
Cardiomyocyte Damage: Ferroptosis Relation to Ischemia-Reperfusion Injury and Future Treatment Options

Abstract
:1. Introduction
2. Iron Metabolism in the Heart
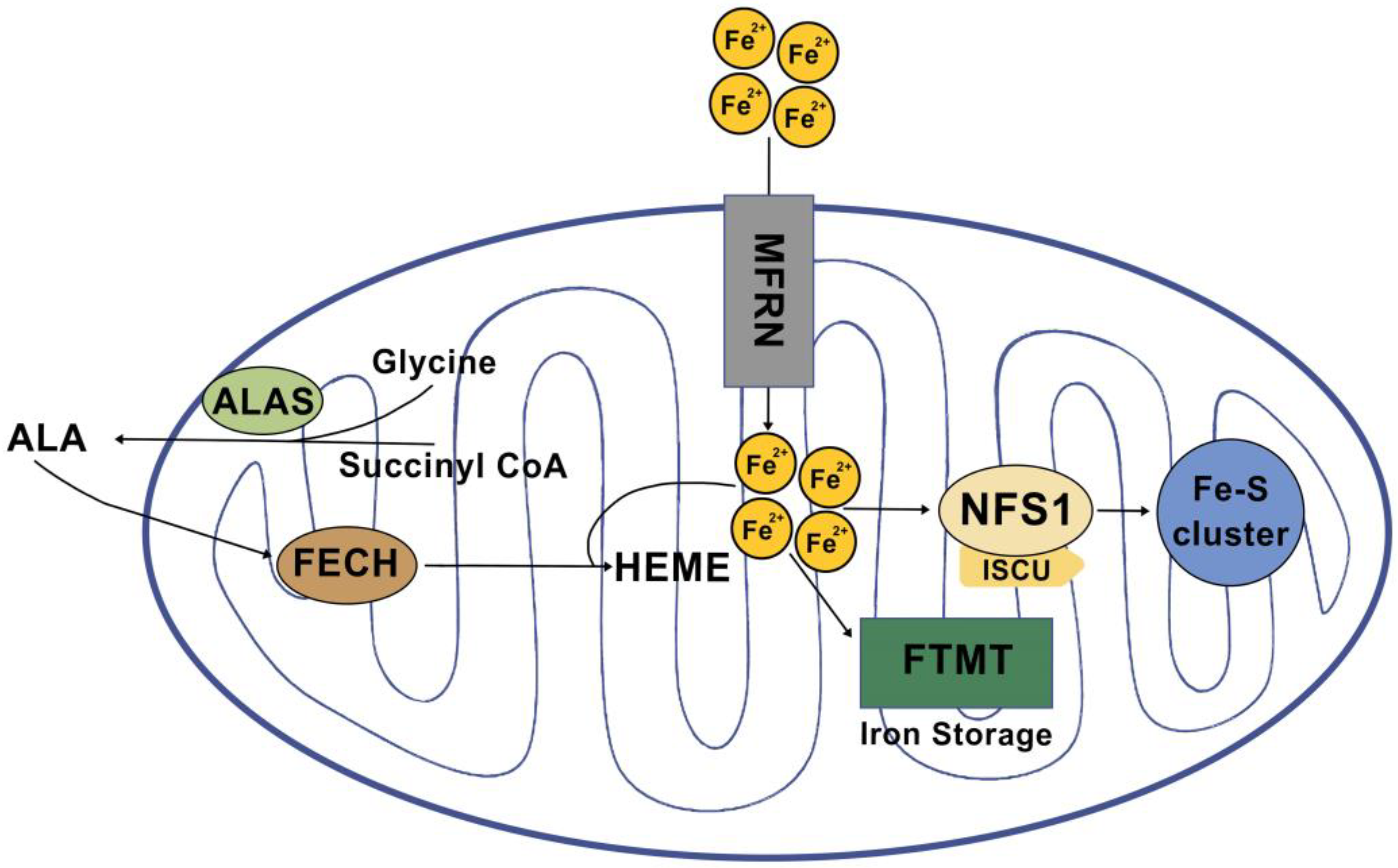
3. Mitochondrial Iron Regulation
3.1. Heme Synthesis
3.2. Fe-S Cluster Biogenesis
3.3. Mitochondrial Iron Storage
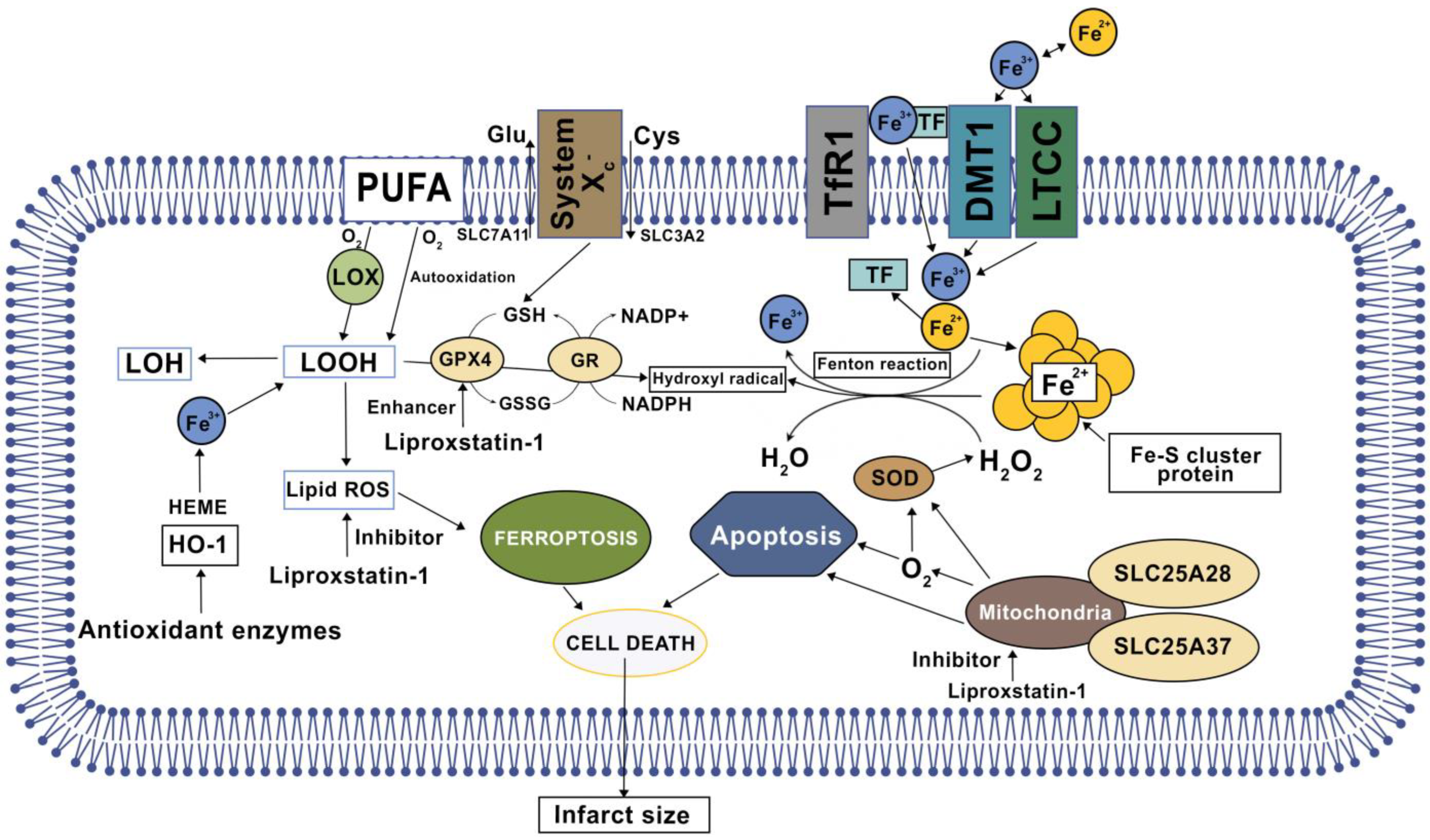
4. Cardiac Ferroptosis
5. Ferroptosis in Medication-Induced Myocardial Injury
6. Ferroptosis Role in Heart Failure
7. Treatment Options

{kind=link}
{kind=link}
| Compound | Source | Testing Model | Mechanism of Action | References |
|---|---|---|---|---|
| N-Acetylcysteine (NAC) | Synthetic antioxidant | RCT | Prodrug to L-cysteine, a precursor to the biologic antioxidant glutathione, an ROS scavenger, indirect action as a metal ion chelator and ability to inhibit NF-_B | [46,47,48,49] |
| Vitamin C (vit C) | Water-soluble vitamin found in citrus and other fruits and vegetables | RCT | Has action as an ROS scavenger | [50,51] |
| Vitamin E (Vit E) | Fat soluble vitamin a group of eight fat-soluble compounds including four tocopherols and four tocotrienols | RCT | Has action as an ROS scavenger | [52,53,54] |
| Deferoxamine (DFO) | Synthetic iron-chelating agent | RCT | Acts by binding free iron in the bloodstream and enhancing its elimination in the urine | [55,56] |
| Liproxstatin-1 (Lip-1) | Synthetic antioxidant | Langendorff model using male C57BL/6J mice hearts (ex vivo) | Radical-trapping antioxidant (RTA) via reduction in voltage-dependent anion channel 1 (VDAC1) | [57,58] |
| Ferrostatin-1 (Fer-1) | Synthetic antioxidant | Cell models (ex vivo) | Radical-trapping antioxidant (RTAs) via GPX4 inhibition, GPX4 deletion, or GSH depletion and ability to inhibit lipid peroxidation directly by trapping chain-carrying radicals | [57,59] |
| Zileuton (ZIL) | Synthetic derivative of hydroxyurea | HepG2 and HL60 cells models (ex vivo) | Leukotriene inhibitor, blocks 5-lipoxygenase and formation of 5-HETE in ACSL4-overexpressed cells | [5,60] |
| Nuclear factor erythroid 2-related factor 2 (NRF2) | Transcription factor in humans encoded by the NFE2L2 gene | HUVECs and EA.hy926 vascular endothelial cell models (ex vivo) | Regulates the expression of antioxidant proteins that protect against oxidative damage | [25,61] |
| Xanthohumol (XH) | A natural product found in the female inflorescences of Humulus lupulus, also known as hops | Neonatal rat cardiomyocytes (NRCMs) model (ex vivo) | Has action as an ROS scavenger, chelate Fe2+, and regulates NRF2 and GPX4 protein levels in cardiomyocytes during Fe-SP and RSL3-induced ferroptosis | [29] |
| Ferulic acid (FA) | A natural organic polyphenol derived from the genus Ferula | Murine MIN6 cells models (ex vivo) | Enhances the activity of antioxidant enzymes (SOD, GSH-Px, and CAT) | [62,63] |
| Resveratrol (Res) | A stilbenoid, a type of natural phenol, and a phytoalexin produced by several plants in response to injury | Male rats (Sprague-Dawley) (in vivo) | Blocks oxidative stress and Fe2+ levels in IR models and regulates USP19-Beclin 1 autophagy | [21,64] |
| Gossypol acetic acid (GAA) | A natural product isolated from cottonseeds and roots | mitochondria from the Mcu−/−, Mcufl/fl–MCM, and DN-Mcu mouse models (ex vivo) | Downregulates PTGS2 and ACSL4 levels in both mRNA and protein | [65,66] |
| Naringenin (NAR) | A natural flavanone compound, widely distributed in several citrus fruits | AKI mouse model (ex vivo) | Downregulates NRF2 | [61] |
| Cyanidin-3-glucoside (C3G) | A natural anthocyanin polyphenol with polyphenolic structure widely occurring in plants | H9c2 cells (ex vivo) | Can relieve oxidative stress, downregulate LC3II/LC3I and TFR1 levels, and upregulate FTH1 and GPX4 expression | [67,68] |
| Histochrome (HC) | Isolated from sea urchin Scaphechinus and standardized echinochrome | AKI mouse model (ex vivo) | Reduces cytosolic and mitochondrial ROS, maintains intracellular GSH levels, and elevates GPX4 activity | [17,69] |
| Dexmedetomidine (Dex) | A synthetic sympatholytic medicine agonist of α2-adrenergic receptors in the brain | EA.hy926 vascular endothelial cells (ex vivo) | Chelates iron and activates NRF2 through the AMPK/GSK-3β pathway | [28,61,65] |
| Dexrazoxane (DXZ), | A derivative of Ethylenediaminetetraacetic acid (EDTA) | (Mlkl−/− and Fadd−/−Mlkl−/−) mice model (in vivo) | Chelates iron and regulates the level of Ptgs2 mRNA | [70] |
| XJB-5-131 | A synthetic antioxidant | HT-1080, BJeLR, and panc-1 cells (ex vivo) | Nitroxide-based lipid peroxidation, ROS scavengers | [71] |
| JP4-039 | A synthetic antioxidant | HT-1080, BJeLR, and panc-1 cells (ex vivo) | Nitroxide-based lipid peroxidation, ROS scavengers | [71] |
8. Antioxidants
8.1. Liproxstatin-1 (Lip-1)
8.2. Ferrostatin-1 (Fer-1)
8.3. Zileuton (ZIL)
8.4. Nuclear Factor Erythroid 2-Related Factor 2 (NRF2)
8.5. Vitamin E
8.6. Vitamin C
9. Natural Herbal Phytochemicals
9.1. Xanthohumol (XH)
9.2. Ferulic Acid (FA)
9.3. Cyanidin-3-Glucoside (C3G)
9.4. Gossypol Acetic Acid (GAA)
9.5. Naringenin (NAR)
10. Iron Chelators
11. ROS Scavengers
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-TOH | α-tocopherol |
| 4HNE | 4-hydroxynonenal |
| 5-FU | Fluorouracil |
| ABCB10 | ATP-binding cassette subfamily B member 10 |
| ABCB7 | ATP-binding cassette transporter of the inner mitochondrial membrane |
| ABCB8 | Mitochondrial potassium channel ATP-binding subunit |
| ACSL4 | Acyl-CoA synthetase long chain family member 4 |
| AIFM2 | Apoptosis-inducing factor mitochondria-associated 2 |
| AMI | Acute myocardial infarction |
| AMP | Adenosine monophosphate |
| AMPK/GSK-3β | AMP-activated protein kinase/Glycogen synthase kinase-3beta |
| AMPKα2 | AMP-activated protein kinase, alpha 2 catalytic subunit |
| ALAS | 5-aminolevulinic acid synthase |
| ATP | Adenosine triphosphate |
| C3G | Cyanidin-3-glucoside |
| CK | Creatine kinase |
| DAMP | damage-associated molecular patterns |
| Dex | Dexmedetomidine |
| DMT1 | Divalent metal transporter 1 |
| DOX | Doxorubicin |
| ESCRT-III | Endosomal sorting complex required for transport-III |
| FA | Ferulic acid |
| Fe2+ | Ferrous iron |
| Fe3+ | Ferric iron |
| Fer-1 | Ferrostatin-1 |
| Fe-S | Iron–sulfur |
| FLVCR1B | Feline leukemia virus subgroup C receptor-related protein 1B |
| FPN | Ferroportin |
| FTH | Heavy chain of ferritin |
| FTMT | Mitochondria-specific ferritin |
| GAA | Gossypol acetic acid |
| GPX4 | Glutathione peroxidase 4 |
| GSH | Glutathione |
| HC | Histochrome |
| IRI | Ischemia-reperfusion injury |
| IRP-IRE | Iron regulatory protein (IRP)-iron responsive element |
| IscU | Scaffold protein |
| LC3I | Cystolic form of microtubule-associated protein light chain 3 that is soluble |
| LC3II | Membrande bound microtubule-associated protein light chain 3 |
| Lip-1 | Liproxstatin-1 |
| L-OOHs | Lipid hydroperoxides |
| LVEF | Left ventricular ejection fraction |
| MDA | Malondialdehyde |
| mPTP | Mitochondrial permeability transition pore |
| mRNA | Messenger ribonucleic acid |
| NAR | Naringenin |
| NCOA4 | Nuclear receptor coactivator 4 |
| NFS1 | Cysteine desulfurase |
| NRAMP2 | Natural resistance-associated macrophage protein 2 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NRF2/HO-1 | Nuclear factor erythroid 2-related factor 2/Heme oxygenase 1 |
| OGD/R | Oxygen–glucose deprivation/reoxygenation |
| PPIX | Protoporphyrin IX |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| PUFA | Polyunsaturated fatty acids |
| Res | Resveratrol |
| ROS | Reactive oxygen species |
| RSL3 | RAS selective lethal 3 |
| RTA | Radical trapping antioxidants |
| SLC25A28 | Mitoferrin-2 |
| SLC25A37 | Mitoferrin-1 |
| SLC3A2 | Solute carrier family 3 member 2 |
| SLC7A11/xCT | Heterodimeric transmembrane complex composed of light chain, solute carrier family 7 member 11 |
| STEAP | Six-transmembrane epithelial antigen of prostate |
| TF | Transferrin |
| TFR1 | Transferrin receptor protein 1 |
| USP19 | Ubiquitin specific peptidase 19 |
| 5′ UTR | 5′ untranslated region |
| VDAC1 | Voltage-dependent anion channel 1 |
| VDAC2/3 | Voltage-dependent anion channel’s 2 and 3 |
| XH | Xanthohumol |
| ZIL | Zileuton |
References
- Bulluck, H.; Yellon, D.M.; Hausenloy, D.J. Reducing myocardial infarct size: Challenges and future opportunities. Heart 2015, 102, 341–348. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Lillo-Moya, J.; Rojas-Solé, C.; Muñoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants 2021, 10, 667. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Yan, H.-F.; Tuo, Q.-Z.; Yin, Q.-Z.; Lei, P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool. Res. 2020, 41, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Donegan, R.K.; Moore, C.; Hanna, D.; Reddi, A.R. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 2018, 133, 88–100. [Google Scholar] [CrossRef]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial iron metabolism and its role in diseases. Clin. Chim. Acta 2020, 513, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef]
- He, J.; Liu, Q.; Wang, J.; Xu, F.; Fan, Y.; He, R.; Yan, R.; Zhu, L. Identification of the metabolic remodeling profile in the early-stage of myocardial ischemia and the contributory role of mitochondrion. Bioengineered 2022, 13, 11106–11121. [Google Scholar] [CrossRef]
- Murphy, E.; Liu, J.C. Mitochondrial calcium and reactive oxygen species in cardiovascular disease. Cardiovasc. Res. 2022, 119, 1105–1116. [Google Scholar] [CrossRef]
- Corsi, B.; Cozzi, A.; Arosio, P.; Drysdale, J.; Santambrogio, P.; Campanella, A.; Biasiotto, G.; Albertini, A.; Levi, S. Human Mitochondrial Ferritin Expressed in HeLa Cells Incorporates Iron and Affects Cellular Iron Metabolism. J. Biol. Chem. 2002, 277, 22430–22437. [Google Scholar] [CrossRef]
- Ali, Y.; Oliva, C.R.; Flor, S.; Griguer, C.E. Mitoferrin, Cellular and Mitochondrial Iron Homeostasis. Cells 2022, 11, 3464. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Li, K.; Xing, W.; Dong, M.; Yi, M.; Zhang, H. Role of Iron-Related Oxidative Stress and Mitochondrial Dysfunction in Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2022, 2022, 5124553. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhou, L.; Han, S.; Guo, J.; Qiu, T.; Zhou, J.; Shen, L. Ferroptosis—A New Dawn in the Treatment of Organ Ischemia–Reperfusion Injury. Cells 2022, 11, 3653. [Google Scholar] [CrossRef]
- Dong, H.; Qiang, Z.; Chai, D.; Peng, J.; Xia, Y.; Hu, R.; Jiang, H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 2020, 12, 12943–12959. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2023, 24, 449. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2018, 133, 144–152. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.-I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, Y.-L.; Mao, J.-A.; Tang, L.-F.; Xu, J.; Wang, Z.-X.; He, Y.; Li, M. NCOA4-mediated ferritinophagy is involved in ionizing radiation-induced ferroptosis of intestinal epithelial cells. Redox Biol. 2022, 55, 102413. [Google Scholar] [CrossRef]
- Tuo, Q.-Z.; Liu, Y.; Xiang, Z.; Yan, H.-F.; Zou, T.; Shu, Y.; Ding, X.-L.; Zou, J.-J.; Xu, S.; Tang, F.; et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct. Target. Ther. 2022, 7, 59. [Google Scholar] [CrossRef]
- Chen, J.; Yang, L.; Geng, L.; He, J.; Chen, L.; Sun, Q.; Zhao, J.; Wang, X. Inhibition of Acyl-CoA Synthetase Long-Chain Family Member 4 Facilitates Neurological Recovery After Stroke by Regulation Ferroptosis. Front. Cell. Neurosci. 2021, 15, 632354. [Google Scholar] [CrossRef]
- Li, X.; Liang, J.; Qin, A.; Wang, T.; Liu, S.; Li, W.; Yuan, C.; Qu, L.; Zou, W. Protective effect of Di’ao Xinxuekang capsule against doxorubicin-induced chronic cardiotoxicity. J. Ethnopharmacol. 2021, 287, 114943. [Google Scholar] [CrossRef]
- Fu, D.; Wang, C.; Yu, L.; Yu, R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell. Mol. Biol. Lett. 2021, 26, 26. [Google Scholar] [CrossRef]
- He, S.; Li, R.; Peng, Y.; Wang, Z.; Huang, J.; Meng, H.; Min, J.; Wang, F.; Ma, Q. ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J. Cachexia. Sarcopenia Muscle 2022, 13, 1717–1730. [Google Scholar] [CrossRef]
- Wang, T.; Yuan, C.; Liu, J.; Deng, L.; Li, W.; He, J.; Liu, H.; Qu, L.; Wu, J.; Zou, W. Targeting Energy Protection as a Novel Strategy to Disclose Di’ao Xinxuekang against the Cardiotoxicity Caused by Doxorubicin. Int. J. Mol. Sci. 2023, 24, 897. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Prasad, S.V.N.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Koleini, N.; Shapiro, J.S.; Geier, J.; Ardehali, H. Ironing out mechanisms of iron homeostasis and disorders of iron deficiency. J. Clin. Investig. 2021, 131, 148671. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Nakagawa, Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003, 34, 145–169. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zou, Q.; Tang, H.; Liu, J.; Zhang, S.; Fan, C.; Zhang, J.; Liu, R.; Liu, Y.; Liu, R.; et al. Melanin nanoparticles alleviate sepsis-induced myocardial injury by suppressing ferroptosis and inflammation. Bioact. Mater. 2023, 24, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Sun, K.; Wu, J.-H.; Zhong, Z.-H.; Xu, S.-L.; Zhang, H.-R.; Gu, Y.-H.; Lu, S.-F. Proteomic and metabolomic characterization of cardiac tissue in acute myocardial ischemia injury rats. PLoS ONE 2020, 15, e0231797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, Y.; Li, Y.; Lin, H.-B.; Fang, X. Iron homeostasis in the heart: Molecular mechanisms and pharmacological implications. J. Mol. Cell. Cardiol. 2022, 174, 15–24. [Google Scholar] [CrossRef]
- Li, D.; Pi, W.; Sun, Z.; Liu, X.; Jiang, J. Ferroptosis and its role in cardiomyopathy. Biomed. Pharmacother. 2022, 153, 113279. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, Z.; Zhou, X.; Yan, Y.; Shi, L.; Chen, J.; Fu, B.; Mao, J. Ferroptosis: The Potential Target in Heart Failure with Preserved Ejection Fraction. Cells 2022, 11, 2842. [Google Scholar] [CrossRef]
- Warbrick, I.; Rabkin, S.W. Effect of the peptides Relaxin, Neuregulin, Ghrelin and Glucagon-like peptide-1, on cardiomyocyte factors involved in the molecular mechanisms leading to diastolic dysfunction and/or heart failure with preserved ejection fraction. Peptides 2019, 111, 33–41. [Google Scholar] [CrossRef]
- Runte, K.E.; Bell, S.P.; Selby, D.E.; Häußler, T.N.; Ashikaga, T.; LeWinter, M.M.; Palmer, B.M.; Meyer, M. Relaxation and the Role of Calcium in Isolated Contracting Myocardium from Patients with Hypertensive Heart Disease and Heart Failure with Preserved Ejection Fraction. Circ. Heart Fail. 2017, 10, e004311. [Google Scholar] [CrossRef]
- Mancardi, D.; Mezzanotte, M.; Arrigo, E.; Barinotti, A.; Roetto, A. Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver. Antioxidants 2021, 10, 1864. [Google Scholar] [CrossRef]
- Yang, X.; Kawasaki, N.K.; Min, J.; Matsui, T.; Wang, F. Ferroptosis in heart failure. J. Mol. Cell. Cardiol. 2022, 173, 141–153. [Google Scholar] [CrossRef]
- Zhao, W.-K.; Zhou, Y.; Xu, T.-T.; Wu, Q. Ferroptosis: Opportunities and Challenges in Myocardial Ischemia-Reperfusion Injury. Oxidative Med. Cell. Longev. 2021, 2021, 9929687. [Google Scholar] [CrossRef]
- Hong, M.; Rong, J.; Tao, X.; Xu, Y. The Emerging Role of Ferroptosis in Cardiovascular Diseases. Front. Pharmacol. 2022, 13, 822083. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lu, C.; Hu, K.; Cai, C.; Wang, W. Ferroptosis in Cardiovascular Diseases: Current Status, Challenges, and Future Perspectives. Biomolecules 2022, 12, 390. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.-F.; Zou, T.; Tuo, Q.-Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Ju, J.; Song, Y.-N.; Wang, K. Mechanism of Ferroptosis: A Potential Target for Cardiovascular Diseases Treatment. Aging Dis. 2021, 12, 261–276. [Google Scholar] [CrossRef]
- Lu, S.; Wang, X.-Z.; He, C.; Wang, L.; Liang, S.-P.; Wang, C.-C.; Li, C.; Luo, T.-F.; Feng, C.-S.; Wang, Z.-C.; et al. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron. Acta Pharmacol. Sin. 2021, 42, 1690–1702. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.B.; Aliagan, A.D.I.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Dong, H.-Q.; Liang, S.-J.; Xu, Y.-L.; Dai, Y.; Sun, N.; Deng, D.-H.; Cheng, P. Liproxstatin-1 induces cell cycle arrest, apoptosis, and caspase-3/GSDME-dependent secondary pyroptosis in K562 cells. Int. J. Oncol. 2022, 61, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhang, J.; Wang, B.; Xu, G.; Yang, X.; Zou, Z.; Yu, C. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy 2021, 17, 4266–4285. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Weinstein, S.J.; Yu, K.; Männistö, S.; Albanes, D. Association between serum retinol and overall and cause-specific mortality in a 30-year prospective cohort study. Nat. Commun. 2021, 12, 6418. [Google Scholar] [CrossRef]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. α-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.K.; Saleh, H.A. Protective effects of vitamin E against myocardial ischemia/reperfusion injury in rats. Saudi Med. J. 2010, 31, 142–147. [Google Scholar]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef]
- Davis, J.L.; Paris, H.L.; Beals, J.W.; Binns, S.E.; Giordano, G.R.; Scalzo, R.L.; Schweder, M.M.; Blair, E.; Bell, C. Liposomal-encapsulated Ascorbic Acid: Influence on Vitamin C Bioavailability and Capacity to Protect against Ischemia–Reperfusion Injury. Nutr. Metab. Insights 2016, 9, NMI-S39764. [Google Scholar] [CrossRef]
- Rodrigo, R.; Prieto, J.C.; Aguayo, R.; Ramos, C.; Puentes, Á.; Gajardo, A.; Panieri, E.; Rojas-Solé, C.; Lillo-Moya, J.; Saso, L. Joint Cardioprotective Effect of Vitamin C and Other Antioxidants against Reperfusion Injury in Patients with Acute Myocardial Infarction Undergoing Percutaneous Coronary Intervention. Molecules 2021, 26, 5702. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Shan, X.; Lv, Z.-Y.; Yin, M.-J.; Chen, J.; Wang, J.; Wu, Q.-N. The Protective Effect of Cyanidin-3-Glucoside on Myocardial Ischemia-Reperfusion Injury through Ferroptosis. Oxidative Med. Cell. Longev. 2021, 2021, 8880141. [Google Scholar] [CrossRef]
- Raj, P.; McCallum, J.L.; Kirby, C.; Grewal, G.; Yu, L.; Wigle, J.T.; Netticadan, T. Effects of cyanidin 3-0-glucoside on cardiac structure and function in an animal model of myocardial infarction. Food Funct. 2017, 8, 4089–4099. [Google Scholar] [CrossRef]
- Liu, H.; Mo, H.; Yang, C.; Mei, X.; Song, X.; Lu, W.; Xiao, H.; Yan, J.; Wang, X.; Yan, J.; et al. A novel function of ATF3 in suppression of ferroptosis in mouse heart suffered ischemia/reperfusion. Free Radic. Biol. Med. 2022, 189, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-P.; Zheng, Z.; Hu, S.; Ru, X.; Fan, Z.; Qiu, L.; Zhang, Y. Unification of Opposites between Two Antioxidant Transcription Factors Nrf1 and Nrf2 in Mediating Distinct Cellular Responses to the Endoplasmic Reticulum Stressor Tunicamycin. Antioxidants 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef] [PubMed]
- Sookprasert, A.; Khamnongkhu, B.; Chindaprasirt, J.; Wirasorn, K. Prognostic factors of hypercalcemia of malignancy. J. Med. Assoc. Thail. 2018, 101, S197–S201. [Google Scholar] [CrossRef]
- Jiang, G.-P.; Liao, Y.-J.; Huang, L.-L.; Zeng, X.-J.; Liao, X.-H. Effects and molecular mechanism of pachymic acid on ferroptosis in renal ischemia reperfusion injury. Mol. Med. Rep. 2020, 23, 9. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Chan, W.; Taylor, A.J.; Ellims, A.H.; Lefkovits, L.; Wong, C.; Kingwell, B.A.; Natoli, A.; Croft, K.D.; Mori, T.; Kaye, D.M.; et al. Effect of Iron Chelation on Myocardial Infarct Size and Oxidative Stress in ST-Elevation–Myocardial Infarction. Circ. Cardiovasc. Interv. 2012, 5, 270–278. [Google Scholar] [CrossRef]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Cent. Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laukaitiene, J.; Gujyte, G.; Kadusevicius, E. Cardiomyocyte Damage: Ferroptosis Relation to Ischemia-Reperfusion Injury and Future Treatment Options. Int. J. Mol. Sci. 2023, 24, 12846. https://doi.org/10.3390/ijms241612846
Laukaitiene J, Gujyte G, Kadusevicius E. Cardiomyocyte Damage: Ferroptosis Relation to Ischemia-Reperfusion Injury and Future Treatment Options. International Journal of Molecular Sciences. 2023; 24(16):12846. https://doi.org/10.3390/ijms241612846
Chicago/Turabian StyleLaukaitiene, Jolanta, Greta Gujyte, and Edmundas Kadusevicius. 2023. "Cardiomyocyte Damage: Ferroptosis Relation to Ischemia-Reperfusion Injury and Future Treatment Options" International Journal of Molecular Sciences 24, no. 16: 12846. https://doi.org/10.3390/ijms241612846
APA StyleLaukaitiene, J., Gujyte, G., & Kadusevicius, E. (2023). Cardiomyocyte Damage: Ferroptosis Relation to Ischemia-Reperfusion Injury and Future Treatment Options. International Journal of Molecular Sciences, 24(16), 12846. https://doi.org/10.3390/ijms241612846