
Identification of an Additional Metal-Binding Site in Human Dipeptidyl Peptidase III
 , , , and
, , , and
Abstract
1. Introduction
2. Results
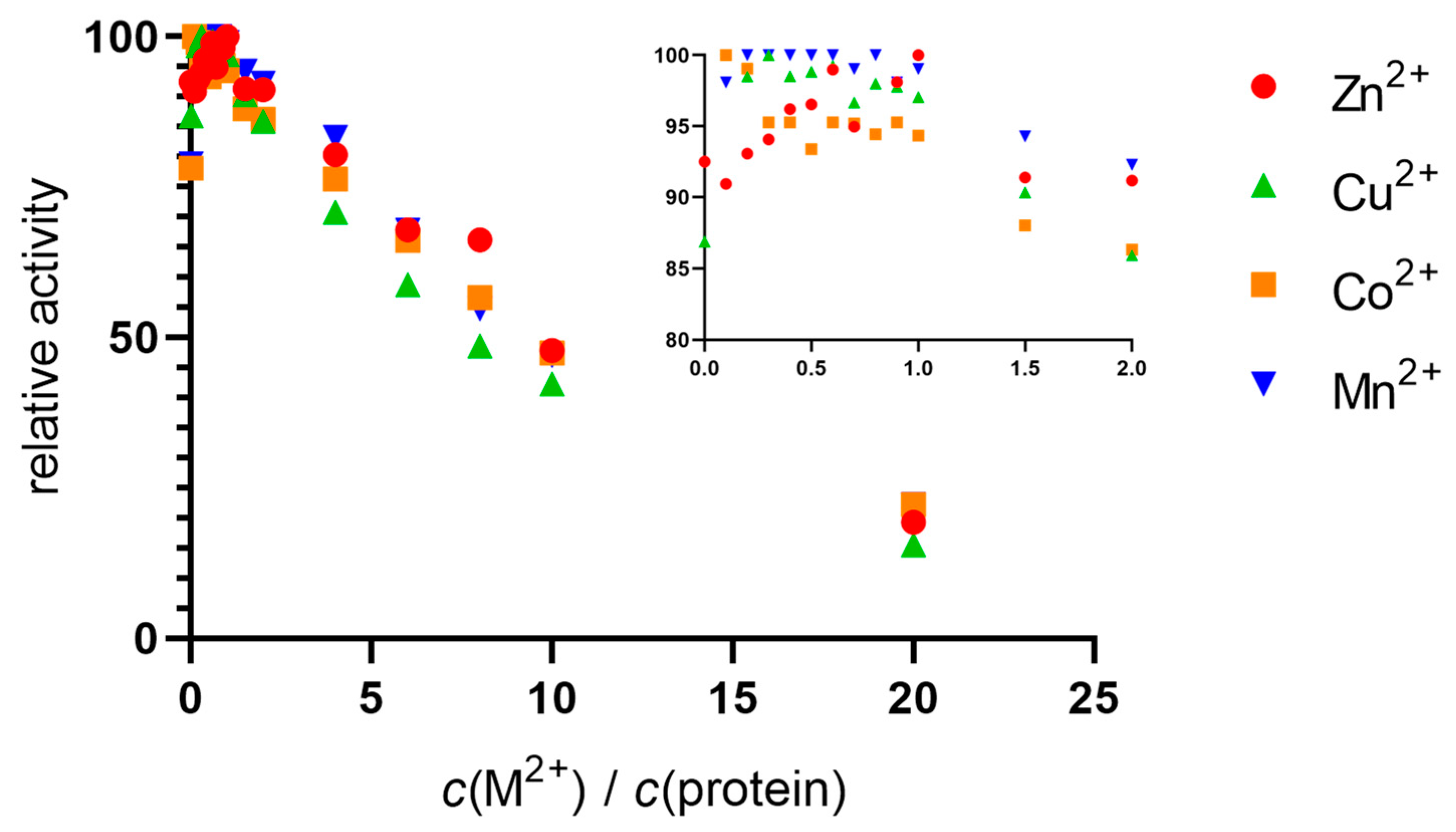
2.1. Metals in Excess Inactivate DPP III in Stopped-Flow Experiments
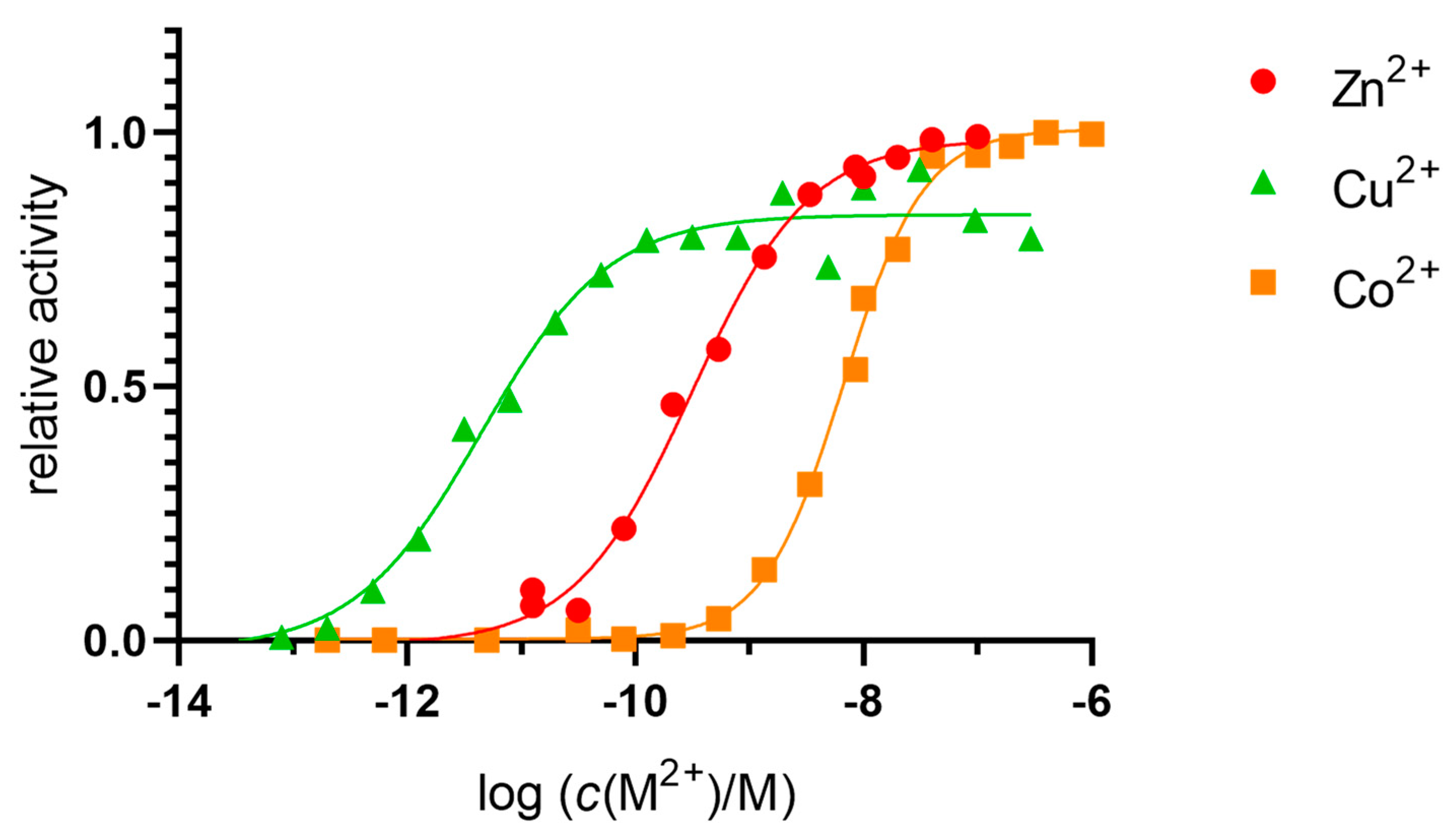
2.2. Dissociation Constants of the Catalytic Binding Site
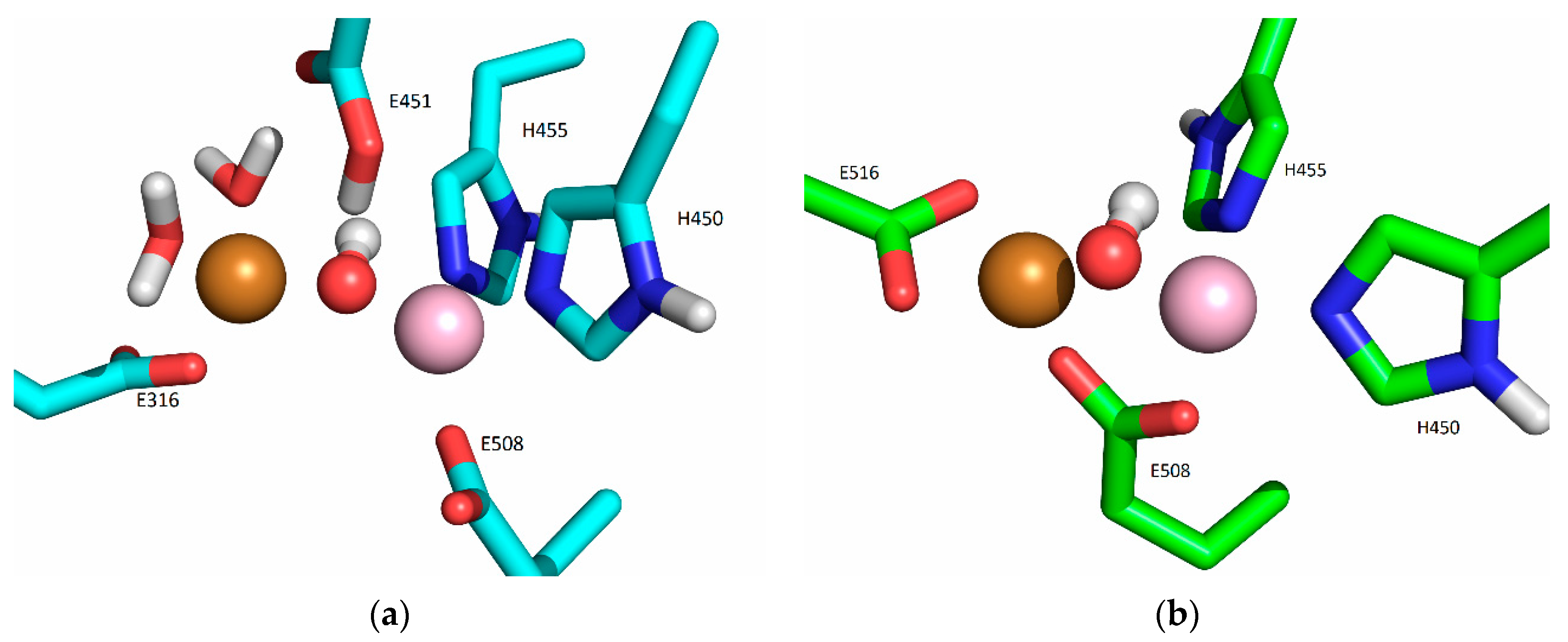
2.3. Additional Metal-Binding Site Confirmed Using ICP-MS
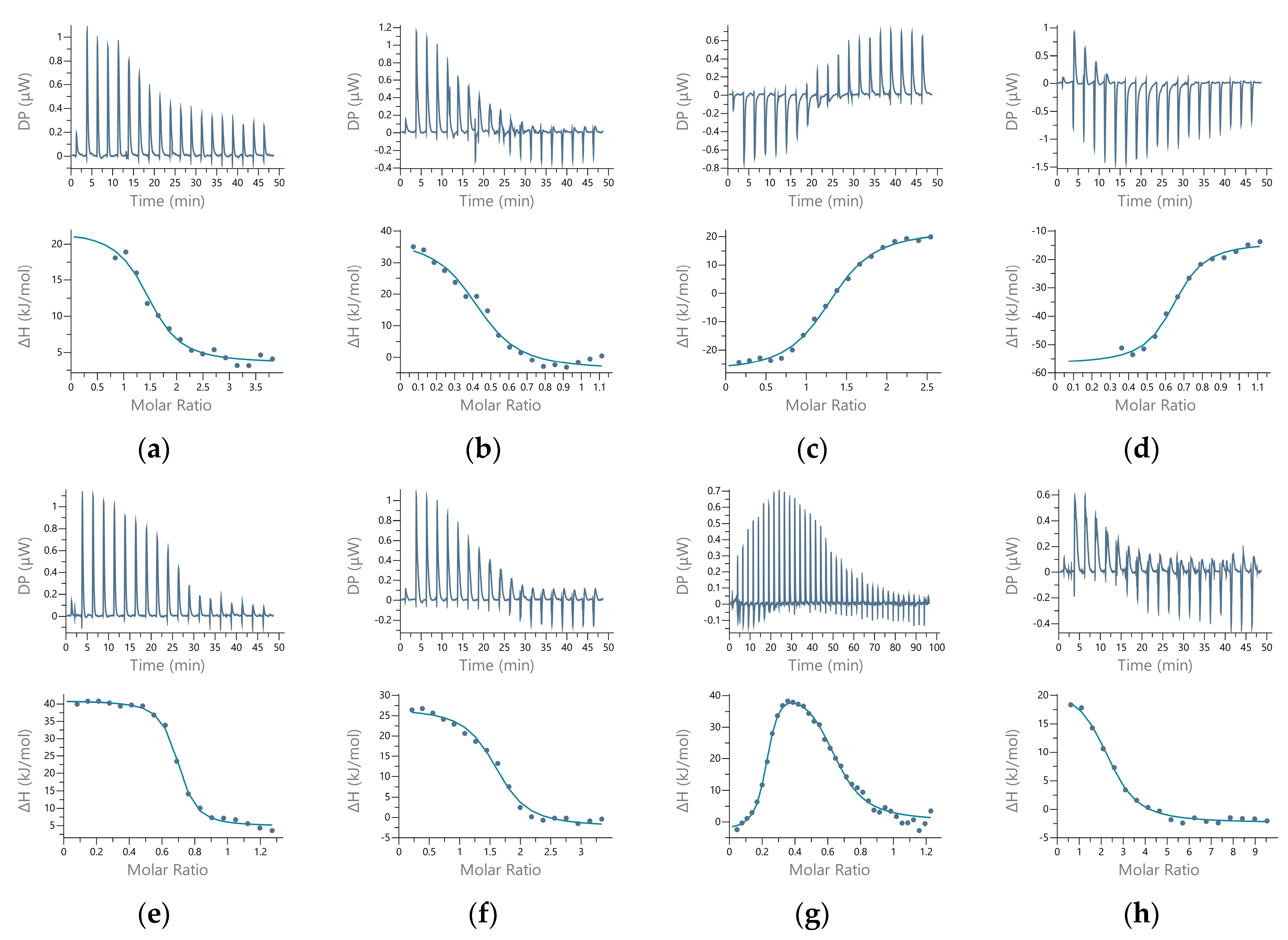
2.4. Dissociation Constants of the Additional Metal-Binding Side
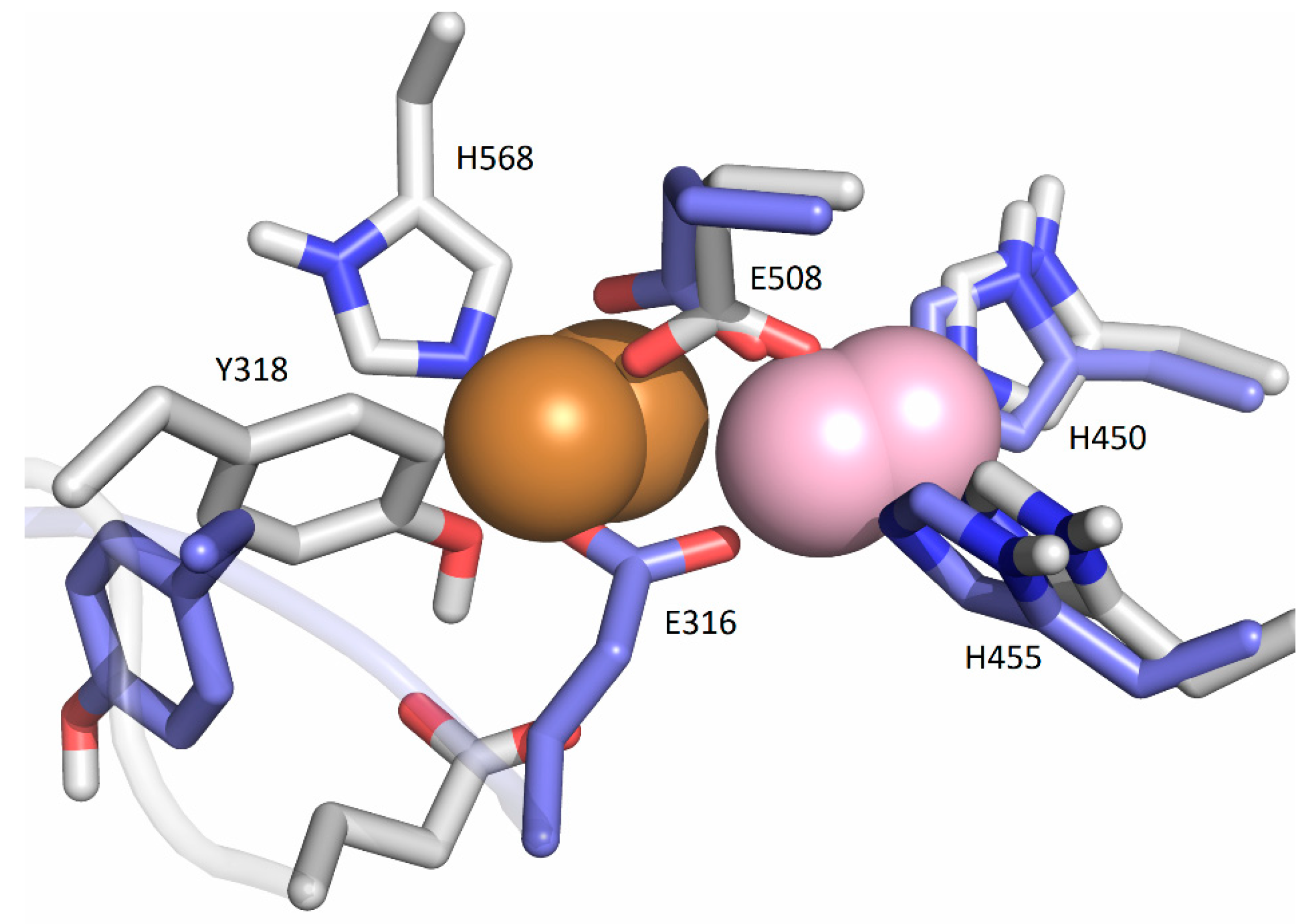
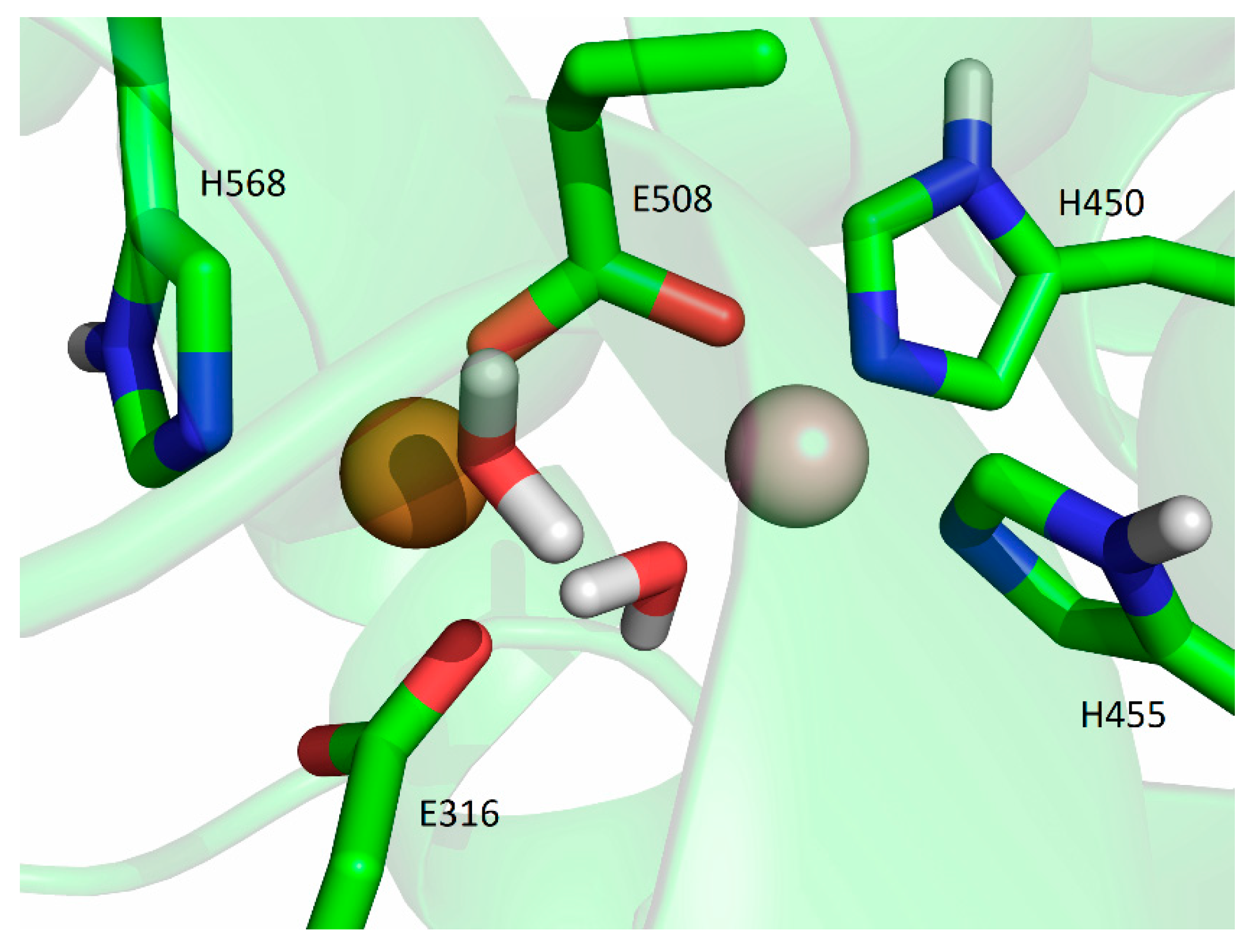
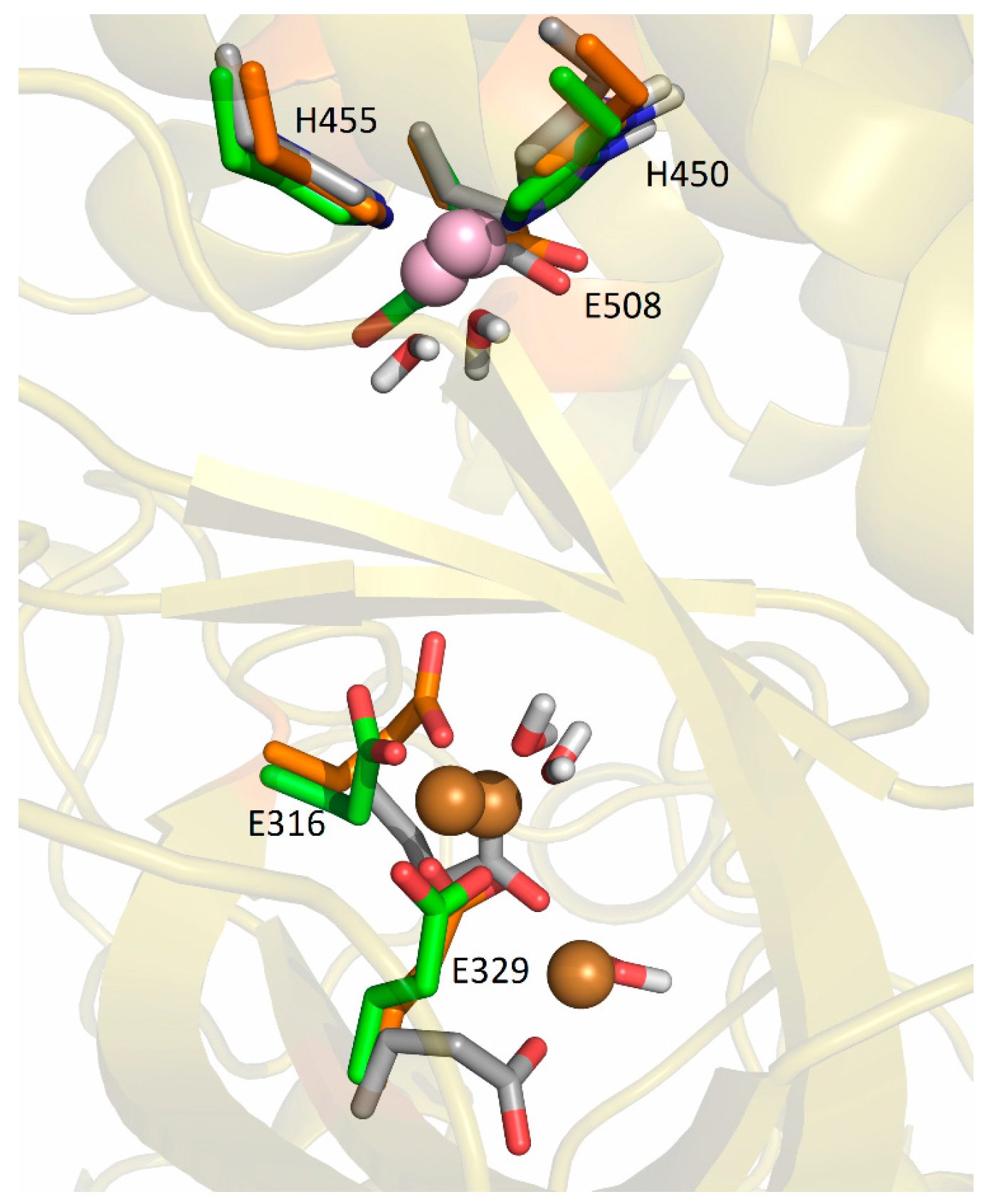
2.5. Computational Approach
2.5.1. Affinity of Different Metal Cations for the Catalytically Active Metal-Binding Site: Quantum Mechanical (QM) Calculations
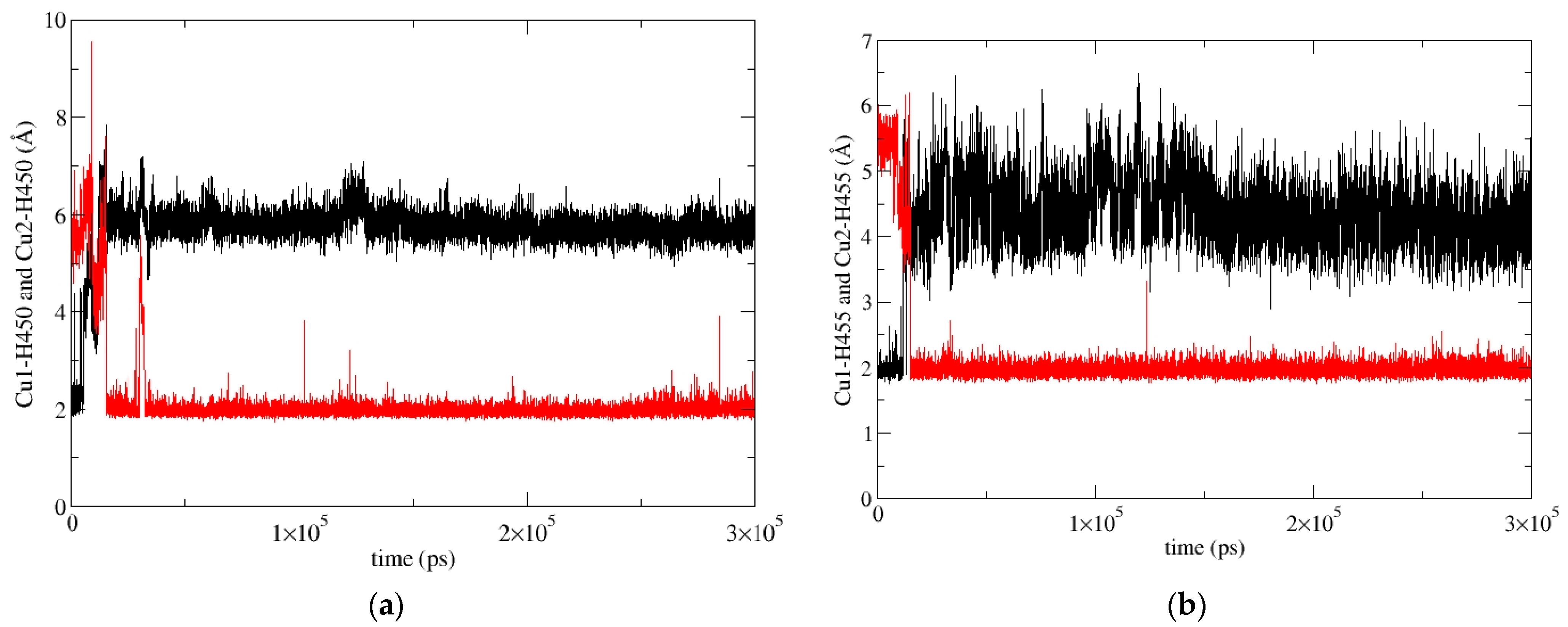
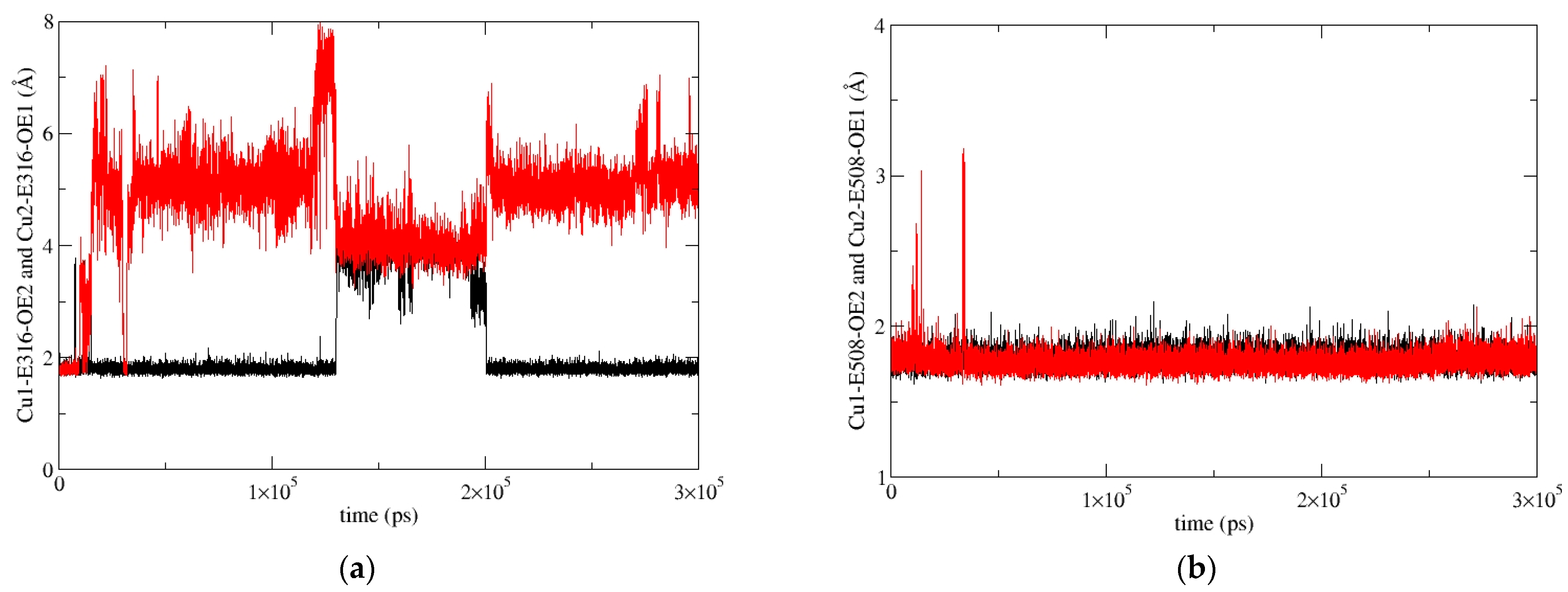
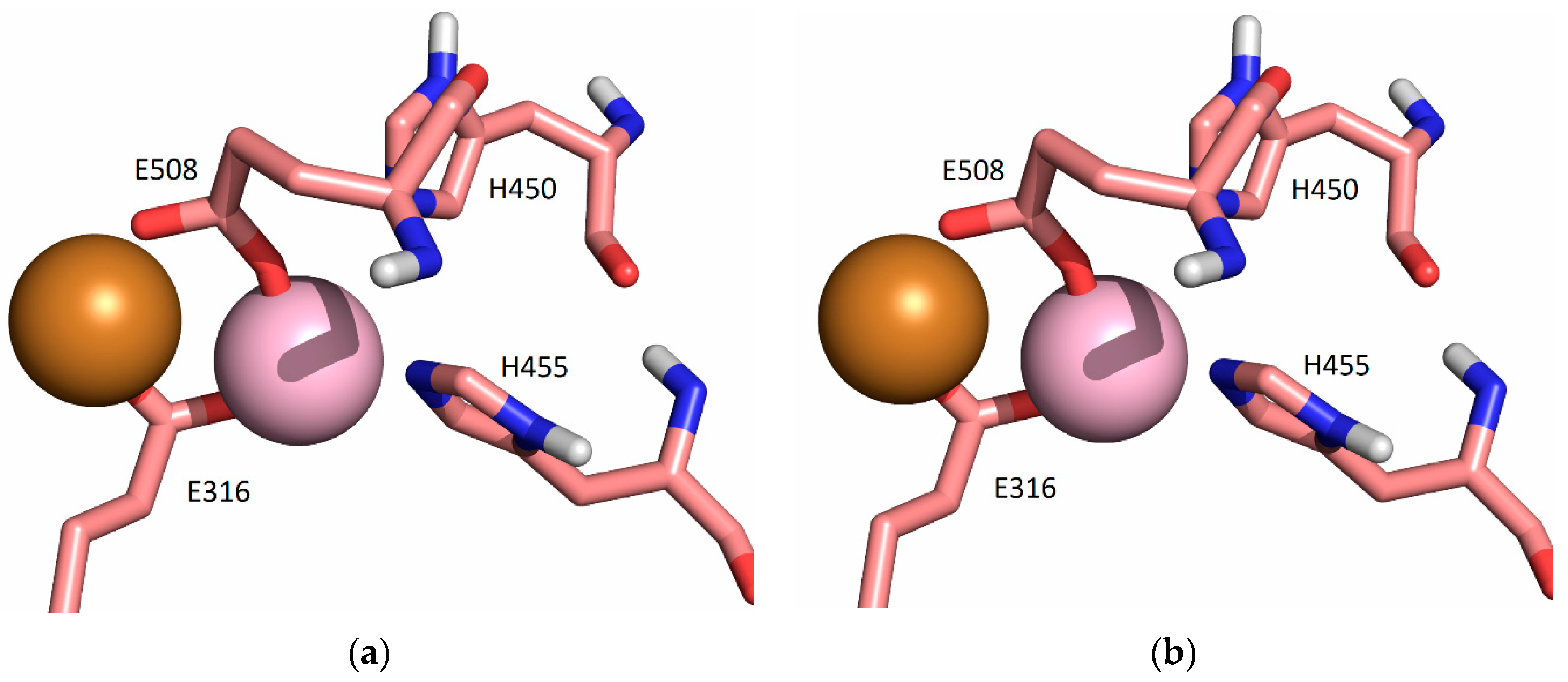
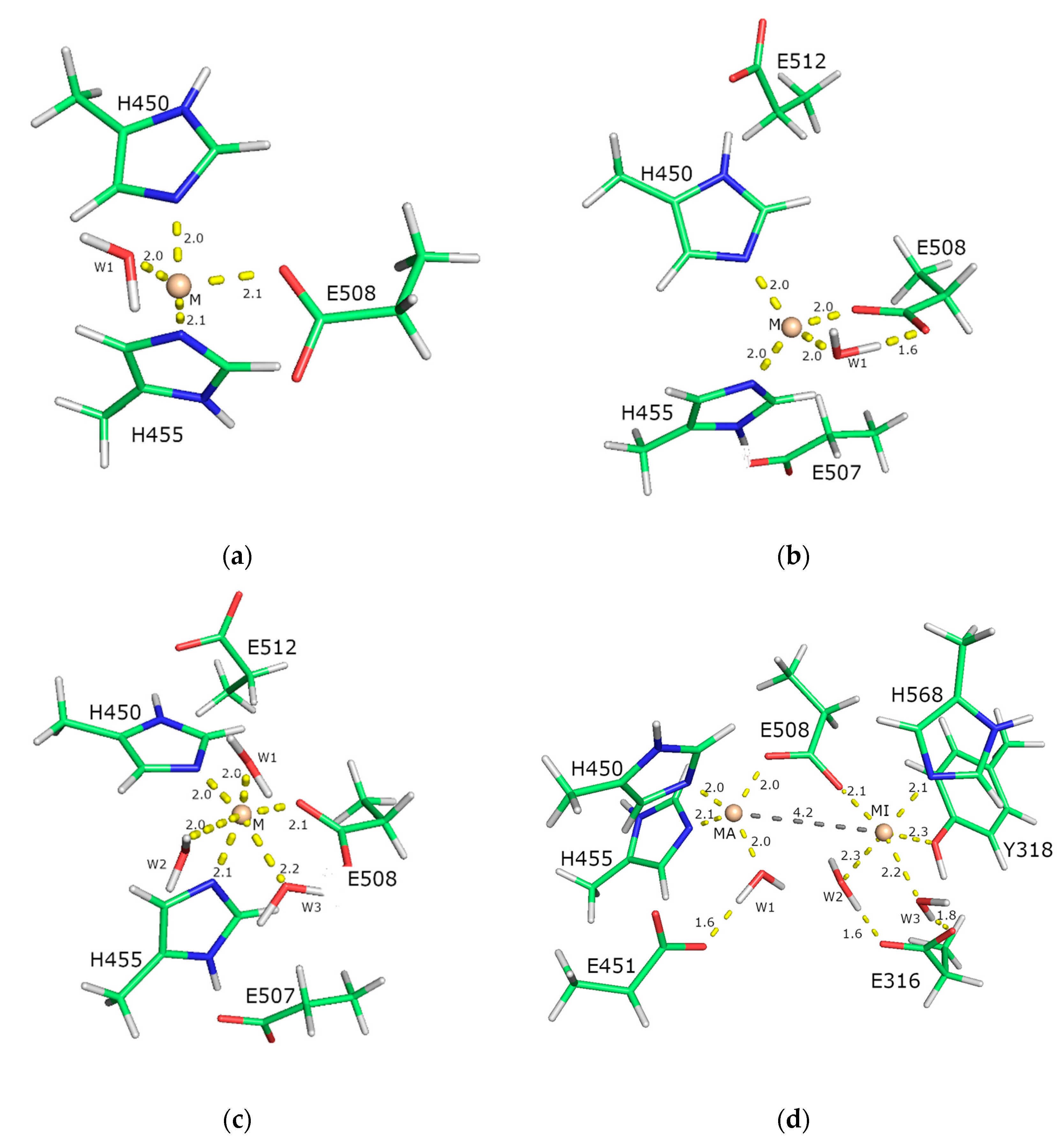
2.5.2. Molecular-Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Experimental Methods
4.1.1. Chemicals
4.1.2. General
4.1.3. Bacterial Transformation and Protein Expression
4.1.4. Site-Directed Mutagenesis
| hDPP III_H455Y_F | CACGAGCTGCTGGGTtAtGGCTCCGGCAAACTG |
| hDPP III_H455Y_R | CAGTTTGCCGGAGCCaTaACCCAGCAGCTCGTG |
| hDPP III_H568Y_F | CAACTGGCGCCAAGCCtAtATGCAGGCCCGTTTCG |
| hDPP III_H568Y_R | CGAAACGGGCCTGCATaTaGGCTTGGCGCCAGTTG |
| hDPP III_E316A_F | GTCTTACATTGGTTTCATCGcgTCTTACCGTGATCCTTTCG |
| hDPP III_E316A_R | CGAAAGGATCACGGTAAGAcgCGATGAAACCAATGTAAGAC |
| hDPP III_E508D_F | CGAGCTCCTACGAGGAtTGTCGTGCAGAGTCTG |
| hDPP III_E508D_R | CAGACTCTGCACGACAaTCCTCGTAGGAGCTCG |
4.1.5. Immunoassays/Western Blot
4.1.6. Protein Purification
4.1.7. Preparation of Apo hDPP III
4.1.8. Preparation of Holoprotein
4.1.9. Determination of Zinc, Copper, Cobalt, and Manganese by Stopped-Flow Spectrophotometry
4.1.10. Determination of Dissociation Constant—Fluorimetric Measurements
4.1.11. Analysis of Metal-Ion Content by ICP-MS
4.1.12. Isothermal Titration Calorimetry (ITC)
4.2. Computational Methods
4.2.1. Quantum Mechanical Calculations (QM)
- Model preparation
- 2.
- Details of QM calculations
4.2.2. Molecular-Dynamics Simulations (MD)
- System Preparations
- 2.
- Classical MD simulations
- 3.
- Data analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.M.; Barrett, A.J. Dipeptidyl-Peptidase III. In Handbook of Proteolytic Enzymes, 2nd ed.; Academic Press: Cambridge, MA, USA, 2004; Volume 1, pp. 809–812. ISBN 9780080984155. [Google Scholar]
- Cruz-Diaz, N.; Wilson, B.A.; Pirro, N.T.; Brosnihan, K.B.; Marshall, A.C.; Chappell, M.C. Identification of Dipeptidyl Peptidase 3 as the Angiotensin-(1–7) Degrading Peptidase in Human HK-2 Renal Epithelial Cells. Peptides 2016, 83, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Shimizu, A.; Kurita, S.; Zankov, D.P.; Takeuchi, K.; Yasuda-Yamahara, M.; Kume, S.; Ishida, T.; Ogita, H. Novel Therapeutic Role for Dipeptidyl Peptidase III in the Treatment of Hypertension. Hypertension 2016, 68, 630–641. [Google Scholar] [CrossRef]
- Komeno, M.; Pang, X.; Shimizu, A.; Molla, M.R.; Yasuda-Yamahara, M.; Kume, S.; Rahman, N.I.A.; Soh, J.E.C.; Nguyen, L.K.C.; Amin, M.K.B.A.; et al. Cardio- And Reno-Protective Effects of Dipeptidyl Peptidase Iii in Diabetic Mice. J. Biol. Chem. 2021, 296, 100761. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Prausmüller, S.; Spinka, G.; Goliasch, G.; Bartko, P.E.; Arfsten, H.; Santos, K.; Strunk, G.; Hülsmann, M. Circulating Dipeptidyl Peptidase (CDPP3)—A Marker for End-Stage Heart Failure? J. Intern. Med. 2022, 291, 886–890. [Google Scholar] [CrossRef]
- Menale, C.; Robinson, L.J.; Palagano, E.; Rigoni, R.; Erreni, M.; Almarza, A.J.; Strina, D.; Mantero, S.; Lizier, M.; Forlino, A.; et al. Absence of Dipeptidyl Peptidase 3 Increases Oxidative Stress and Causes Bone Loss. J. Bone Miner. Res. 2019, 34, 2133–2148. [Google Scholar] [CrossRef]
- Ren, X.; Yu, J.; Guo, L.; Ma, H. Dipeptidyl-Peptidase 3 Protects Oxygen-Glucose Deprivation/Reoxygenation-Injured Hippocampal Neurons by Suppressing Apoptosis, Oxidative Stress and Inflammation via Modulation of Keap1/Nrf2 Signaling. Int. Immunopharmacol. 2021, 96, 107595. [Google Scholar] [CrossRef] [PubMed]
- Matić, S.; Paić, A.T.; Sobočanec, S.; Pinterić, M.; Pipalović, G.; Martinčić, M.; Matovina, M.; Tomić, S. Interdisciplinary Study of the Effects of Dipeptidyl-Peptidase III Cancer Mutations on the KEAP1-NRF2 Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 1994. [Google Scholar] [CrossRef]
- Abramić, M.; Zubanović, M.; Vitale, L. Dipeptidyl Peptidase III from Human Erythrocytes. Biol. Chem. Hoppe. Seyler. 1988, 369, 29–38. [Google Scholar] [CrossRef]
- Abramić, M.; Schleuder, D.; Dolovčak, L.; Schröder, W.; Strupat, K.; Šagi, D.; Peter-Katalinić, J.; Vitale, L. Human and Rat Dipeptidy Peptidase III: Biochemical and Mass Spectrometric Arguments for Similarities and Differences. Biol. Chem. 2000, 381, 1233–1243. [Google Scholar] [CrossRef]
- Hirose, J.; Iwamoto, H.; Nagao, I.; Enmyo, K.; Sugao, H.; Kanemitu, N.; Ikeda, K.; Takeda, M.; Inoue, M.; Ikeda, T.; et al. Characterization of the Metal-Substituted Dipeptidyl Peptidase III (Rat Liver). Biochemistry 2001, 40, 11860–11865. [Google Scholar] [CrossRef]
- Bezerra, G.A.; Dobrovetsky, E.; Viertlmayr, R.; Dong, A.; Binter, A.; Abramić, M.; Macheroux, P.; Dhe-Paganon, S.; Gruber, K. Entropy-Driven Binding of Opioid Peptides Induces a Large Domain Motion in Human Dipeptidyl Peptidase III. Proc. Natl. Acad. Sci. USA 2012, 109, 6525–6530. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Reithofer, V.; Reisinger, M.; Wallner, S.; Pavkov-Keller, T.; Macheroux, P.; Gruber, K. Substrate Complexes of Human Dipeptidyl Peptidase III Reveal the Mechanism of Enzyme Inhibition. Sci. Rep. 2016, 6, 23787. [Google Scholar] [CrossRef]
- Baral, P.K.; Jajčanin-Jozić, N.; Deller, S.; Macheroux, P.; Abramić, M.; Gruber, K. The First Structure of Dipeptidyl-Peptidase III Provides Insight into the Catalytic Mechanism and Mode of Substrate Binding. J. Biol. Chem. 2008, 283, 22316–22324. [Google Scholar] [CrossRef]
- Xu, T.; Xie, C.; Yao, D.; Zhou, C.-Z.; Liu, J. Crystal Structures Reveal a Dual Activity Enzyme from Armillariella Tabescens. Biochem. Biophys. Res. Commun. 2017, 494, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Sabljić, I.; Meštrović, N.; Vukelić, B.; Macheroux, P.; Gruber, K.; Luić, M.; Abramić, M. Crystal Structure of Dipeptidyl Peptidase III from the Human Gut Symbiont Bacteroides Thetaiotaomicron. PLoS ONE 2017, 12, e0187295. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.B.; Zhang, H.; Chen, S.; Ye, X.F.; Li, Z.K.; Liu, W.D.; Cui, Z.L.; Huang, Y. Structural and Functional Characterization of a New Bacterial Dipeptidyl Peptidase III Involved in Fruiting Body Formation in Myxobacteria. Int. J. Mol. Sci. 2023, 24, 631. [Google Scholar] [CrossRef]
- Sabljić, I.; Tomin, M.; Matovina, M.; Sučec, I.; Tomašić Paić, A.; Tomić, A.; Abramić, M.; Tomić, S. The First Dipeptidyl Peptidase III from a Thermophile: Structural Basis for Thermal Stability and Reduced Activity. PLoS ONE 2018, 13, e0192488. [Google Scholar] [CrossRef]
- Tomić, A.; Brkić, H.; Matić, A.; Tomić, S. Unravelling the Inhibitory Zinc Ion Binding Site and the Metal Exchange Mechanism in Human DPP III. Phys. Chem. Chem. Phys. 2021, 23, 13267–13275. [Google Scholar] [CrossRef]
- Young, C.J.; Siemann, S. Highly Dynamic Metal Exchange in Anthrax Lethal Factor Involves the Occupation of an Inhibitory Metal Binding Site. Chem. Commun. 2016, 52, 11748–11751. [Google Scholar] [CrossRef]
- Maret, W. Inhibitory Zinc Sites in Enzymes. BioMetals 2013, 26, 197–204. [Google Scholar] [CrossRef]
- Bukrinsky, J.T.; Bjerrum, M.J.; Kadziola, A. Native Carboxypeptidase A in a New Crystal Environment Reveals a Different Conformation of the Important Tyrosine 248. Biochemistry 1998, 37, 16555–16564. [Google Scholar] [CrossRef]
- Holland, D.R.; Hausrath, A.C.; Juers, D.; Matthews, B.W. Structural Analysis of Zinc Substitutions in the Active Site of Thermolysin. Protein Sci. 1995, 4, 1955–1965. [Google Scholar] [CrossRef]
- Whittington, D.A.; Rusche, K.M.; Shin, H.; Fierke, C.A.; Christianson, D.W. Crystal Structure of LpxC, a Zinc-Dependent Deacetylase Essential for Endotoxin Biosynthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8146–8150. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K.M.; Hata, T.; Ono, Y.; Hirose, J. Metal Preferences of Zinc-Binding Motif on Metalloproteases. J. Amino Acids 2011, 2011, 574816. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.Y.; Säbel, C.E.; Webb, M.I.; Walsby, C.J.; Siemann, S. High Metal Substitution Tolerance of Anthrax Lethal Factor and Characterization of Its Active Copper-Substituted Analogue. J. Inorg. Biochem. 2014, 140, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Säbel, C.E.; Carbone, R.; Dabous, J.R.; Lo, S.Y.; Siemann, S. Preparation and Characterization of Cobalt-Substituted Anthrax Lethal Factor. Biochem. Biophys. Res. Commun. 2011, 416, 106–110. [Google Scholar] [CrossRef]
- Fukasawa, K.M.; Hirose, J.; Hata, T.; Ono, Y. In Rat Dipeptidyl Peptidase III, His568 Is Essential for Catalysis, and Glu507 or Glu512 Stabilizes the Coordination Bond between His455 or His450 and Zinc Ion. Biochim. Biophys. Acta 2010, 1804, 2063–2069. [Google Scholar] [CrossRef]
- Säbel, C.E.; St-Denis, S.; Neureuther, J.M.; Carbone, R.; Siemann, S. Alkaline Earth Metals Are Not Required for the Restoration of the Apoform of Anthrax Lethal Factor to Its Holoenzyme State. Biochem. Biophys. Res. Commun. 2010, 403, 209–213. [Google Scholar] [CrossRef]
- Chao, Y.; Fu, D. Thermodynamic Studies of the Mechanism of Metal Binding to the Escherichia Coli Zinc Transporter YiiP. J. Biol. Chem. 2004, 279, 17173–17180. [Google Scholar] [CrossRef]
- Briknarová, K.; Thomas, C.J.; York, J.; Nunberg, J.H. Structure of a Zinc-Binding Domain in the Junín Virus Envelope Glycoprotein. J. Biol. Chem. 2011, 286, 1528–1536. [Google Scholar] [CrossRef]
- Smolko, A.; Šupljika, F.; Martinčić, J.; Jajčanin-Jozić, N.; Grabar-Branilović, M.; Tomić, S.; Ludwig-Müller, J.; Piantanida, I.; Salopek-Sondi, B. The Role of Conserved Cys Residues in Brassica rapa Auxin Amidohydrolase: Cys139 Is Crucial for the Enzyme Activity and Cys320 Regulates Enzyme Stability. Phys. Chem. Chem. Phys. 2016, 18, 8890–8900. [Google Scholar] [CrossRef] [PubMed]
- Motara, H.; Mistry, D.; Brown, D.R.; Cryan, R.A.; Nigen, M.; Page, M.I. PH and Basicity of Ligands Control the Binding of Metal-Ions to B. Cereus B1 β-Lactamase. Chem. Sci. 2014, 5, 3120–3129. [Google Scholar] [CrossRef]
- Cheng, Z.; Thomas, P.W.; Ju, L.; Bergstrom, A.; Mason, K.; Clayton, D.; Miller, C.; Bethel, C.R.; VanPelt, J.; Tierney, D.L.; et al. Evolution of New Delhi Metallo-β-Lactamase (NDM) in the Clinic: Effects of NDM Mutations on Stability, Zinc Affinity, and Mono-Zinc Activity. J. Biol. Chem. 2018, 293, 12606–12618. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K.; Fukasawa, M.K.; Kanai, M.; Fujii, S.; Hirose, J.; Harada, M. Dipeptidyl Peptidase III Is a Zinc Metallo-Exopeptidase: Molecular Cloning and Expression. Biochem. J. 1998, 329, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K.; Fukasawa, M.K.; Kanai, M.; Fujii, S.; Hirose, J.; Harada, M. The HELLGH Motif of Rat Liver Dipeptidyl Peptidase III Is Involved in Zinc Coordination and the Catalytic Activity of the Enzyme. Biochemistry 1999, 38, 8299–8303. [Google Scholar] [CrossRef] [PubMed]
- Irving, H.; Williams, R.J.P. 637. The Stability of Transition-Metal Complexes. J. Chem. Soc. 1953, 3192–3210. [Google Scholar] [CrossRef]
- Osman, D.; Robinson, N.J. Protein Metalation in a Nutshell. FEBS Lett. 2023, 597, 141–150. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Freire, E. ITC in the Post-Genomic Era...? Priceless. Biophys. Chem. 2005, 115, 115–124. [Google Scholar] [CrossRef]
- Xiao, C.Q.; Huang, Q.; Zhang, Y.; Zhang, H.Q.; Lai, L. Binding Thermodynamics of Divalent Metal Ions to Several Biological Buffers. Thermochim. Acta 2020, 691, 178721. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelint, T.; Gordon, J. Electrophoretic Transfer of Proteins from Polyacrylamide Gels to Nitrocellulose Sheets: Procedure and Some Applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef]
- Quinn, C.F.; Carpenter, M.C.; Croteau, M.L.; Wilcox, D.E. Isothermal Titration Calorimetry Measurements of Metal Ions Binding to Proteins, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 567. [Google Scholar]
- Wyrzykowski, D.; Pilarski, B.; Jacewicz, D.; Chmurzyński, L. Investigation of metal–buffer interactions using isothermal titration calorimetry. J. Therm. Anal. Calorim. 2012, 111, 1829–1836. [Google Scholar] [CrossRef]
- Wyrzykowski, D.; Tesmar, A.; Jacewicz, D.; Pranczk, J.; Chmurzyński, L. Zinc(II) complexation by some biologically relevant pH buffers. J. Mol. Recognit. 2014, 27, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; John Wiley and Sons, Ltd.: Hoboken, NJ, USA, 2004; ISBN 978-0-470-09182-1. [Google Scholar]
- Kircheva, N.; Dobrev, S.; Nikolova, V.; Angelova, S.; Dudev, T. Zinc and Its Critical Role in Retinitis Pigmentosa: Insights from DFT/SMD Calculations. Inorg. Chem. 2020, 59, 17347–17355. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, S.; Kircheva, N.; Nikolova, V.; Angelova, S.; Dudev, T. Competition between Ag+ and Ni2+ in Nickel Enzymes: Implications for the Ag+ Antibacterial Activity. Comput. Biol. Chem. 2022, 101, 107785. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Brothers, E.N.; Suarez, D.; Deerfield II, D.W.; Merz, K.M., Jr. PM3-Compatible Zinc Parameters Optimized for Metalloenzyme Active Sites. J. Comput. Chem. 2004, 25, 1677–1692. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Irmer, G. Hydration and Speciation Studies of Mn2+ in Aqueous Solution with Simple Monovalent Anions (ClO4−, NO3−, Cl−, Br−). Dalt. Trans. 2013, 42, 14460. [Google Scholar] [CrossRef]
- Daily, M.D.; Baer, M.D.; Mundy, C.J. Divalent Ion Parameterization Strongly Affects Conformation and Interactions of an Anionic Biomimetic Polymer. J. Phys. Chem. B 2016, 120, 2198–2208. [Google Scholar] [CrossRef] [PubMed]
- Monika; Ansari, A. Electronic Structures and Energetic of Metal(II)-Superoxo Species: A DFT Exploration. Struct. Chem. 2023, 34, 825–835. [Google Scholar] [CrossRef]
- Tomić, A.; Tomić, S. Hunting the Human DPP III Active Conformation: Combined Thermodynamic and QM/MM Calculations. Dalt. Trans. 2014, 43, 15503–15514. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M. Systematic Parameterization of Monovalent Ions Employing the Nonbonded Model. J. Chem. Theory Comput. 2015, 11, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin Dynamics of Peptides: The Frictional Dependence of Isomerization Rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hDPP III | N (Zn2+) | N (Cu2+) | N (Co2+) | N (Mn2+) |
|---|---|---|---|---|
| native | 0.07 ± 0.04 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.00 ± 0.00 |
| apo | 0.02 ± 0.01 | 0.00 ± 0.00 | 0.02 ± 0.00 | 0.00 ± 0.00 |
| apo + Zn2+ | 1.07 ± 0.16 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.01 ± 0.02 |
| apo + Cu2+ | 0.05 ± 0.05 | 2.03 ± 0.09 | 0.02 ± 0.01 | 0.01 ± 0.01 |
| apo + Co2+ | 0.37 ± 0.17 | 0.03 ± 0.03 | 0.33 ± 0.01 | 0.00 ± 0.00 |
| apo + Mn2+ | 0.32 ± 0.03 | 0.04 ± 0.02 | 0.05 ± 0.04 | 0.03 ± 0.01 |
| napp | Kd,app/μM | ΔrHapp/kJ mol−1 | ΔrGapp/kJ mol−1 | −T ΔrSapp/kJ mol−1 | Supposed Binding Site | |
|---|---|---|---|---|---|---|
| Zn2+ | 1.46 ± 0.07 | 1.0 ± 0.8 | 18 ± 2 | −34 ± 1 | −52 ± 1 | additional |
| Cu2+ | 1.2 ± 0.1 | 2.4 ± 0.7 | −49.8 ± 0.3 | −32.2 ± 0.7 | 17.6 ± 0.5 | additional |
| Co2+ | 0.63 ± 0.04 | 0.13 ± 0.02 | 39 ± 1 | −36.7 ± 0.5 | −78 ± 1 | active |
| Mn2+ (1) | 0.22 ± 0.01 | 0.23 ± 0.05 | −42 ± 1 | −38.0 ± 0.5 | 4 ± 2 | active |
| Mn2+ (2) | 0.44 ± 0.04 | 1.0 ± 0.1 | 47.0 ± 0.5 | −34.3 ± 0.4 | −81.2 ± 0.5 | additional |
| Metal Ion | Buffer | Kd,app/M | logKMB | QMB | Kd/M | Binding Site |
|---|---|---|---|---|---|---|
| Zn2+ | Na-cacodylate | 9.6 × 10−7 | 2.14 | 8.0 | 1.2 × 10−7 | additional |
| Zn2+ | MOPS-NaOH | 8.8 × 10−6 | 3.22 | 84 | 1.0 × 10−7 | additional |
| Cu2+ | MOPS-NaOH | 5.5 × 10−6 | 3.8 | 313 | 1.8 × 10−8 | additional |
| Co2+ | Na-cacodylate | 1.3 × 10−7 | 2.27 | 10.4 | 1.3 × 10−8 | active |
| Metal Cation (M2+) | Relative Affinities/kcal mol−1 | ||
|---|---|---|---|
| (A) | |||
| Model 1 | Model 2 | Model 3 | |
| Cu2+ | −5.65 | 0.63 | −2.51 |
| Co2+ | 3.77 | 4.39 | −31.38 |
| Mn2+ | 11.30 | 15.06 | 33.89 |
| (B) | |||
| Cu2+ | −10.67 | −4.39 | 2.51 |
| Co2+ | 5.02 | −1.26 | 9.41 |
| Mn2+ | 12.55 | 13.18 | 25.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matić, A.; Šupljika, F.; Brkić, H.; Jurasović, J.; Karačić, Z.; Tomić, S. Identification of an Additional Metal-Binding Site in Human Dipeptidyl Peptidase III. Int. J. Mol. Sci. 2023, 24, 12747. https://doi.org/10.3390/ijms241612747
Matić A, Šupljika F, Brkić H, Jurasović J, Karačić Z, Tomić S. Identification of an Additional Metal-Binding Site in Human Dipeptidyl Peptidase III. International Journal of Molecular Sciences. 2023; 24(16):12747. https://doi.org/10.3390/ijms241612747
Chicago/Turabian StyleMatić, Antonia, Filip Šupljika, Hrvoje Brkić, Jasna Jurasović, Zrinka Karačić, and Sanja Tomić. 2023. "Identification of an Additional Metal-Binding Site in Human Dipeptidyl Peptidase III" International Journal of Molecular Sciences 24, no. 16: 12747. https://doi.org/10.3390/ijms241612747
APA StyleMatić, A., Šupljika, F., Brkić, H., Jurasović, J., Karačić, Z., & Tomić, S. (2023). Identification of an Additional Metal-Binding Site in Human Dipeptidyl Peptidase III. International Journal of Molecular Sciences, 24(16), 12747. https://doi.org/10.3390/ijms241612747