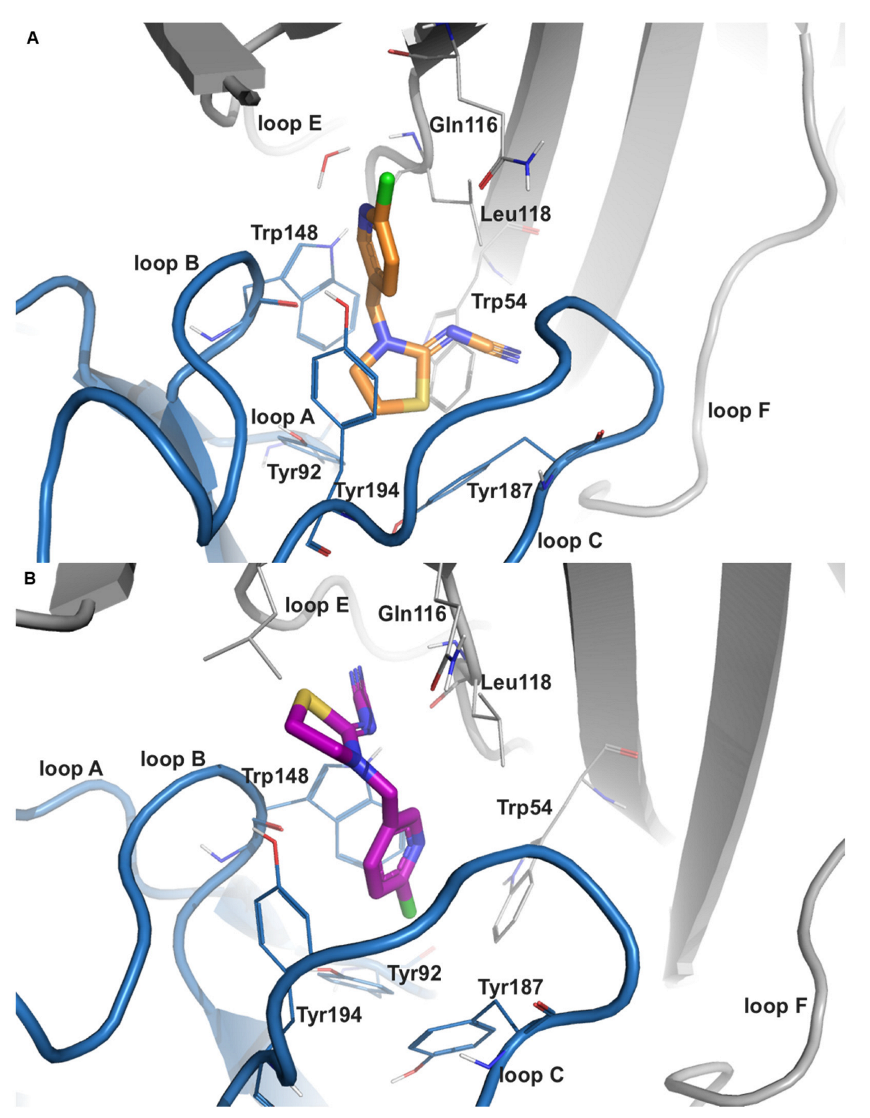
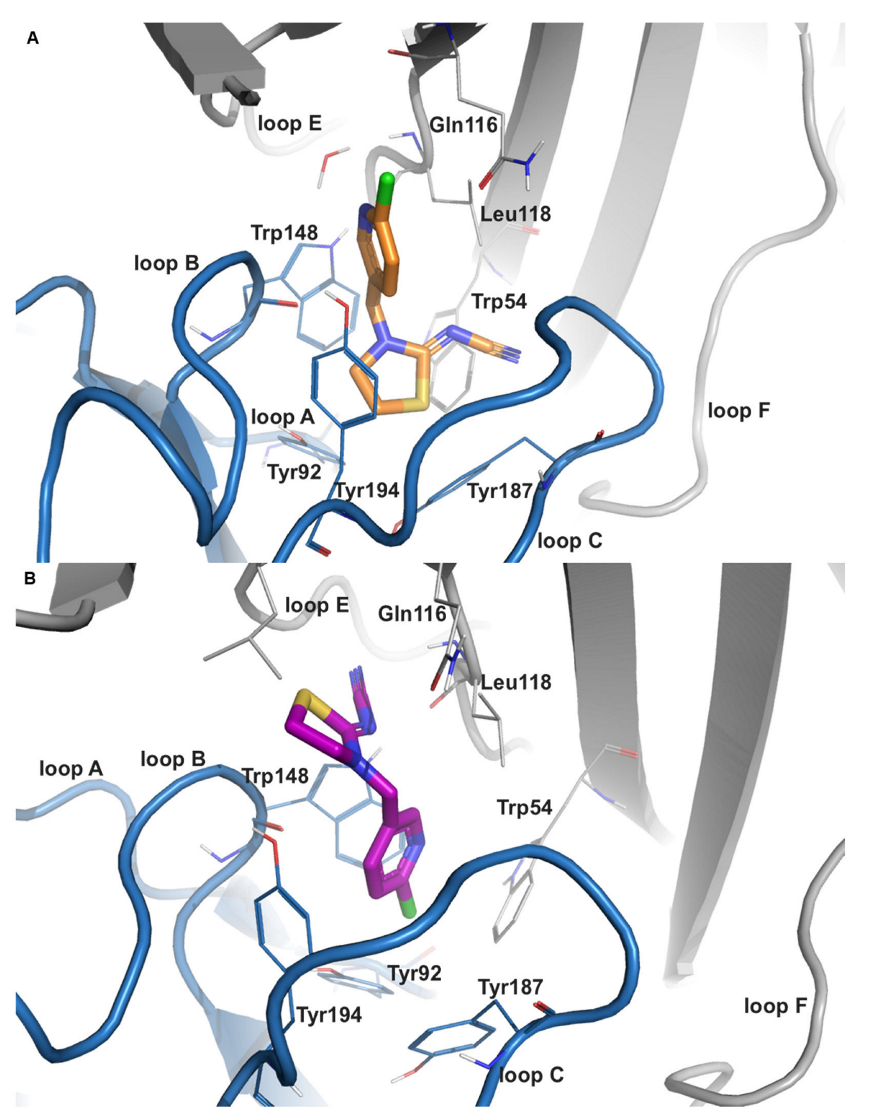
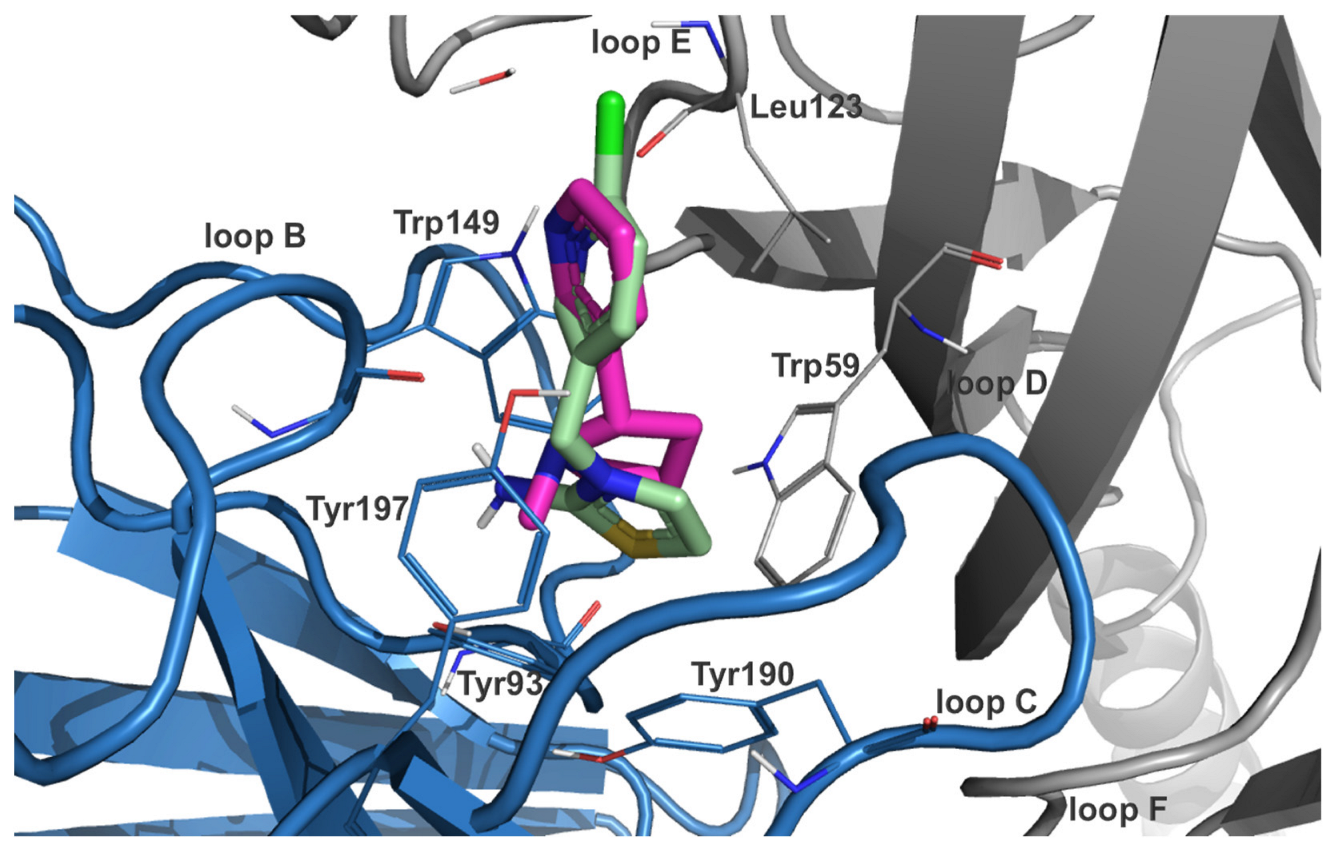
2.2.1. Imidacloprid (IMI)
2.2.2. Desnitro-Imidacloprid (DNIMI)
2.2.3. Thiacloprid (THIAC)
2.2.4. Descyano-Thiacloprid (DCNT) and Descyano-Thiacloprid-Olefin (DCNTO)
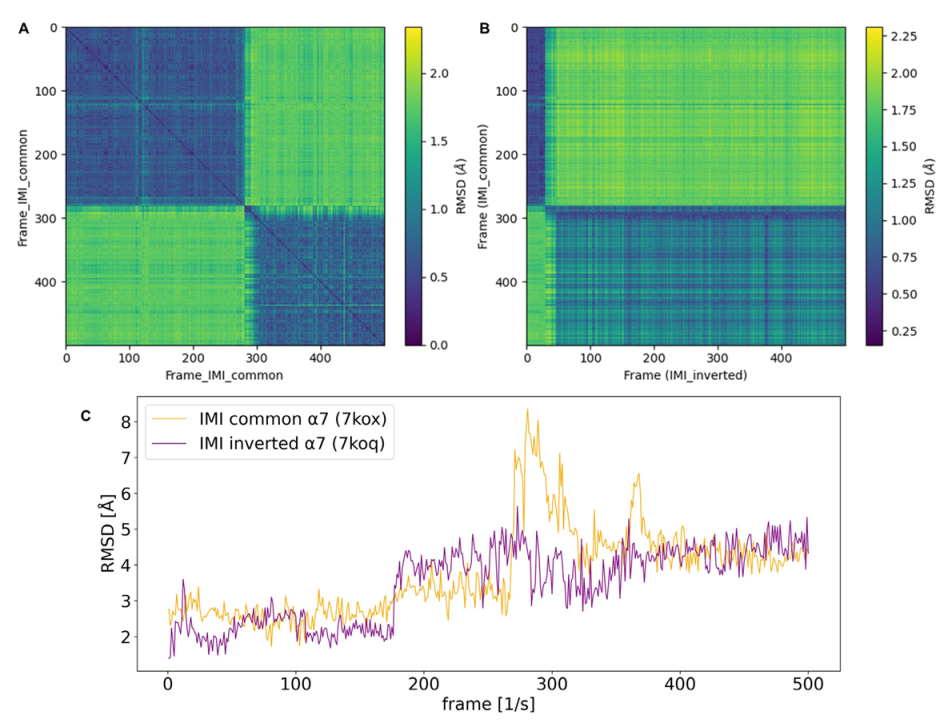
2.3. Molecular Dynamics (MD) Simulation Analysis
RMSD Calculations
2.4. nAChR Subtype Selectivity
2.5. MM-GBSA Binding Free Energy Calculations
2.5.1. Postprocessing of Ensemble Docking Approach
2.5.2. Postprocessing of MD Simulations
2.5.3. Quantification of Uncertainty for Binding Energy Predictions
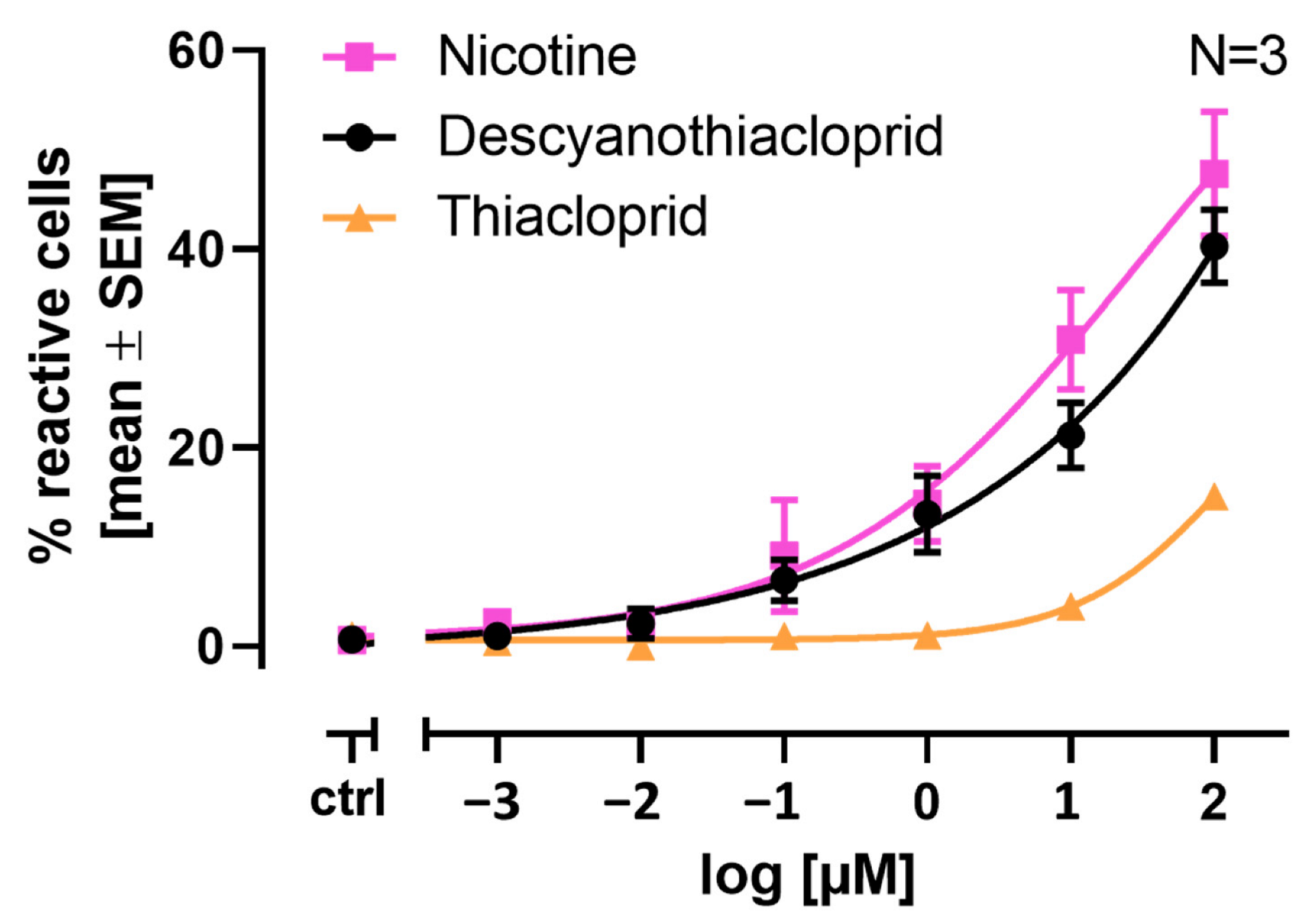
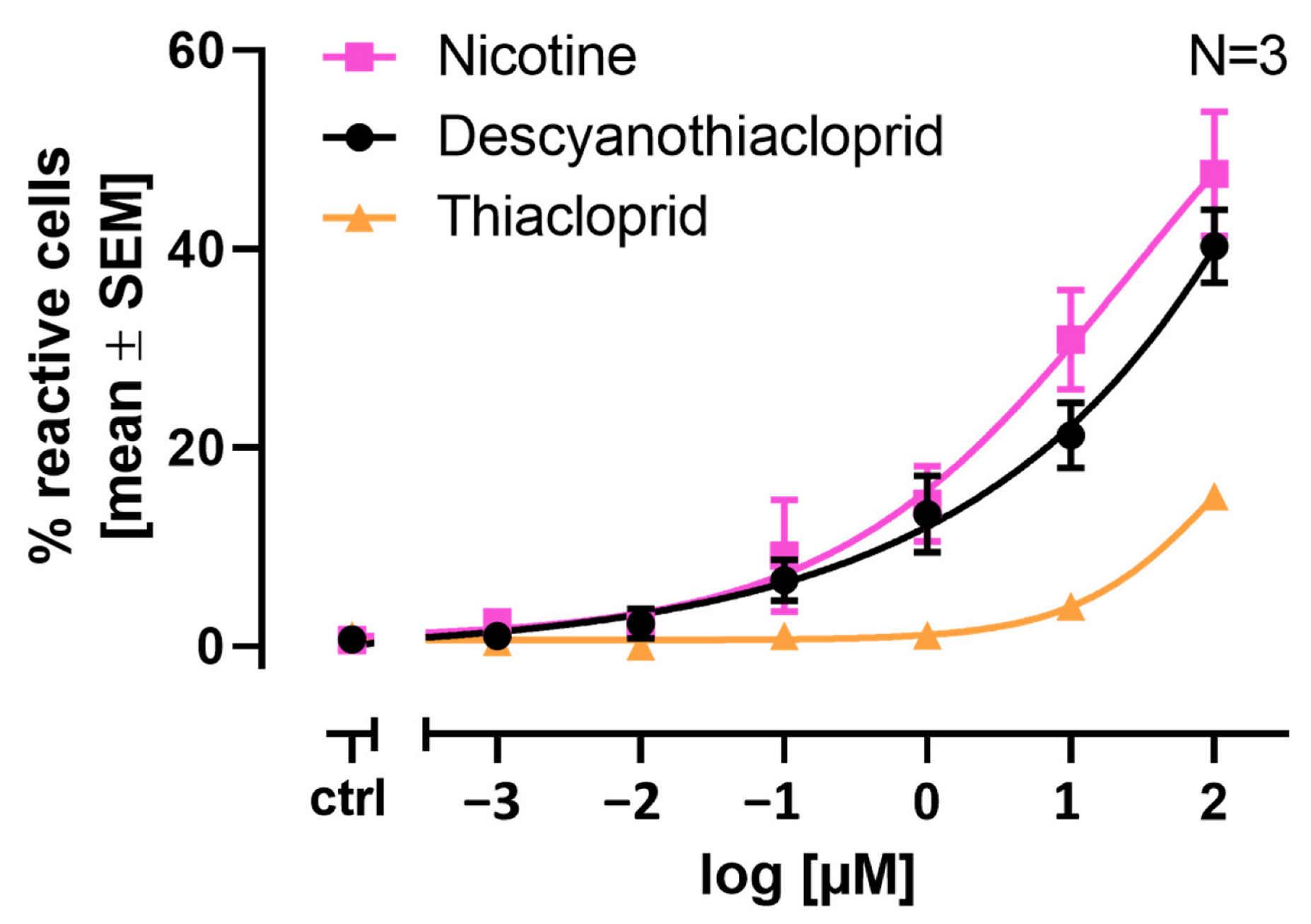
2.6. Single Cell Calcium Measurements
3. Materials and Methods
3.1. Molecular Docking Studies
3.2. Binding Free Energy Calculations of Docking Results
3.3. Molecular Dynamics (MD) Simulation
Postprocessing Analysis of MD Simulations
3.4. In Vitro Approach
3.4.1. LUHMES Cell Culture
3.4.2. LUHMES Ca2+ Imaging
3.4.3. Calcium Fluorescent Flash Evaluating Engine (CaFFEE)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gadaleta, D.; Spînu, N.; Roncaglioni, A.; Cronin, M.T.D.; Benfenati, E. Prediction of the Neurotoxic Potential of Chemicals Based on Modelling of Molecular Initiating Events Upstream of the Adverse Outcome Pathways of (Developmental) Neurotoxicity. Int. J. Mol. Sci. 2022, 23, 3053. [Google Scholar] [CrossRef] [PubMed]
- Spînu, N.; Cronin, M.T.D.; Lao, J.; Bal-Price, A.; Campia, I.; Enoch, S.J.; Madden, J.C.; Mora Lagares, L.; Novič, M.; Pamies, D.; et al. Probabilistic Modelling of Developmental Neurotoxicity Based on a Simplified Adverse Outcome Pathway Network. Comput. Toxicol. 2022, 21, 100206. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Klambauer, G.; Clevert, D.-A.; Shah, I.; Benfenati, E. Artificial Intelligence Meets Toxicology. Chem. Res. Toxicol. 2022, 35, 1289–1290. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural Effects of Developmental Toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Kimura-Kuroda, J.; Komuta, Y.; Kuroda, Y.; Hayashi, M.; Kawano, H. Nicotine-Like Effects of the Neonicotinoid Insecticides Acetamiprid and Imidacloprid on Cerebellar Neurons from Neonatal Rats. PLoS ONE 2012, 7, e32432. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Lein, P.J.; Keil, K.P.; Sethi, S.; Shafer, T.; Barenys, M.; Fritsche, E.; Sachana, M.; Meek, M.E.B. Developing and Applying the Adverse Outcome Pathway Concept for Understanding and Predicting Neurotoxicity. Neurotoxicology 2017, 59, 240–255. [Google Scholar] [CrossRef]
- Blum, J.; Masjosthusmann, S.; Bartmann, K.; Bendt, F.; Dolde, X.; Dönmez, A.; Förster, N.; Holzer, A.-K.; Hübenthal, U.; Keßel, H.E.; et al. Establishment of a Human Cell-Based in Vitro Battery to Assess Developmental Neurotoxicity Hazard of Chemicals. Chemosphere 2022, 311, 137035. [Google Scholar] [CrossRef]
- Spinu, N.; Bal-Price, A.; Cronin, M.T.D.; Enoch, S.J.; Madden, J.C.; Worth, A.P. Development and Analysis of an Adverse Outcome Pathway Network for Human Neurotoxicity. Arch. Toxicol. 2019, 93, 2759–2772. [Google Scholar] [CrossRef] [PubMed]
- Crofton, K.M.; Mundy, W.R. External Scientific Report on the Interpretation of Data from the Developmental Neurotoxicity In Vitro Testing Assays for Use in Integrated Approaches for Testing and Assessment. EFSA Support. Publ. 2021, 18, 6924E. [Google Scholar] [CrossRef]
- Janowska-Sejda, E.I.; Adeleye, Y.; Currie, R.A. Exploration of the DARTable Genome- a Resource Enabling Data-Driven NAMs for Developmental and Reproductive Toxicity Prediction. Front. Toxicol. 2022, 3, 806311. [Google Scholar] [CrossRef]
- Noviello, C.M.; Gharpure, A.; Mukhtasimova, N.; Cabuco, R.; Baxter, L.; Borek, D.; Sine, S.M.; Hibbs, R.E. Structure and Gating Mechanism of the A7 Nicotinic Acetylcholine Receptor. Cell 2021, 184, 2121–2134.e13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, W.; Zhang, L.; Cao, L.; Ling, J.; Liao, K.; Shen, G.; Du, W.; Chen, K.; Zhao, M.; et al. First Evidence of Neonicotinoid Insecticides in Human Bile and Associated Hepatotoxicity Risk. J. Hazard. Mater. 2023, 446, 130715. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Tian, Y.; Shen, X. Human Exposure to Neonicotinoid Insecticides and the Evaluation of Their Potential Toxicity: An Overview. Chemosphere 2018, 192, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Mahai, G.; Wan, Y.; Yang, Z.; He, Z.; Xu, S.; Xia, W. Assessment of Imidacloprid Related Exposure Using Imidacloprid-Olefin and Desnitro-Imidacloprid: Neonicotinoid Insecticides in Human Urine in Wuhan, China. Environ. Int. 2020, 141, 105785. [Google Scholar] [CrossRef] [PubMed]
- Sheets, L.P.; Li, A.A.; Minnema, D.J.; Collier, R.H.; Creek, M.R.; Peffer, R.C. A Critical Review of Neonicotinoid Insecticides for Developmental Neurotoxicity. Crit. Rev. Toxicol. 2016, 46, 153–190. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Lehmler, H.-J.; Kolpin, D.W.; Hladik, M.L.; Vargo, J.D.; Schilling, K.E.; LeFevre, G.H.; Peeples, T.L.; Poch, M.C.; LaDuca, L.E.; et al. A Critical Review on the Potential Impacts of Neonicotinoid Insecticide Use: Current Knowledge of Environmental Fate, Toxicity, and Implications for Human Health. Environ. Sci. Process. Impacts 2020, 22, 1315–1346. [Google Scholar] [CrossRef]
- Loser, D.; Grillberger, K.; Hinojosa, M.G.; Blum, J.; Haufe, Y.; Danker, T.; Johansson, Y.; Möller, C.; Nicke, A.; Bennekou, S.H.; et al. Acute Effects of the Imidacloprid Metabolite Desnitro-Imidacloprid on Human NACh Receptors Relevant for Neuronal Signaling. Arch. Toxicol. 2021, 95, 3695–3716. [Google Scholar] [CrossRef]
- Loser, D.; Hinojosa, M.G.; Blum, J.; Schaefer, J.; Brüll, M.; Johansson, Y.; Suciu, I.; Grillberger, K.; Danker, T.; Möller, C.; et al. Functional Alterations by a Subgroup of Neonicotinoid Pesticides in Human Dopaminergic Neurons. Arch. Toxicol. 2021, 95, 2081–2107. [Google Scholar] [CrossRef]
- Integrated Approaches to Testing and Assessment (IATA)-OECD. Available online: https://www.oecd.org/chemicalsafety/risk-assessment/iata/ (accessed on 6 June 2023).
- Dahlin, D.C.; Miwa, G.T.; Lu, A.Y.; Nelson, S.D. N-Acetyl-p-Benzoquinone Imine: A Cytochrome P-450-Mediated Oxidation Product of Acetaminophen. Proc. Natl. Acad. Sci. USA 1984, 81, 1327–1331. [Google Scholar] [CrossRef]
- Svennebring, A. The Role of Intramolecular Self-Destruction of Reactive Metabolic Intermediates in Determining Toxicity. J. Appl. Toxicol. 2016, 36, 483–500. [Google Scholar] [CrossRef]
- Williams, D.P.; Park, B.K. Idiosyncratic Toxicity: The Role of Toxicophores and Bioactivation. Drug Discov. Today 2003, 8, 1044–1050. [Google Scholar] [CrossRef]
- AOP-Wiki AOP 12. Available online: https://aopwiki.org/aops/12 (accessed on 20 June 2023).
- AOP-Wiki AOP 13. Available online: https://aopwiki.org/aops/13 (accessed on 20 June 2023).
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A Practical Guide to Large-Scale Docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Tomizawa, M.; Casida, J.E. Minor Structural Changes in Nicotinoid Insecticides Confer Differential Subtype Selectivity for Mammalian Nicotinic Acetylcholine Receptors. Br. J. Pharmacol. 1999, 127, 115–122. [Google Scholar] [CrossRef]
- Tomizawa, M.; Lee, D.L.; Casida, J.E. Neonicotinoid Insecticides: Molecular Features Conferring Selectivity for Insect versus Mammalian Nicotinic Receptors. J. Agric. Food Chem. 2000, 48, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 2.5; Schrödinger, LLC.: New York, NY, USA, 2021. [Google Scholar]
- Taly, A.; Colas, C.; Malliavin, T.; Blondel, A.; Nilges, M.; Corringer, P.-J.; Joseph, D. Discrimination of Agonists versus Antagonists of Nicotinic Ligands Based on Docking onto AChBP Structures. J. Mol. Graph. Model. 2011, 30, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Okajima, T.; Yamashita, A.; Oda, T.; Hirata, K.; Nishiwaki, H.; Morimoto, T.; Akamatsu, M.; Ashikawa, Y.; Kuroda, S.; et al. Crystal Structures of Lymnaea Stagnalis AChBP in Complex with Neonicotinoid Insecticides Imidacloprid and Clothianidin. Invert. Neurosci. 2008, 8, 71–81. [Google Scholar] [CrossRef]
- Tomizawa, M.; Maltby, D.; Talley, T.T.; Durkin, K.A.; Medzihradszky, K.F.; Burlingame, A.L.; Taylor, P.; Casida, J.E. Atypical Nicotinic Agonist Bound Conformations Conferring Subtype Selectivity. Proc. Natl. Acad. Sci. USA 2008, 105, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Xiu, X.; Puskar, N.L.; Shanata, J.A.P.; Lester, H.A.; Dougherty, D.A. Nicotine Binding to Brain Receptors Requires a Strong Cation–π Interaction. Nature 2009, 458, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.I.; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, A.; Teng, J.; Zhuang, Y.; Noviello, C.M.; Walsh, R.M.; Cabuco, R.; Howard, R.J.; Zaveri, N.T.; Lindahl, E.; Hibbs, R.E. Agonist Selectivity and Ion Permeation in the A3β4 Ganglionic Nicotinic Receptor. Neuron 2019, 104, 501–511.e6. [Google Scholar] [CrossRef] [PubMed]
- Grutter, T.; De Carvalho, L.P.; Le Novère, N.; Corringer, P.J.; Edelstein, S.; Changeux, J.-P. An H-Bond between Two Residues from Different Loops of the Acetylcholine Binding Site Contributes to the Activation Mechanism of Nicotinic Receptors. EMBO J. 2003, 22, 1990–2003. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Bargen, C.D.V.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, Z.; Wang, L.; Tian, S.; Hou, T.; Sun, H. Predicting the Mutation Effects of Protein–Ligand Interactions via End-Point Binding Free Energy Calculations: Strategies and Analyses. J. Cheminform. 2022, 14, 56. [Google Scholar] [CrossRef]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The Normal-Mode Entropy in the MM/GBSA Method: Effect of System Truncation, Buffer Region, and Dielectric Constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef]
- Grossfield, A.; Patrone, P.N.; Roe, D.R.; Schultz, A.J.; Siderius, D.W.; Zuckerman, D.M. Best Practices for Quantification of Uncertainty and Sampling Quality in Molecular Simulations [Article v1.0]. Living J. Comput. Mol. Sci. 2018, 1, 5067. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PKaprediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, E.S.D.; Anderson, A.C. In Pursuit of Virtual Lead Optimization: Pruning Ensembles of Receptor Structures for Increased Efficiency and Accuracy during Docking. Proteins Struct. Funct. Bioinform. 2009, 75, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 Model: A next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz Jr., K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cisneros, G.A.; Cruzeiro, V.D.W.; et al. Amber2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Brünger, A.T. X-PLOR: Version 3.1: A System for X-Ray Crystallography and NMR; Yale University Press: London, UK, 1993. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 56–61. [Google Scholar]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E.I. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Pöltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, Complete and Large-Scale Generation of Post-Mitotic Neurons from the Human LUHMES Cell Line. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Karreman, C.; Klima, S.; Holzer, A.-K.; Leist, M. CaFFEE: A Program for Evaluating Time Courses of Ca2+ Dependent Signal Changes of Complex Cells Loaded with Fluorescent Indicator Dyes. ALTEX-Altern. Anim. Exp. 2020, 37, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Bolz, S.N.; Al-Fatlawi, A.; Schroeder, M. Decomposing Compounds Enables Reconstruction of Interaction Fingerprints for Structure-Based Drug Screening. J. Cheminform. 2022, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Ozekin, Y.H.; Saal, M.L.; Pineda, R.H.; Moehn, K.; Ordonez-Erives, M.A.; Delgado Figueroa, M.F.; Frazier, C.; Korth, K.M.; Königshoff, M.; Bates, E.A.; et al. Intrauterine Exposure to Nicotine through Maternal Vaping Disrupts Embryonic Lung and Skeletal Development via the Kcnj2 Potassium Channel. Dev. Biol. 2023, 501, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.B.; McQuown, S.C.; Leslie, F.M. The Dynamic Effects of Nicotine on the Developing Brain. Pharmacol. Ther. 2009, 122, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Hsieh, L.S.; Lee, A.M.; Zhou, Z.; Coman, D.; Heath, C.J.; Hyder, F.; Mineur, Y.S.; Yuan, Q.; Goldman, D.; et al. An Epigenetic Mechanism Mediates Developmental Nicotine Effects on Neuronal Structure and Behavior. Nat. Neurosci. 2016, 19, 905–914. [Google Scholar] [CrossRef]
- Levin, E.D.; Briggs, S.J.; Christopher, N.C.; Rose, J.E. Prenatal Nicotine Exposure and Cognitive Performance in Rats. Neurotoxicol. Teratol. 1993, 15, 251–260. [Google Scholar] [CrossRef]
- Muhammad, A.; Mychasiuk, R.; Nakahashi, A.; Hossain, S.R.; Gibb, R.; Kolb, B. Prenatal Nicotine Exposure Alters Neuroanatomical Organization of the Developing Brain. Synapse 2012, 66, 950–954. [Google Scholar] [CrossRef]
- Slikker, W.; Xu, Z.A.; Levin, E.D.; Slotkin, T.A. Mode of Action: Disruption of Brain Cell Replication, Second Messenger, and Neurotransmitter Systems During Development Leading to Cognitive Dysfunction—Developmental Neurotoxicity of Nicotine. Crit. Rev. Toxicol. 2005, 35, 703–711. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Tate, C.A.; Cousins, M.M.; Seidler, F.J. Prenatal Nicotine Exposure Alters the Responses to Subsequent Nicotine Administration and Withdrawal in Adolescence: Serotonin Receptors and Cell Signaling. Neuropsychopharmacology 2006, 31, 2462–2475. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Ceccatelli, S.; Daneshian, M.; Fritsche, E.; Hasiwa, N.; Hartung, T.; Hogberg, H.T.; Leist, M.; Li, A.; Mundy, W.R.; et al. Reference Compounds for Alternative Test Methods to Indicate Developmental Neurotoxicity (DNT) Potential of Chemicals: Example Lists and Criteria for Their Selection and Use. ALTEX-Altern. Anim. Exp. 2017, 34, 49–74. [Google Scholar] [CrossRef]
- Smirnova, L.; Hogberg, H.T.; Leist, M.; Hartung, T. Developmental Neurotoxicity—Challenges in the 21st Century and in Vitro Opportunities. ALTEX 2014, 31, 129–156. [Google Scholar] [CrossRef]
- Bal-Price, A.; Crofton, K.M.; Leist, M.; Allen, S.; Arand, M.; Buetler, T.; Delrue, N.; FitzGerald, R.E.; Hartung, T.; Heinonen, T.; et al. International STakeholder NETwork (ISTNET): Creating a Developmental Neurotoxicity (DNT) Testing Road Map for Regulatory Purposes. Arch. Toxicol. 2015, 89, 269–287. [Google Scholar] [CrossRef] [PubMed]
- AOP-Wiki AOP 3. Available online: https://aopwiki.org/aops/3 (accessed on 14 July 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α7 | α3β4 | |||||||
|---|---|---|---|---|---|---|---|---|
| PDB-ID-water set | 7kox-ws1 | 7kox-ws2 | 7koq | 7koo | 6pv7-ws1 | 6pv7-ws2 | 6pv8-ws1 | 6pv8-ws2 |
| Docking score | 0.75 | 0.7 | 0.64 | 0.0089 | 0.75 | 0.66 | 0.81 | 0.74 |
| dG bind | 0.57 | 0.54 | 0.6 | 0.3 | 0.53 | 0.38 | 0.43 | 0.45 |
| Compound | PDB-ID | Mean RMSD | Average Mean RMSD | |
| α7 | IMI | 7kox-ws2 | 3.78 | 2.96 |
| 7koq | 2.14 | |||
| DNIMI | 7kox-ws1 | 2.34 | 2.03 | |
| 7koq | 1.73 | |||
| THIAC | 7kox-ws1 | 3.87 | 3.15 | |
| 7koq | 2.43 | |||
| DCNT | 7kox-ws1 | 1.91 | 1.60 | |
| 7koq | 1.29 | |||
| DCNTO | 7kox-ws1 | 1.73 | 1.78 | |
| 7koq | 1.83 | |||
| NIC | 7kox-ws2 | 1.86 | 1.82 | |
| 7koq | 1.78 | |||
| α3β4 | IMI | 6pv7-ws2 | 3.47 | 4.15 |
| 6pv8-ws2 | 4.82 | |||
| DNIMI | 6pv7-ws2 | 1.87 | 2.17 | |
| 6pv8-ws1 | 2.46 | |||
| THIAC | 6pv7-ws1 | 2.53 | 2.78 | |
| 6pv8-ws2 | 3.03 | |||
| DCNT | 6pv7-ws2 | 1.73 | 1.82 | |
| 6pv8-ws1 | 1.91 | |||
| DCNTO | 6pv7-ws2 | 1.37 | 1.38 | |
| 6pv8-ws1 | 1.38 | |||
| NIC | 6pv7-ws2 | 1.43 | 1.61 | |
| 6pv8-ws2 | 1.78 |
| Ligand | PDB-ID | dG Binding Energy | vdW Energy | Eel Energy | dG + Entropy | Total Delta S Entropy | |
| α7 | IMI | 7kox-ws2 | −27.08 | −35.13 | −10.15 | 8.43 | −35.51 |
| 7koq | −36.44 | −42.91 | −10.04 | −1.13 | −35.31 | ||
| DNIMI | 7kox-ws1 | −36.31 | −35.66 | −163.17 | −3.34 | −32.96 | |
| 7koq | −36.31 | −36.10 | −169.96 | −3.61 | −32.70 | ||
| THIAC | 7kox-ws1 | −33.77 | −42.55 | −1.12 | −0.83 | −32.94 | |
| 7koq | −33.50 | −39.74 | −12.52 | 0.12 | −33.63 | ||
| DCNT | 7kox-ws1 | −39.72 | −39.44 | −163.88 | −7.15 | −32.57 | |
| 7koq | −37.62 | −37.05 | −156.58 | −4.81 | −32.81 | ||
| DCNTO | 7kox-ws1 | −35.33 | −35.57 | −155.57 | −4.77 | −30.56 | |
| 7koq | −36.77 | −36.91 | −153.18 | −5.14 | −31.63 | ||
| NIC | 7kox-ws2 | −33.72 | −31.41 | −143.67 | −3.29 | −30.42 | |
| 7koq | −35.79 | −31.13 | −165.53 | −6.32 | −29.47 | ||
| α3β4 | IMI | 6pv7-ws2 | −25.34 | −36.44 | −11.51 | 11.86 | −37.20 |
| 6pv8-ws2 | −24.24 | −35.16 | −13.67 | 13.11 | −37.35 | ||
| DNIMI | 6pv7-ws2 | −44.21 | −38.20 | −147.71 | −13.18 | −31.03 | |
| 6pv8-ws1 | −37.48 | −36.30 | −127.50 | −4.52 | −32.96 | ||
| THIAC | 6pv7-ws1 | −28.50 | −34.97 | −22.51 | 4.73 | −33.23 | |
| 6pv8-ws2 | −32.74 | −38.60 | −22.39 | −0.47 | −32.27 | ||
| DCNT | 6pv7-ws2 | −35.63 | −36.20 | −129.06 | −3.15 | −32.47 | |
| 6pv8-ws1 | −36.38 | −35.38 | −124.08 | −3.84 | −32.54 | ||
| DCNTO | 6pv7-ws2 | −37.23 | −37.45 | −119.56 | −7.83 | −29.40 | |
| 6pv8-ws1 | −37.74 | −36.85 | −135.41 | −6.96 | −30.78 | ||
| NIC | 6pv7-ws2 | −36.62 | −31.06 | −120.78 | −6.58 | −30.04 | |
| 6pv8-ws2 | −33.42 | −28.69 | −120.71 | −4.42 | −30.00 |
| Ligand | Mean MM-GBSA dG | 95% Confidence Interval | Standard Uncertainty | |
| NIC | −34.89 | −37.39 | −32.38 | 0.55 |
| DCNT | −37.34 | −40.18 | −34.49 | 1.110 |
| DCNTO | −36.77 | −38.41 | −35.12 | 1.198 |
| DNIMI | −38.58 | −37.80 | −35.86 | 1.374 |
| IMI | −28.27 | −37.13 | −19.42 | 1.953 |
| THIAC | −32.13 | −35.04 | −29.22 | 1.243 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grillberger, K.; Cöllen, E.; Trivisani, C.I.; Blum, J.; Leist, M.; Ecker, G.F. Structural Insights into Neonicotinoids and N-Unsubstituted Metabolites on Human nAChRs by Molecular Docking, Dynamics Simulations, and Calcium Imaging. Int. J. Mol. Sci. 2023, 24, 13170. https://doi.org/10.3390/ijms241713170
Grillberger K, Cöllen E, Trivisani CI, Blum J, Leist M, Ecker GF. Structural Insights into Neonicotinoids and N-Unsubstituted Metabolites on Human nAChRs by Molecular Docking, Dynamics Simulations, and Calcium Imaging. International Journal of Molecular Sciences. 2023; 24(17):13170. https://doi.org/10.3390/ijms241713170
Chicago/Turabian StyleGrillberger, Karin, Eike Cöllen, Claudia Immacolata Trivisani, Jonathan Blum, Marcel Leist, and Gerhard F. Ecker. 2023. "Structural Insights into Neonicotinoids and N-Unsubstituted Metabolites on Human nAChRs by Molecular Docking, Dynamics Simulations, and Calcium Imaging" International Journal of Molecular Sciences 24, no. 17: 13170. https://doi.org/10.3390/ijms241713170
APA StyleGrillberger, K., Cöllen, E., Trivisani, C. I., Blum, J., Leist, M., & Ecker, G. F. (2023). Structural Insights into Neonicotinoids and N-Unsubstituted Metabolites on Human nAChRs by Molecular Docking, Dynamics Simulations, and Calcium Imaging. International Journal of Molecular Sciences, 24(17), 13170. https://doi.org/10.3390/ijms241713170