

Amyloid-β Tetramers and Divalent Cations at the Membrane/Water Interface: Simple Models Support a Functional Role

Abstract
1. Introduction
2. Results
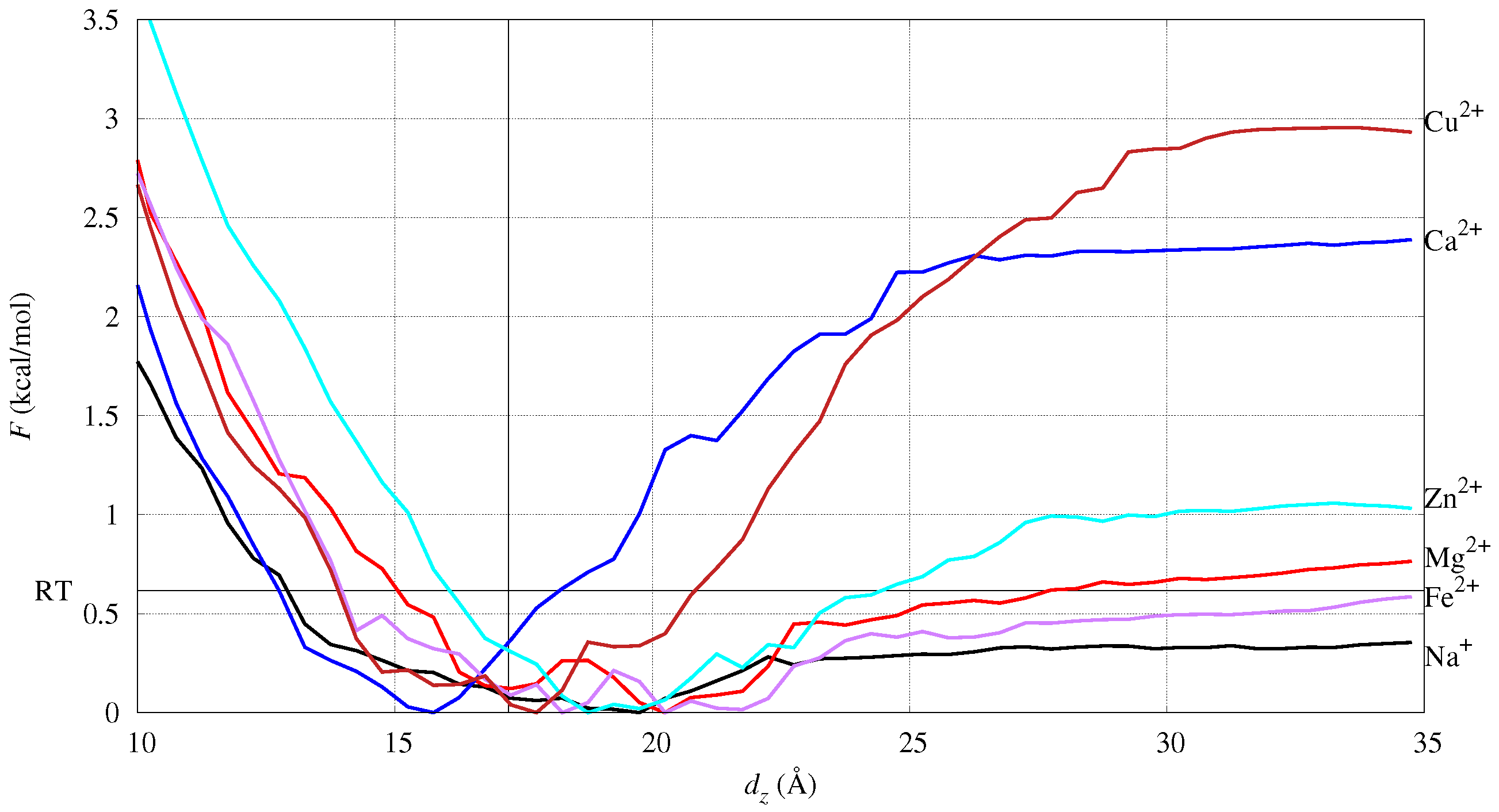
2.1. Free Energy of Cation Absorption to DMPC (Model M/DMPC)
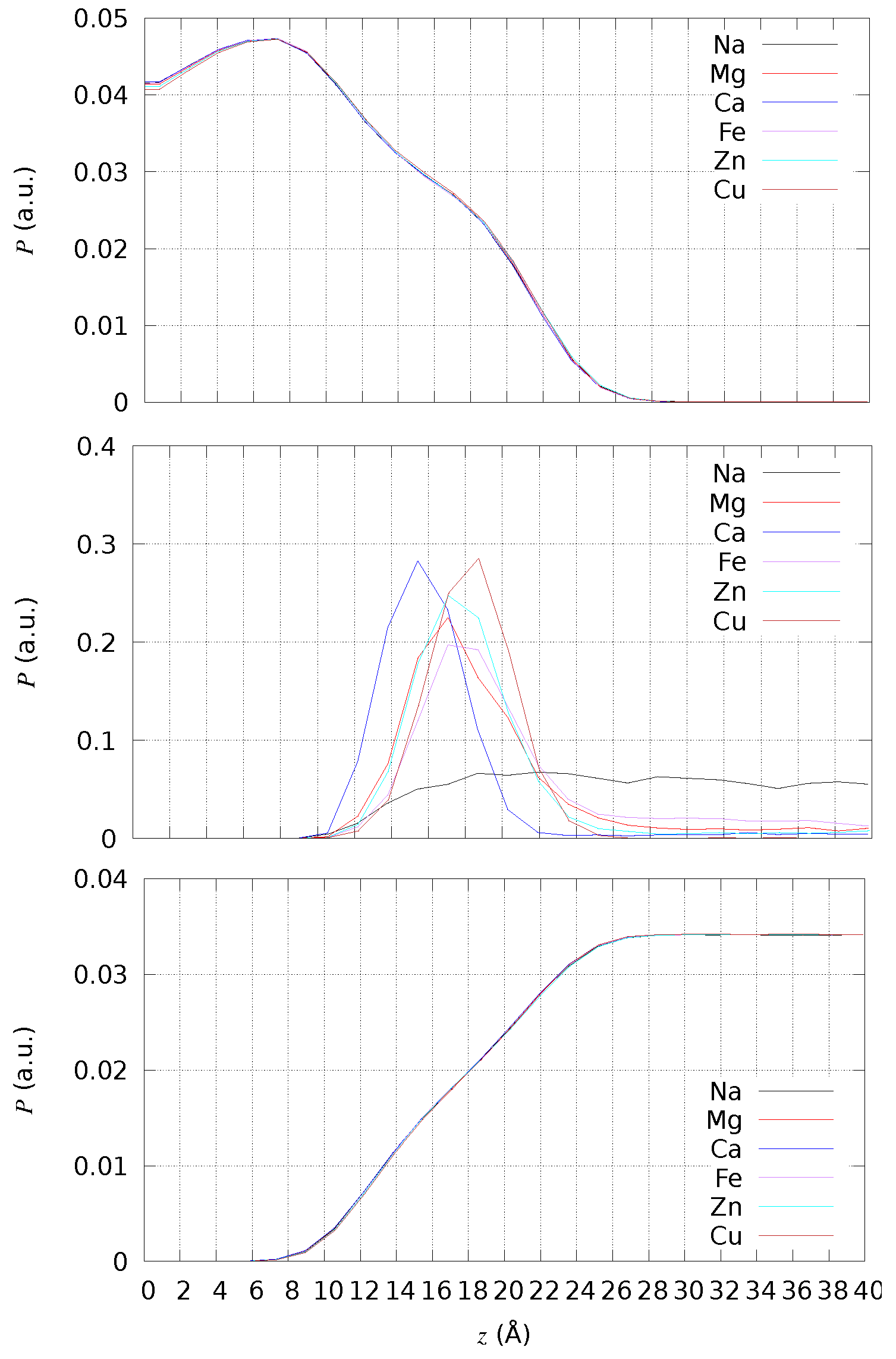
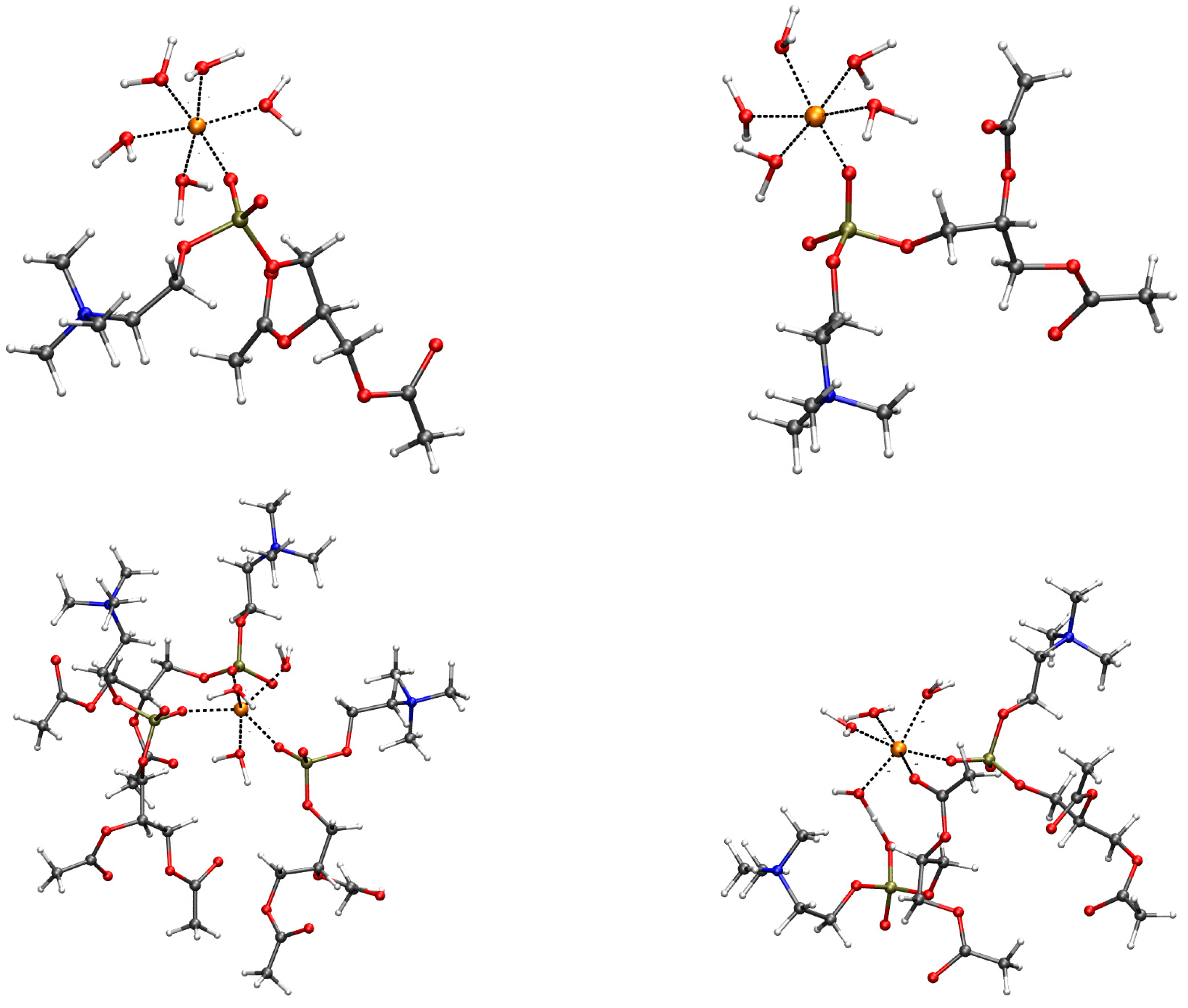
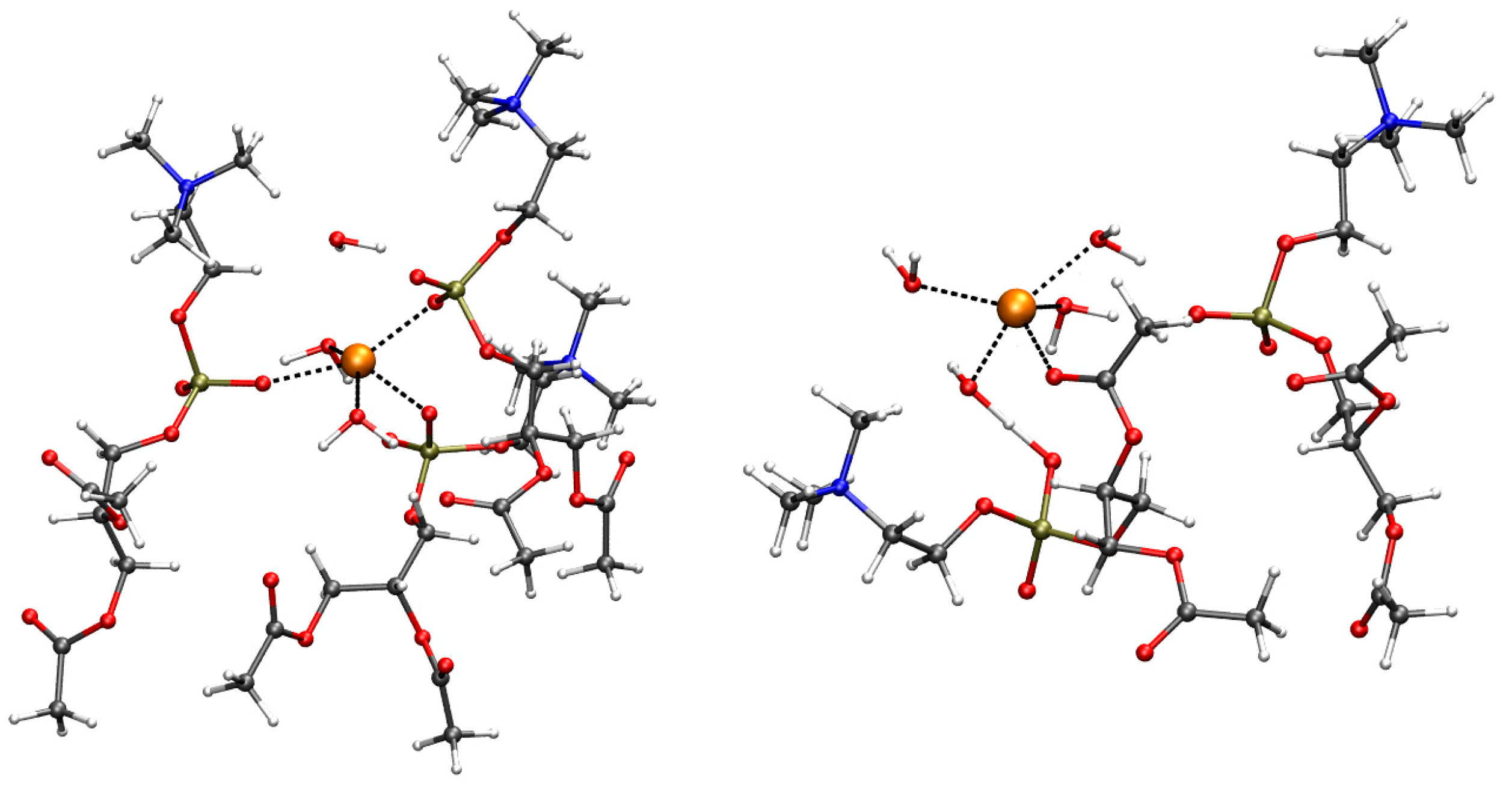
2.2. Geometry of Divalent Cation Binding Site
2.3. Assessing the Geometry of Divalent Cation Binding Site
- y is the number of water molecules in the bulk hydrated M cation;
- x is the number of PC headgroups assembled around each cation;
- z is the number of water molecules released by the hydrated cation when bonding the assembled PC headgroups.
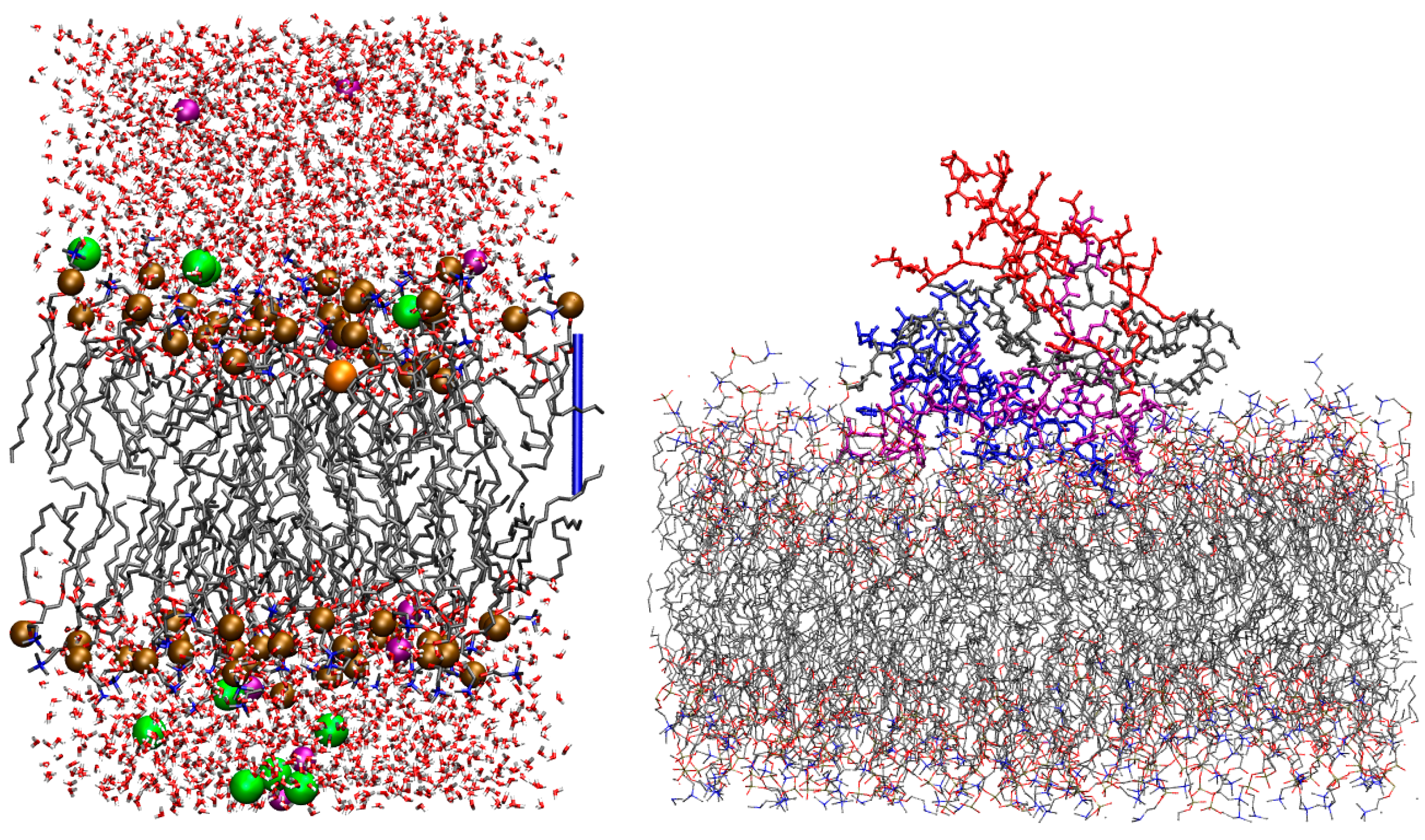
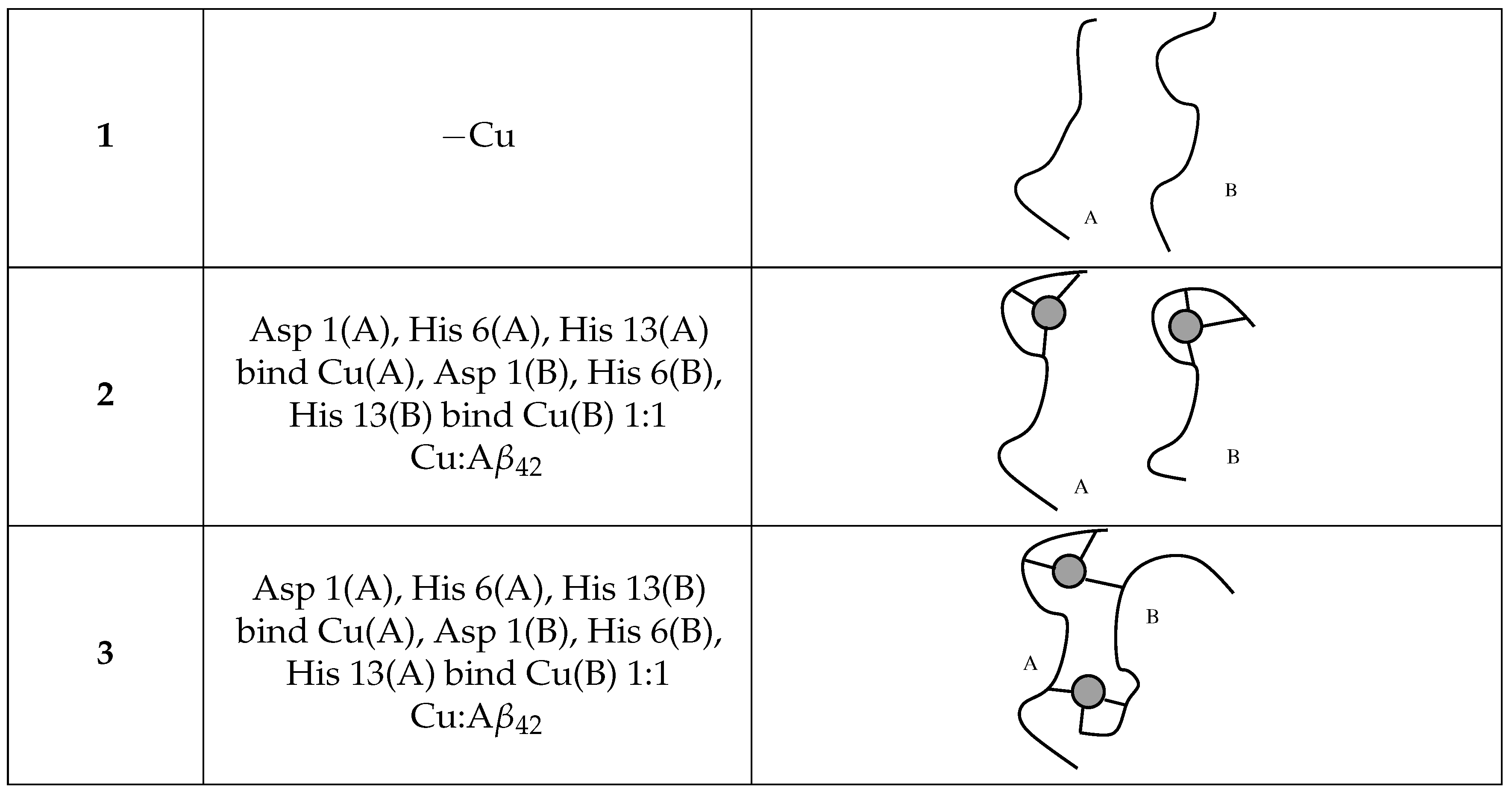
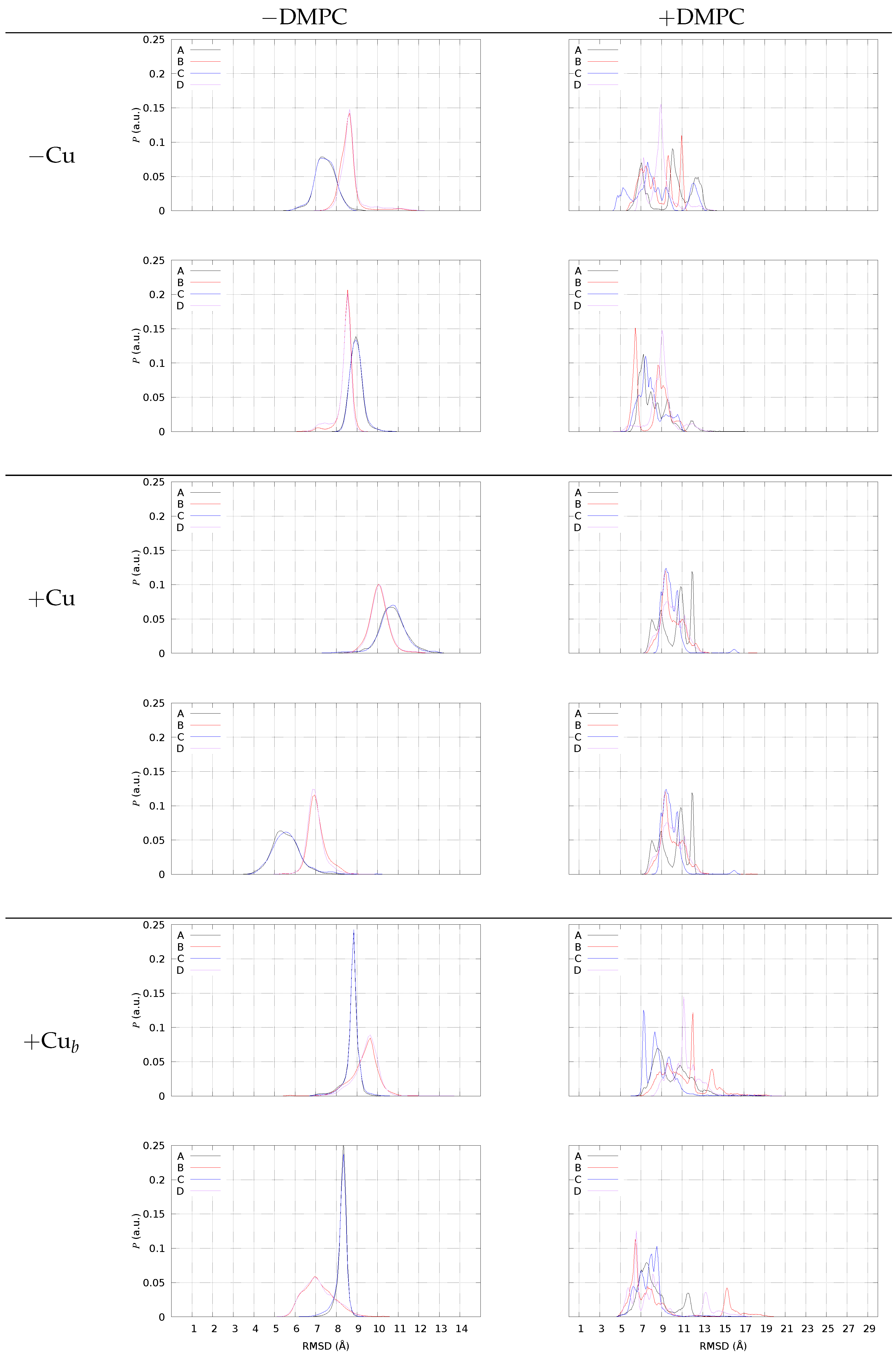
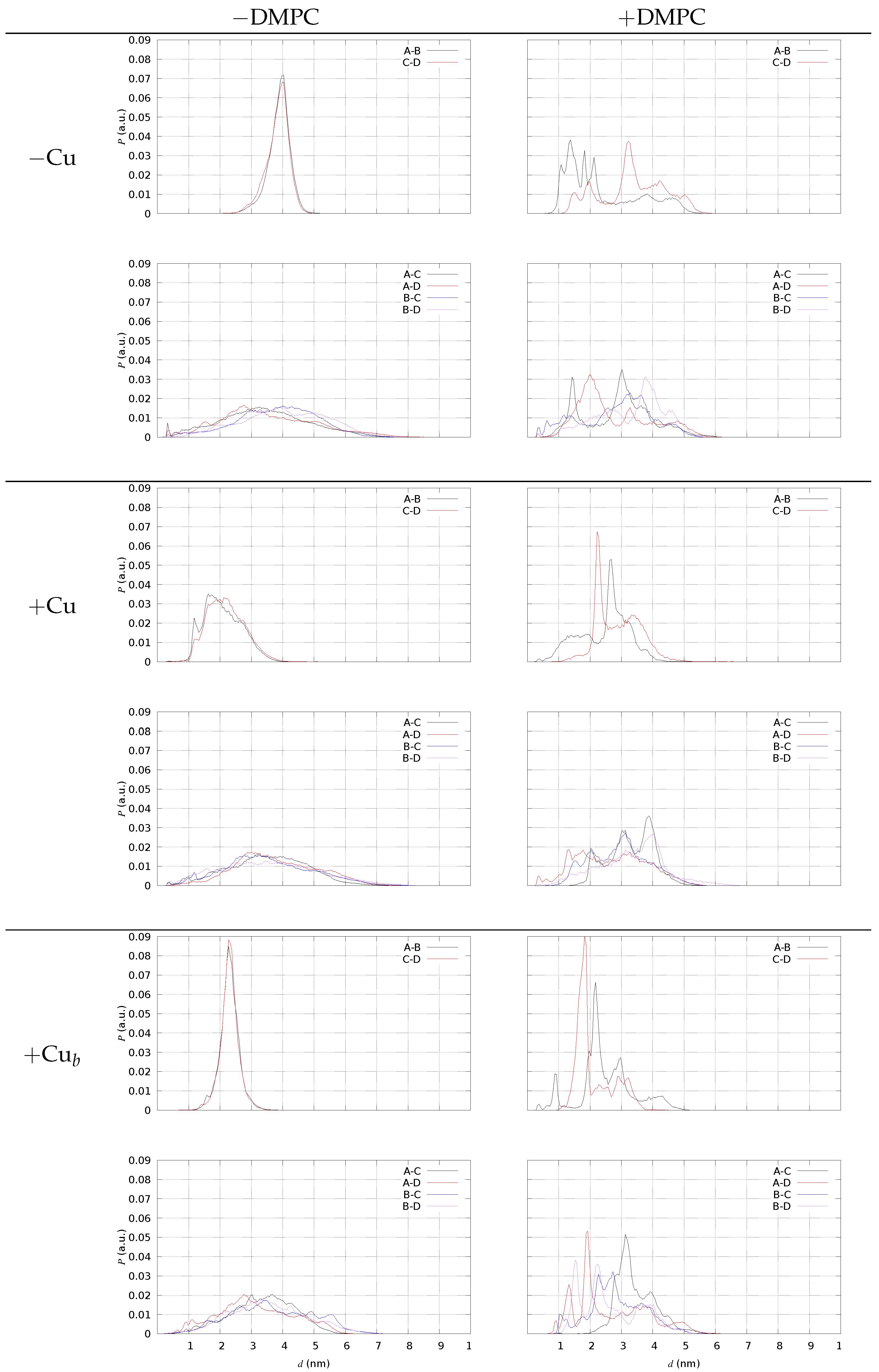
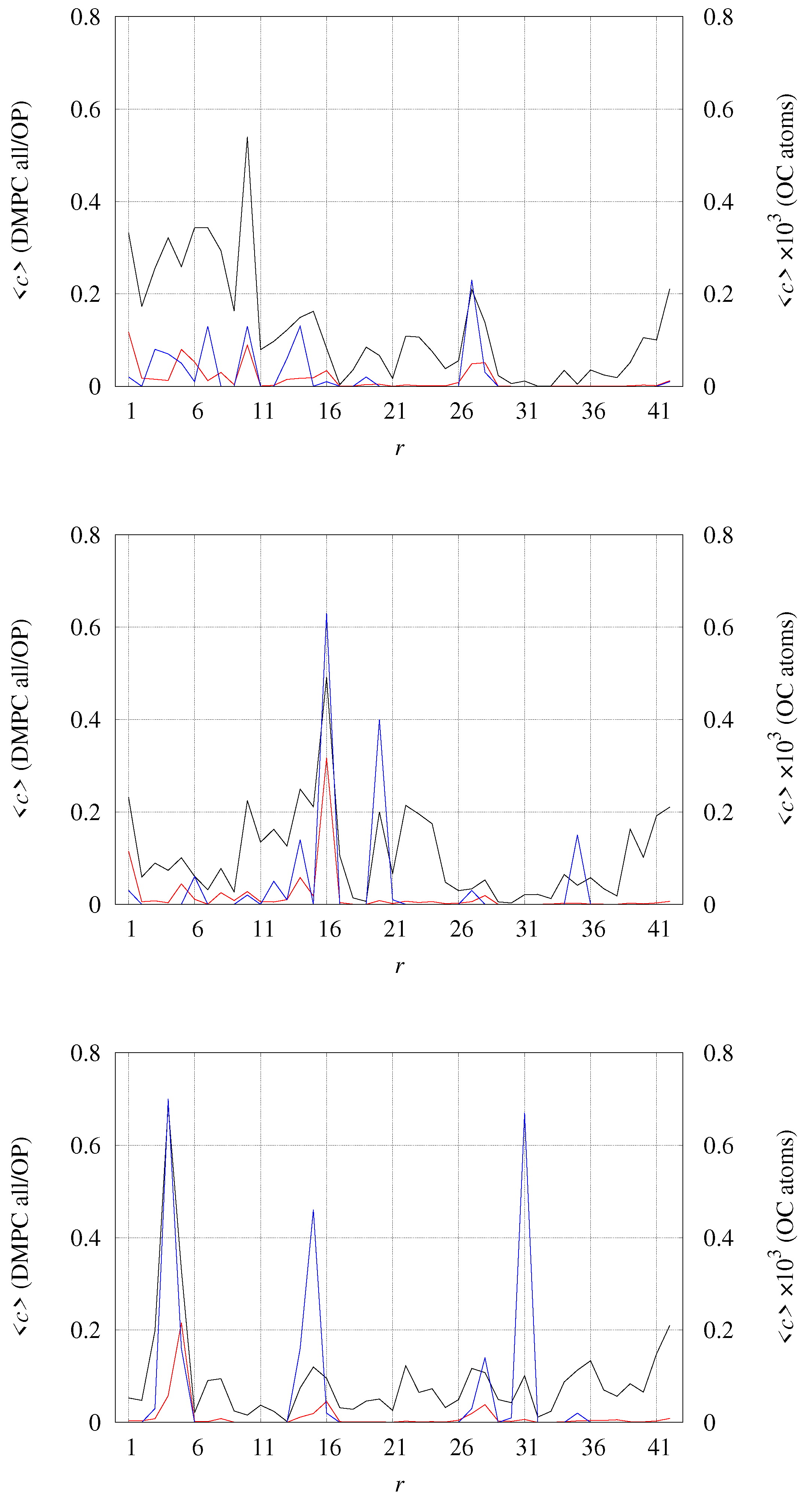
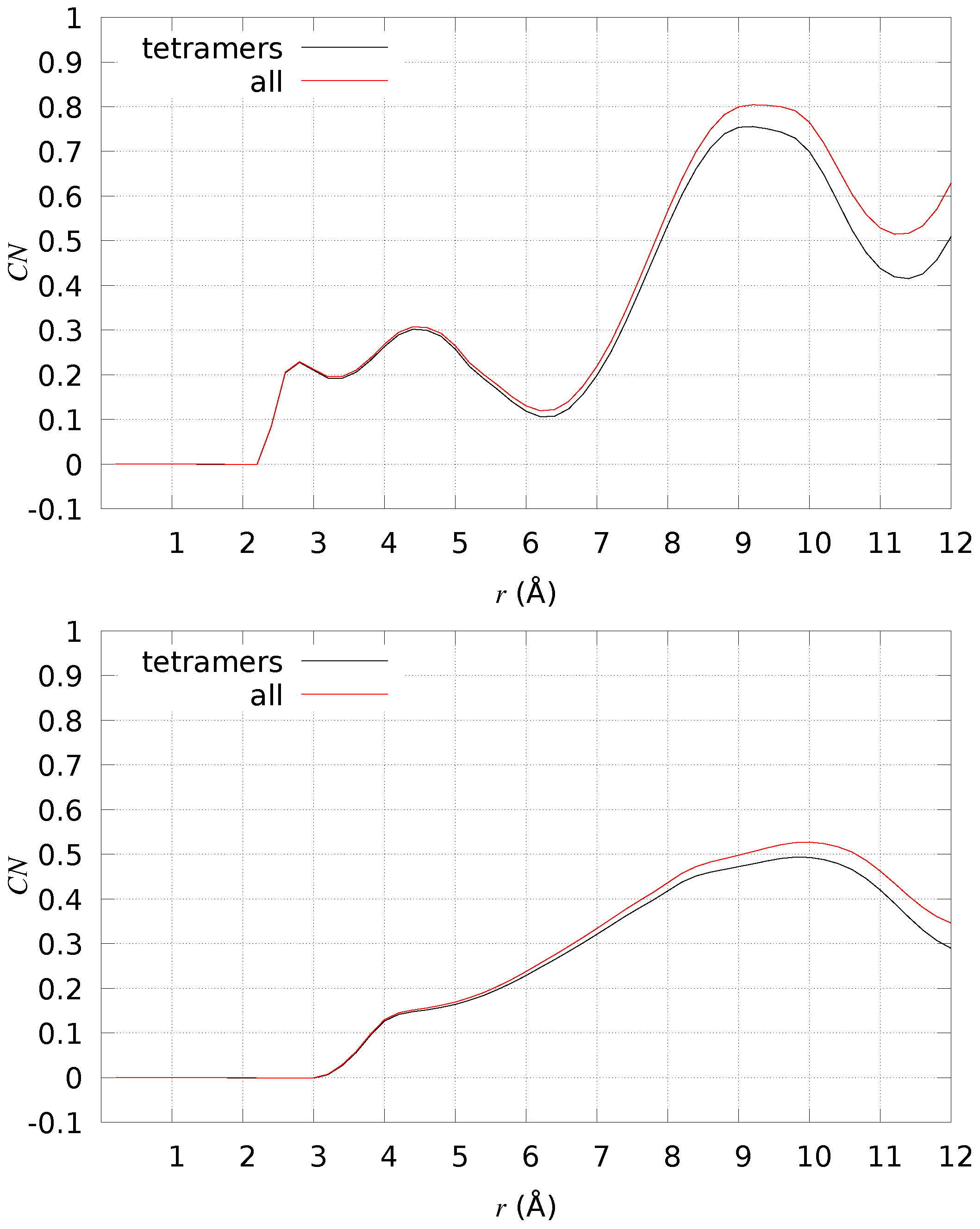
2.4. A Tetramer and DMPC (Models 1–3/DMPC)
- The U-like shape of the single-strand fiber (PDB 2BEG [58], solution NMR).
3. Discussion
- Amyloid oligomers start perturbing lipid bilayers in computational models, including realistic membrane models, when dodecamers [50].
4. Methods
- Cations situated near the DMPC lipid bilayer (models M/DMPC);
- Pairs of docked A dimers merged in water, simulated both with and without the presence of copper ions (models 1–3);
- Tetrameric A structures, also with and without copper ions, positioned in close proximity to a DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) lipid bilayer (models 1–3/DMPC).
4.1. Divalent Cations and DMPC (Models M/DMPC)
4.2. Molecular Dynamics Parameters
4.3. Refining DMPC Ion-Binding Sites
4.4. A Tetramers in a Water Solution (Models 1–3)
- 2 × A (model 1, hereafter);
- 2 × Cu-A (model 2);
- [Cu-A] (model 3);
4.5. Dimer/Dimer Contact
4.6. A Tetramers and DMPC Bilayer (Models 1–3/DMPC)
4.7. Protein/Lipid Contact
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oyane, A.; Kim, H.M.; Furuya, T.; Kokubo, T.; Miyazaki, T.; Nakamura, T. Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. A 2003, 65A, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R. (Ed.) Biological Inorganic Chemistry, 3rd ed.; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar] [CrossRef]
- Andersson, J.; Fuller, M.A.; Wood, K.; Holt, S.A.; Köper, I. A tethered bilayer lipid membrane that mimics microbial membranes. Phys. Chem. Chem. Phys. 2018, 20, 12958–12969. [Google Scholar] [CrossRef] [PubMed]
- Deplazes, E.; Tafalla, B.D.; Cranfield, C.G.; Garcia, A. Role of Ion-Phospholipid Interactions in Zwitterionic Phospholipid Bilayer Ion Permeation. J. Phys. Chem. Lett. 2020, 11, 6353–6358. [Google Scholar] [CrossRef] [PubMed]
- John, L.H.; Preston, G.M.; Sansom, M.S.; Clifton, L.A. Large scale model lipid membrane movement induced by a cation switch. J. Coll. Interf. Sci. 2021, 596, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Wei, D.Q.; Hu, D. Free Energy Calculations on the Water-Chain-Assisted and the Dehydration Mechanisms of Transmembrane Ion Permeation. J. Chem. Theory Comput. 2020, 16, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Vorobyov, I.; Roux, B.; Allen, T.W. Molecular Dynamics Simulations Based on Polarizable Models Show that Ion Permeation Interconverts between Different Mechanisms as a Function of Membrane Thickness. J. Phys. Chem. B 2021, 125, 1020–1035. [Google Scholar] [CrossRef]
- Graber, Z.T.; Shi, Z.; Baumgart, T. Cations induce shape remodeling of negatively charged phospholipid membranes. Phys. Chem. Chem. Phys. 2017, 19, 15285–15295. [Google Scholar] [CrossRef]
- Huy, P.D.Q.; Krupa, P.; Nguyen, H.L.; La Penna, G.; Li, M.S. Computational Model to Unravel the Function of Amyloid-beta Peptides in Contact with a Phospholipid Membrane. J. Phys. Chem. B 2020, 124, 3300–3314. [Google Scholar] [CrossRef]
- Hunter, G.K.; O’Young, J.; Grohe, B.; Karttunen, M.; Goldberg, H.A. The flexible polyelectrolyte hypothesis of protein-biomineral interaction. Langmuir 2010, 26, 18639–18646. [Google Scholar] [CrossRef]
- Kalmar, L.; Homola, D.; Varga, G.; Tompa, P. Structural disorder in proteins brings order to crystal growth in biomineralization. Bone 2012, 51, 528–534. [Google Scholar] [CrossRef]
- La Penna, G.; Andreussi, O. When water plays an active role in electronic structure. Insights from first-principles molecular dynamics simulations of biological systems. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes, 2nd ed.; Liwo, A.J., Ed.; Springer Series in Bio- and Neurosystems; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1, pp. 715–753. [Google Scholar] [CrossRef]
- Reybier, K.; Ayala, S.; Alies, B.; Rodrigues, J.A.V.; Bustos Rodriguez, S.; La Penna, G.; Collin, F.; Gomes, C.M.; Hureau, C.; Faller, P. Free Superoxide is an Intermediate in the Production of H2O2 by Copper(I)-Aβ Peptide and O2. Angew. Chem. Intl. Ed. 2016, 55, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- La Penna, G.; Li, M.S. Towards a High-throughput Modelling of Copper Reactivity Induced by Structural Disorder in Amyloid Peptides. Chem. Eur. J. 2018, 24, 5259–5270. [Google Scholar] [CrossRef]
- Lynch, T.; Cherny, R.; Bush, A.I. Oxidative Processes in Alzheimer’s Disease: The Role of Aβ-metal Interactions. Experim. Gerontol. 2000, 35, 445–451. [Google Scholar] [CrossRef]
- Perry, G.; Sayre, L.M.; Atwood, C.S.; Castellani, R.J.; Cash, A.D.; Rottkamp, C.A.; Smith, M.A. The Role of Iron and Copper in the Aetiology of Neurodegenerative Disorders. CNS Drugs 2002, 16, 339–352. [Google Scholar] [CrossRef]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Widomska, J.; Raguz, M.; Subczynski, W.K. Oxygen Permeability of the Lipid Bilayer Membrane Made of Calf Lens Lipids. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Hiramatsu, M.; Edamatsu, R.; Mori, I.; Mori, A. Metal Ions Affect Neuronal Membrane Fluidity of Rat Cerebral Cortex. Neurochem. Res. 1994, 19, 237–241. [Google Scholar] [CrossRef]
- Suwalsky, M.; Ungerer, B.; Quevedo, L.; Aguilar, F.; Sotomayor, C. Cu2+ Ions Interact with Cell Membranes. J. Inorg. Biochem. 1998, 70, 233–238. [Google Scholar] [CrossRef]
- García, J.J.; Martínez-Ballarín, E.; Millán-Plano, S.; Allué, J.L.; Albendea, C.; Fuentes, L.; Escanero, J.F. Effects of Trace Elements on Membrane Fluidity. J. Trace Elem. Med. Biol. 2005, 19, 19–22. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, J.; Zhou, B.; Li, P.; Hu, X.; Zhu, Z.; Tan, Y.; Chang, C.; Lü, J.; Song, B. Anomalous Behavior of Membrane Fluidity Caused by Copper-copper Bond Coupled Phospholipids. Sci. Rep. 2018, 8, 14093–15002. [Google Scholar] [CrossRef]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid Ion Channels: A Common Structural Link for Protein-misfolding Disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-Amyloid Peptides with Cholesterol: Mechanistic Insights into Amyloid Pore Formation. Biochemistry 2014, 53, 4489–4502. [Google Scholar] [CrossRef]
- Maynard, C.J.; Bush, A.I.; Masters, C.L.; Cappai, R.; Lin, Q.X. Metals and amyloid-β in Alzheimer’s disease. Int. J. Exp. Path. 2005, 86, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Hartter, D.E.; Barnea, A. Evidence for Release of Copper in the Brain: Depolarization-induced Release of Newly Taken-up 67Copper. Synapse 1988, 2, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, N.; Herms, J. Cellular Prion Protein Function in Copper Homeostasis and Redox Signalling at the Synapse. J. Neurochem. 2003, 86, 538–544. [Google Scholar] [CrossRef]
- Wild, K.; August, A.; Pietrzik, C.U.; Kins, S. Structure and Synaptic Function of Metal Binding to the Amyloid Precursor Protein and its Proteolytic Fragments. Front. Mol. Neurosci. 2017, 10, 21–32. [Google Scholar] [CrossRef]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable Intracellular Free Copper: The Requirement of a Copper Chaperone for Superoxide Dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef]
- Kepp, K.P. Alzheimer’s Disease: How Metal Ions Define β-amyloid Function. Coord. Chem. Rev. 2017, 351, 127–159. [Google Scholar] [CrossRef]
- Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.; Masters, C.L.; Beyreuther, K. The Amyloid Precursor Protein of Alzheimer’s Disease in the Reduction of Copper(II) to Copper(I). Science 1996, 271, 1406–1409. [Google Scholar] [CrossRef]
- Strausak, D.; Mercer, J.F.B.; Dieter, H.H.; Stremmel, W.; Multhaup, G. Copper in Disorders with Neurological Symptoms: Alzheimer’s, Menkes, and Wilson Diseases. Brain Res. Bull. 2001, 55, 175–185. [Google Scholar] [CrossRef]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper Homeostasis and Neurodegenerative Disorders (Alzheimer’s, Prion, and Parkinson’s Diseases and Amyotrophic Lateral Sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, C.M.; Chang, C.J. Copper Signaling in the Brain and Beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef]
- Opazo, C.M.; Greenough, M.A.; Bush, A.I. Copper: From neurotransmission to neuroproteostasis. Front. Aging Neurosci. 2014, 6, 143. [Google Scholar] [CrossRef]
- Marrink, S.; Corradi, V.; Souza, P.; Ingólfsson, H.; Tieleman, D.; Sansom, M. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Bevan, D.R. A Comparative Molecular Dynamics Analysis of the Amyloid β-peptide in a Lipid Bilayer. Arch. Biochem. Biophys. 2008, 470, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, C.; Klimov, D.K. Alzheimer’s Aβ10-40 Peptide Binds and Penetrates DMPC Bilayer: An Isobaric-Isothermal Replica Exchange Molecular Dynamics Study. J. Phys. Chem. B 2014, 118, 2638–2648. [Google Scholar] [CrossRef]
- Friedman, R.; Pellarin, R.; Caflisch, A. Amyloid Aggregation on Lipid Bilayers and Its Impact on Membrane Permeability. J. Mol. Biol. 2009, 387, 407–415. [Google Scholar] [CrossRef]
- Liu, L.; Hyeon, C. Contact Statistics Highlight Distinct Organizing Principles of Proteins and RNA. Biophys. J. 2016, 110, 2320–2327. [Google Scholar] [CrossRef]
- Tofoleanu, F.; Buchete, N.V. Molecular Interactions of Alzheimer’s Aβ Protofilaments with Lipid Membranes. J. Mol. Biol. 2012, 421, 572–586. [Google Scholar] [CrossRef]
- Poojari, C.; Kukol, A.; Strödel, B. How the Amyloid-beta Peptide and Membranes Affect each Other: An Extensive Simulation Study. Biochim. Biophys. Acta Biomembr. 2013, 1828, 327–339. [Google Scholar] [CrossRef]
- Tofoleanu, F.; Brooks, B.R.; Buchete, N.V. Modulation of Alzheimer’s Aβ Protofilament-Membrane Interactions by Lipid Headgroups. ACS Chem. Neurosci. 2015, 6, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Bevan, D. Molecular Dynamics Simulations of Amyloid β-Peptide(1–42): Tetramer Formation and Membrane Interactions. Biophys. J. 2016, 111, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Press-Sandler, O.; Miller, Y. Molecular Mechanisms of Membrane-associated Amyloid Aggregation: Computational Perspective and Challenges. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1889–1905. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R. Membrane–Ion Interactions. J. Membr. Biol. 2018, 251, 453–460. [Google Scholar] [CrossRef]
- Javanainen, M.; Melcrová, A.; Magarkar, A.; Jurkiewicz, P.; Hof, M.; Jungwirth, P.; Martinez-Seara, H. Two Cations, Two Mechanisms: Interactions of Sodium and Calcium with Zwitterionic Lipid Membranes. Chem. Commun. 2017, 53, 5380–5383. [Google Scholar] [CrossRef]
- Bilkova, E.; Pleskot, R.; Rissanen, S.; Sun, S.; Czogalla, A.; Cwiklik, L.; Róg, T.; Vattulainen, I.; Cremer, P.S.; Jungwirth, P.; et al. Calcium Directly Regulates Phosphatidylinositol 4,5-Bisphosphate Headgroup Conformation and Recognition. J. Am. Chem. Soc. 2017, 139, 4019–4024. [Google Scholar] [CrossRef] [PubMed]
- Melcr, J.; Martinez-Seara, H.; Nencini, R.; Kolafa, J.; Jungwirth, P.; Ollila, O.H.S. Accurate Binding of Sodium and Calcium to a POPC Bilayer by Effective Inclusion of Electronic Polarization. J. Phys. Chem. B 2018, 122, 4546–4557. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Linh, H.Q.; Krupa, P.; La Penna, G.; Li, M.S. Amyloid beta Dodecamer Disrupts the Neuronal Membrane More Strongly than the Mature Fibril: Understanding the Role of Oligomers in Neurotoxicity. J. Phys. Chem. B 2022, 126, 3659–3672. [Google Scholar] [CrossRef]
- Li, Z.; Song, L.F.; Li, P.; Merz, K.M. Systematic Parametrization of Divalent Metal Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Theory Comput. 2020, 16, 4429–4442. [Google Scholar] [CrossRef]
- Banchelli, M.; Cascella, R.; D’Andrea, C.; Cabaj, L.; Osticioli, I.; Ciofini, D.; Li, M.S.; Skupień, K.; de Angelis, M.; Siano, S.; et al. Nanoscopic insights into the surface conformation of neurotoxic amyloid beta oligomers. RSC Adv. 2020, 10, 21907–21913. [Google Scholar] [CrossRef]
- Banchelli, M.; Cascella, R.; D’Andrea, C.; La Penna, G.; Li, M.S.; Machetti, F.; Matteini, P.; Pizzanelli, S. Probing the Structure of Toxic Amyloid-beta Oligomers with Electron Spin Resonance and Molecular Modeling. ACS Chem. Neurosci. 2021, 12, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD visual molecular dynamics. J. Molec. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, H.; Seelig, J. Interaction of metal ions with phosphatidylcholine bilayer membranes. Biochemistry 1981, 20, 7366–7373. [Google Scholar] [CrossRef] [PubMed]
- Catte, A.; Girych, M.; Javanainen, M.; Loison, C.; Melcr, J.; Miettinen, M.S.; Monticelli, L.; Määttä, J.; Oganesyan, V.S.; Ollila, O.H.S.; et al. Molecular electrometer and binding of cations to phospholipid bilayers. Phys. Chem. Chem. Phys. 2016, 18, 32560–32569. [Google Scholar] [CrossRef]
- Ferreira, T.M.; Coreta-Gomes, F.; Ollila, O.H.S.; Moreno, M.J.; Vaz, W.L.C.; Topgaard, D. Cholesterol and POPC segmental order parameters in lipid membranes: Solid state 1H–13C NMR and MD simulation studies. Phys. Chem. Chem. Phys. 2013, 15, 1976–1989. [Google Scholar] [CrossRef]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-beta(1–42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Abeta(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499. [Google Scholar] [CrossRef]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef]
- Helm, L.; Nicolle, G.M.; Merbach, A.E. Water and Proton Exchange Processes on Metal Ions. Adv. Inorg. Chem. 2005, 57, 327–380. [Google Scholar]
- Suwalsky, M.; Bolognin, S.; Zatta, P. Interaction between Alzheimer’s Amyloid-beta and Amyloid-beta-Metal Complexes with Cell Membranes. J. Alzheimer Dis. 2009, 17, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kurakin, S.A.; Ermakova, E.V.; Ivankov, A.I.; Smerdova, S.G.; Kuĉerka, N. The Effect of Divalent Ions on the Structure of Bilayers in the Dimyristoylphosphatidylcholine Vesicles. J. Surf. Investig. X-ray Synchr. Neutr. Tech. 2021, 15, 211–220. [Google Scholar] [CrossRef]
- Lau, T.L.; Ambroggio, E.E.; Tew, D.J.; Cappai, R.; Masters, C.L.; Fidelio, G.D.; Barnham, K.J.; Separovic, F. Amyloid-β Peptide Disruption of Lipid Membranes and the Effect of Metal Ions. J. Mol. Biol. 2006, 356, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Shalabaeva, V.; Cotte, M.; Burghammer, M.; Krahne, R.; Riekel, C.; Dante, S. Amyloid β Peptide Conformational Changes in the Presence of a Lipid Membrane System. Langmuir 2014, 30, 3191–3198. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.J.; Wesén, E.; Björkeroth, J.; Rocha, S.; Esbjörner, E.K. Lipid Membranes Catalyse the Fibril Formation of the Amyloid-β (1–42) Peptide through Lipid-fibril Interactions that Reinforce Secondary Pathways. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, E.A.; Fernandes, C.B.; Lundsgaard, J.E.; Brakti, I.; Lindorff-Larsen, K.; Langkilde, A.E.; Skriver, K.; Kragelund, B.B. Insight into Calcium-Binding Motifs of Intrinsically Disordered Proteins. Biomolecules 2021, 11, 1173. [Google Scholar] [CrossRef]
- Case, D.; Belfon, K.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Giambasu, G.; et al. AMBER 2020; University of California at San Fransisco: San Francisco, CA, USA, 2020. [Google Scholar]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.J. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Huy, P.D.Q.; Vuong, Q.V.; La Penna, G.; Faller, P.; Li, M.S. Impact of Cu(II) Binding on Structures and Dynamics of Abeta42 Monomer and Dimer: Molecular Dynamics Study. ACS Chem. Neurosci. 2016, 7, 1348–1363. [Google Scholar] [CrossRef] [PubMed]
- La Penna, G.; Li, M.S. Computational Models Explain how Copper Binding to Amyloid-β Peptide Oligomers Enhances Oxidative Pathways. Phys. Chem. Chem. Phys. 2019, 21, 8774–8784. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.Q.H.; Li, M.S.; La Penna, G. Copper Binding Induces Polymorphism in Amyloid-β Peptide: Results of Computational Models. J. Phys. Chem. B 2018, 122, 7243–7252. [Google Scholar] [CrossRef]
- Krupa, P.; Huy, P.D.Q.; Li, M.S. Properties of Monomeric Abeta42 Probed by Different Sampling Methods and Force Fields: Role of Energy Components. J. Chem. Phys. 2019, 151, 55101–55114. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.; Derreumaux, P.; Ji, B.; Xie, X.Q.; Nguyen, P.H.; Wang, J. Effects of All-Atom Molecular Mechanics Force Fields on Amyloid Peptide Assembly: The Case of Aβ(16-22) Dimer. J. Chem. Theory Comput. 2019, 15, 1440–1452. [Google Scholar] [CrossRef]
- Grossfield, A. WHAM: The Weighted Histogram Analysis Method; Version 2.0.9.1; Department of Biochemistry and Biophysics, University of Rochester Medical Center: Rochester, NY, USA, 2020. [Google Scholar]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Espresso: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Andreussi, O.; Dabo, I.; Marzari, N. Revised Self-Consistent Continuum Solvation in Electronic-Structure Calculations. J. Chem. Phys. 2012, 136, 064102. [Google Scholar] [CrossRef] [PubMed]
- Eisenhaber, F.; Lijnzaad, P.; Argos, P.; Sander, C.; Scharf, M. The Double Cubic Lattice Method: Efficient Approaches to Numerical Integration of Surface Area and Volume and to Dot Surface Contouring of Molecular Assemblies. J. Comput. Chem. 1995, 16, 273–284. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Composition |
|---|---|
| Na/DMPC | 1 Na+ + 9 K+ + 10 Cl− + 64 DMPC + 3191 H2O |
| M/DMPC | 1 M2+ + 8 K+ + 10 Cl− + 64 DMPC + 3191 H2O |
| 1 | 2 × [2 × A] + 48 Na+ + 36 Cl− + 25,013 H2PO |
| 2 | 2 × [2 × Cu-A] + 44 Na+ + 36 Cl− + 25,017 H2O |
| 3 | 2 × [Cu-A] + 44 Na+ + 36 Cl− + 25,017 H2O |
| 1/DMPC | 2 × [2 × A] + 63 K+ + 51 Cl− + 320 DMPC + 28,349 H2O |
| 2/DMPC | 2 × [2 × Cu-A] + 59 K+ + 51 Cl− + 320 DMPC + 28,353 H2O |
| 3/DMPC | 2 × [Cu-A] + 59 K+ + 51 Cl− + 320 DMPC + 28,353 H2O |
| System | Type of | Number of | Time Length of |
|---|---|---|---|
| Simulation | Trajectories | Each Trajectory | |
| M/DMPC | cMD | 1 | 1 s |
| M/DMPC | SMD | 1 | 1 s |
| M/DMPC | US | 1 | 0.7 s |
| 1 | cMD | 125 | 120 ns |
| 2 | cMD | 128 | 120 ns |
| 3 | cMD | 126 | 120 ns |
| 1/DMPC | cMD | 5 | 2.5 s |
| 2/DMPC | cMD | 5 | 2.5 s |
| 3/DMPC | cMD | 5 | 2.5 s |
| System | Exp. | Exp. | ||
|---|---|---|---|---|
| Na/DMPC | 0.159730 | −0.001753 | 0.05 | −0.05 |
| Mg/DMPC | 0.158999 | −0.002734 | - | - |
| Ca/DMPC | 0.154460 | −0.003522 | - | - |
| Fe/DMPC | 0.159847 | −0.002685 | - | - |
| Zn/DMPC | 0.158122 | −0.003374 | - | - |
| Cu/DMPC | 0.165765 | −0.002311 | - | - |
| M (Ion Type) | (kJ/mol) |
|---|---|
| Mg | 67.7 |
| Ca | −120.4 |
| Zn | 84.2 |
| Cu | −32.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krupa, P.; La Penna, G.; Li, M.S. Amyloid-β Tetramers and Divalent Cations at the Membrane/Water Interface: Simple Models Support a Functional Role. Int. J. Mol. Sci. 2023, 24, 12698. https://doi.org/10.3390/ijms241612698
Krupa P, La Penna G, Li MS. Amyloid-β Tetramers and Divalent Cations at the Membrane/Water Interface: Simple Models Support a Functional Role. International Journal of Molecular Sciences. 2023; 24(16):12698. https://doi.org/10.3390/ijms241612698
Chicago/Turabian StyleKrupa, Pawel, Giovanni La Penna, and Mai Suan Li. 2023. "Amyloid-β Tetramers and Divalent Cations at the Membrane/Water Interface: Simple Models Support a Functional Role" International Journal of Molecular Sciences 24, no. 16: 12698. https://doi.org/10.3390/ijms241612698
APA StyleKrupa, P., La Penna, G., & Li, M. S. (2023). Amyloid-β Tetramers and Divalent Cations at the Membrane/Water Interface: Simple Models Support a Functional Role. International Journal of Molecular Sciences, 24(16), 12698. https://doi.org/10.3390/ijms241612698