
On the Value of In Vitro Cell Systems for Mechanobiology from the Perspective of Yes-Associated Protein/Transcriptional Co-Activator with a PDZ-Binding Motif and Focal Adhesion Kinase and Their Involvement in Wound Healing, Cancer, Aging, and Senescence

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cell-Culture-Based In Vitro Test Systems for Biomedical Research
2.1. In Vitro Systems That Helped to Identify and Characterize YAP/TAZ and FAK as Mechanotransducers
2.1.1. YAP/TAZ
2.1.2. FAK
2.1.3. Short Summary
2.2. In Vitro Models as Valuable Tools to Elucidate the Role of YAP/TAZ and FAK in Aging and Senescence
2.2.1. YAP/TAZ
2.2.2. FAK
2.2.3. Short Summary
2.3. Cell Systems to Investigate YAP/TAZ and FAK in Wound Healing
2.3.1. YAP/TAZ
2.3.2. FAK
2.3.3. Short Summary
2.4. YAP/TAZ and FAK in Cancer from the In Vitro Point of View Using Different Cell Systems
2.4.1. YAP/TAZ
2.4.2. FAK
2.4.3. Short Summary
2.5. Future Prospects Regarding YAP/TAZ and FAK in Cancer Diagnosis and Therapy
2.5.1. YAP/TAZ
2.5.2. FAK
2.5.3. Summary
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AJs | Adherens junctions |
| AKI | Ischemia-related acute kidney injury |
| ARF | Acute renal failure |
| cGAS-STING | Cyclic GMP–AMP synthase–stimulator of interferon genes |
| EC | Endothelial cell |
| EM | Electron microscopy |
| EMT | Epithelial–mesenchymal transition |
| ERK 1/2 | Extracellular-signal-regulated kinase |
| FAK | Focal adhesion kinase |
| FAs | Focal adhesions |
| FERM | Band 4.1, Ezrin, Radixin, Moesin |
| HFpEF | Heart failure with a preserved ejection fraction |
| IFs | Intermediate filaments |
| IRS | Interference reflection microscopy |
| KO | Knockout |
| LUAD | Lung adenocarcinoma |
| MCP-1 | Monocyte chemoattractant chemokine-1 |
| MDR | Multidrug resistance |
| miR | microRNA |
| MMPs | Metalloproteinases |
| NES | Nuclear export signal |
| NLS | Nuclear localization signal |
| NTRs | Nuclear transport receptors |
| p-GP | Phosphoglycoprotein |
| PAR1 | Protease-activated receptor-1 |
| PD-L1 | Programmed cell death ligand |
| PDMS | Polydimethylsiloxane |
| PI3K | Phosphatidylinositol 3 kinase |
| PPP | Pentose phosphate pathway |
| SASP | Senescence-associated secretory phenotype |
| SCNAs | Somatic copy number alterations |
| TACE | Transarterial chemoembolization |
| TFs | Transcription factor |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| ZO | Zonula occludens |
References
- Jedrusik, N.; Steinberg, T.; Husari, A.; Volk, L.; Wang, X.; Finkenzeller, G.; Strassburg, S.; Tomakidi, P. Gelatin nonwovens-based epithelial morphogenesis involves a signaling axis comprising EGF-receptor, MAP kinases ERK 1/2, and beta1 integrin. J. Biomed. Mater. Res. A 2019, 107, 663–677. [Google Scholar] [CrossRef]
- Wang, X.; Steinberg, T.; Dieterle, M.P.; Ramminger, I.; Husari, A.; Tomakidi, P. FAK Shutdown: Consequences on Epithelial Morphogenesis and Biomarker Expression Involving an Innovative Biomaterial for Tissue Regeneration. Int. J. Mol. Sci. 2021, 22, 9774. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Mussig, E.; Steinberg, T.; Schulz, S.; Spatz, J.P.; Ulmer, J.; Grabe, N.; Kohl, A.; Komposch, G.; Tomakidi, P. Connective-Tissue Fibroblasts Established on Micropillar Interfaces are Pivotal for Epithelial-Tissue Morphogenesis. Adv. Funct. Mater. 2008, 18, 2919–2929. [Google Scholar] [CrossRef]
- Walker, M.; Pringle, E.W.; Ciccone, G.; Tassieri, M.; Gourdon, D.; Cantini, M. Mind the viscous modulus: The mechanotransductive response to the viscous nature of isoelastic matrices regulates stem cell chondrogenesis. bioRxiv 2023. bioRxiv:2023.03.06.530938. [Google Scholar] [CrossRef]
- Husari, A.; Hulter-Hassler, D.; Steinberg, T.; Schulz, S.D.; Tomakidi, P. Disruption of adherens junction and alterations in YAP-related proliferation behavior as part of the underlying cell transformation process of alcohol-induced oral carcinogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 209–219. [Google Scholar] [CrossRef]
- Bennett, M.; Cantini, M.; Reboud, J.; Cooper, J.M.; Roca-Cusachs, P.; Salmeron-Sanchez, M. Molecular clutch drives cell response to surface viscosity. Proc. Natl. Acad. Sci. USA 2018, 115, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Chighizola, M.; Previdi, A.; Dini, T.; Piazzoni, C.; Lenardi, C.; Milani, P.; Schulte, C.; Podesta, A. Adhesion force spectroscopy with nanostructured colloidal probes reveals nanotopography-dependent early mechanotransductive interactions at the cell membrane level. Nanoscale 2020, 12, 14708–14723. [Google Scholar] [CrossRef]
- Oria, R.; Wiegand, T.; Escribano, J.; Elosegui-Artola, A.; Uriarte, J.J.; Moreno-Pulido, C.; Platzman, I.; Delcanale, P.; Albertazzi, L.; Navajas, D.; et al. Force loading explains spatial sensing of ligands by cells. Nature 2017, 552, 219–224. [Google Scholar] [CrossRef]
- Swaminathan, V.; Waterman, C.M. The molecular clutch model for mechanotransduction evolves. Nat. Cell Biol. 2016, 18, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Saraswathibhatla, A.; Indana, D.; Chaudhuri, O. Cell-extracellular matrix mechanotransduction in 3D. Nat. Rev. Mol. Cell Biol. 2023, 24, 495–516. [Google Scholar] [CrossRef]
- Teo, B.K.K.; Wong, S.T.; Lim, C.K.; Kung, T.Y.S.; Yap, C.H.; Ramagopal, Y.; Romer, L.H.; Yim, E.K.F. Nanotopography Modulates Mechanotransduction of Stem Cells and Induces Differentiation through Focal Adhesion Kinase. ACS Nano 2013, 7, 4785–4798. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.E.; Hu, P.; Barker, T.H. Feeling Things Out: Bidirectional Signaling of the Cell-ECM Interface, Implications in the Mechanobiology of Cell Spreading, Migration, Proliferation, and Differentiation. Adv. Healthc. Mater. 2020, 9, e1901445. [Google Scholar] [CrossRef] [PubMed]
- Di Russo, J.; Young, J.L.; Wegner, J.W.; Steins, T.; Kessler, H.; Spatz, J.P. Integrin alpha5beta1 nano-presentation regulates collective keratinocyte migration independent of substrate rigidity. Elife 2021, 10, e69861. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K. Focal adhesions: A personal perspective on a half century of progress. FEBS J. 2017, 284, 3355–3361. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, A.; Duggan, K.; Buck, C.; Beckerle, M.C.; Burridge, K. Interaction of plasma membrane fibronectin receptor with talin—A transmembrane linkage. Nature 1986, 320, 531–533. [Google Scholar] [CrossRef]
- Golji, J.; Lam, J.; Mofrad, M.R. Vinculin activation is necessary for complete talin binding. Biophys. J. 2011, 100, 332–340. [Google Scholar] [CrossRef]
- Januszyk, M.; Kwon, S.H.; Wong, V.W.; Padmanabhan, J.; Maan, Z.N.; Whittam, A.J.; Major, M.R.; Gurtner, G.C. The Role of Focal Adhesion Kinase in Keratinocyte Fibrogenic Gene Expression. Int. J. Mol. Sci. 2017, 18, 1915. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, N.; Sarpal, R.; Wood, M.N.; Barrick, S.K.; Nishikawa, T.; Hayashi, H.; Kobb, A.B.; Flozak, A.S.; Yemelyanov, A.; Fernandez-Gonzalez, R.; et al. Force-dependent allostery of the alpha-catenin actin-binding domain controls adherens junction dynamics and functions. Nat. Commun. 2018, 9, 5121. [Google Scholar] [CrossRef] [PubMed]
- Amit, C.; Padmanabhan, P.; Narayanan, J. Deciphering the mechanoresponsive role of beta-catenin in keratoconus epithelium. Sci. Rep. 2020, 10, 21382. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–518. [Google Scholar] [CrossRef] [PubMed]
- Nava, M.M.; Miroshnikova, Y.A.; Biggs, L.C.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; Garcia Arcos, J.M.; et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020, 181, 800–817.e22. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Bork, P.; Einbond, A.; Kastury, K.; Druck, T.; Negrini, M.; Huebner, K.; Lehman, D. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 1995, 270, 14733–14741. [Google Scholar] [CrossRef]
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. TAZ: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef]
- Meli, V.S.; Atcha, H.; Veerasubramanian, P.K.; Nagalla, R.R.; Luu, T.U.; Chen, E.Y.; Guerrero-Juarez, C.F.; Yamaga, K.; Pandori, W.; Hsieh, J.Y.; et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci. Adv. 2020, 6, abb8471. [Google Scholar] [CrossRef]
- Walker, M.; Luo, J.; Pringle, E.W.; Cantini, M. ChondroGELesis: Hydrogels to harness the chondrogenic potential of stem cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 121, 111822. [Google Scholar] [CrossRef]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L., 3rd; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.L. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef]
- LeBlanc, L.; Ramirez, N.; Kim, J. Context-dependent roles of YAP/TAZ in stem cell fates and cancer. Cell. Mol. Life Sci. 2021, 78, 4201–4219. [Google Scholar] [CrossRef]
- Garcia-Garcia, M.; Sanchez-Perales, S.; Jarabo, P.; Calvo, E.; Huyton, T.; Fu, L.; Ng, S.C.; Sotodosos-Alonso, L.; Vazquez, J.; Casas-Tinto, S.; et al. Mechanical control of nuclear import by Importin-7 is regulated by its dominant cargo YAP. Nat. Commun. 2022, 13, 1174. [Google Scholar] [CrossRef]
- Hu, X.; Kan, H.; Boye, A.; Jiang, Y.; Wu, C.; Yang, Y. Mitogen-activated protein kinase inhibitors reduce the nuclear accumulation of phosphorylated Smads by inhibiting Imp 7 or Imp 8 in HepG2 cells. Oncol. Lett. 2018, 15, 4867–4872. [Google Scholar] [CrossRef]
- Mana-Capelli, S.; McCollum, D. Angiomotins stimulate LATS kinase autophosphorylation and act as scaffolds that promote Hippo signaling. J. Biol. Chem. 2018, 293, 18230–18241. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef]
- Kim, J.; Kwon, H.; Shin, Y.K.; Song, G.; Lee, T.; Kim, Y.; Jeong, W.; Lee, U.; Zhang, X.; Nam, G.; et al. MAML1/2 promote YAP/TAZ nuclear localization and tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 13529–13540. [Google Scholar] [CrossRef]
- Lehal, R.; Zaric, J.; Vigolo, M.; Urech, C.; Frismantas, V.; Zangger, N.; Cao, L.; Berger, A.; Chicote, I.; Loubery, S.; et al. Pharmacological disruption of the Notch transcription factor complex. Proc. Natl. Acad. Sci. USA 2020, 117, 16292–16301. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Kofler, M.; Speight, P.; Little, D.; Di Ciano-Oliveira, C.; Szaszi, K.; Kapus, A. Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat. Commun. 2018, 9, 4966. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.R.; Nestor-Bergmann, A.; Liang, X.; Gupta, S.; Duszyc, K.; Gauquelin, E.; Gomez, G.A.; Budnar, S.; Marcq, P.; Jensen, O.E.; et al. A Mechanosensitive RhoA Pathway that Protects Epithelia against Acute Tensile Stress. Dev. Cell 2018, 47, 439–452.e436. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Campbell, I.D.; Critchley, D.R. Talins and kindlins: Partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.P.; Dunn, G.A. Cell to substratum contacts of chick fibroblasts and their relation to the microfilament system. A correlated interference-reflexion and high-voltage electron-microscope study. J. Cell Sci. 1978, 29, 197–212. [Google Scholar] [CrossRef]
- Guan, J.L. Role of focal adhesion kinase in integrin signaling. Int. J. Biochem. Cell Biol. 1997, 29, 1085–1096. [Google Scholar] [CrossRef]
- Hanks, S.K.; Polte, T.R. Signaling through focal adhesion kinase. Bioessays 1997, 19, 137–145. [Google Scholar] [CrossRef]
- Schaller, M.D. The focal adhesion kinase. J. Endocrinol. 1996, 150, 1–7. [Google Scholar] [CrossRef][Green Version]
- Goñi, G.M.; Epifano, C.; Boskovic, J.; Camacho-Artacho, M.; Zhou, J.; Bronowska, A.; Martin, M.T.; Eck, M.J.; Kremer, L.; Grater, F.; et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. USA 2014, 111, E3177–E3186. [Google Scholar] [CrossRef]
- Chuang, H.H.; Zhen, Y.Y.; Tsai, Y.C.; Chuang, C.H.; Hsiao, M.; Huang, M.S.; Yang, C.J. FAK in Cancer: From Mechanisms to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 1726. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Salasznyk, R.M.; Klees, R.F.; Boskey, A.; Plopper, G.E. Activation of FAK is necessary for the osteogenic differentiation of human mesenchymal stem cells on laminin-5. J. Cell. Biochem. 2007, 100, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.S.; Song, Y.; Hua, K.Q.; Gao, S.J. Involvement of FAK-ERK2 signaling pathway in CKAP2-induced proliferation and motility in cervical carcinoma cell lines. Sci. Rep. 2017, 7, 2117. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Broome, M.A.; Hunter, T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: Involvement of the Grb2, p130cas, and Nck adaptor proteins. Mol. Cell. Biol. 1997, 17, 1702–1713. [Google Scholar] [CrossRef]
- Deramaudt, T.B.; Dujardin, D.; Hamadi, A.; Noulet, F.; Kolli, K.; De Mey, J.; Takeda, K.; Ronde, P. FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol. Biol. Cell 2011, 22, 964–975. [Google Scholar] [CrossRef]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef]
- Ossovskaya, V.; Lim, S.T.; Ota, N.; Schlaepfer, D.D.; Ilic, D. FAK nuclear export signal sequences. FEBS Lett. 2008, 582, 2402–2406. [Google Scholar] [CrossRef]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef]
- La Cour, T.; Gupta, R.; Rapacki, K.; Skriver, K.; Poulsen, F.M.; Brunak, S. NESbase version 1.0: A database of nuclear export signals. Nucleic Acids Res. 2003, 31, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.C.; Pereira, A.H.M.; Ambrosio, A.L.B.; Consonni, S.R.; Rocha de Oliveira, R.; Bajgelman, M.C.; Dias, S.M.G.; Franchini, K.G. FAK Forms a Complex with MEF2 to Couple Biomechanical Signaling to Transcription in Cardiomyocytes. Structure 2016, 24, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Nadruz, W., Jr.; Corat, M.A.; Marin, T.M.; Guimaraes Pereira, G.A.; Franchini, K.G. Focal adhesion kinase mediates MEF2 and c-Jun activation by stretch: Role in the activation of the cardiac hypertrophic genetic program. Cardiovasc. Res. 2005, 68, 87–97. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wang, W.; Zhao, B. YAP/TAZ links mechanosensing to aging. Life Med. 2023, 2, lnac039. [Google Scholar] [CrossRef]
- Shin, E.Y.; Park, J.H.; You, S.T.; Lee, C.S.; Won, S.Y.; Park, J.J.; Kim, H.B.; Shim, J.; Soung, N.K.; Lee, O.J.; et al. Integrin-mediated adhesions in regulation of cellular senescence. Sci. Adv. 2020, 6, eaay3909. [Google Scholar] [CrossRef]
- Alza, L.; Nager, M.; Visa, A.; Canti, C.; Herreros, J. FAK Inhibition Induces Glioblastoma Cell Senescence-Like State through p62 and p27. Cancers 2020, 12, 1086. [Google Scholar] [CrossRef]
- Chuang, H.H.; Huang, M.S.; Zhen, Y.Y.; Chuang, C.H.; Lee, Y.R.; Hsiao, M.; Yang, C.J. FAK Executes Anti-Senescence via Regulating EZH2 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines 2022, 10, 1937. [Google Scholar] [CrossRef]
- Chuang, H.H.; Wang, P.H.; Niu, S.W.; Zhen, Y.Y.; Huang, M.S.; Hsiao, M.; Yang, C.J. Inhibition of FAK Signaling Elicits Lamin A/C-Associated Nuclear Deformity and Cellular Senescence. Front. Oncol. 2019, 9, 22. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
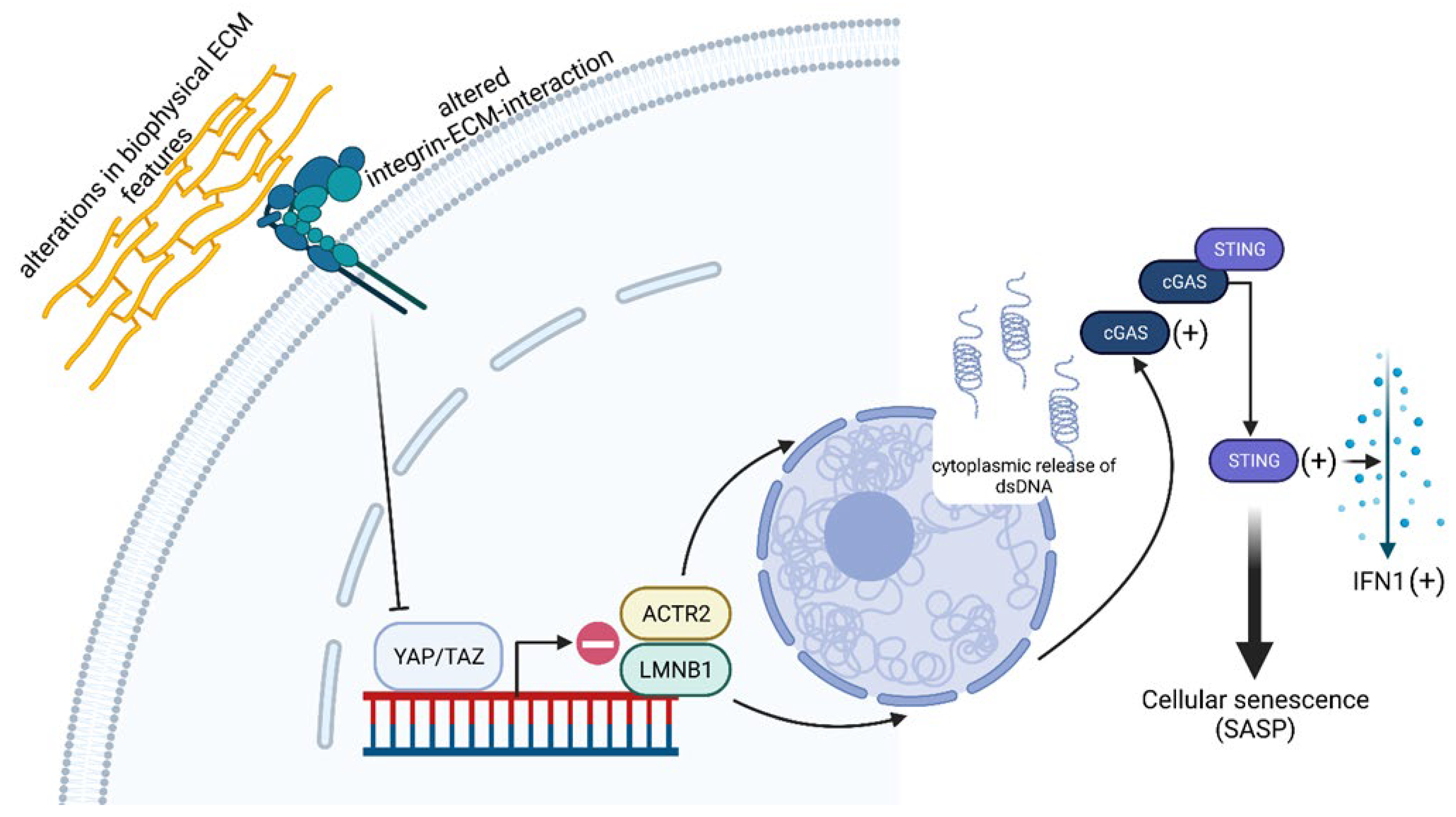
- Sladitschek-Martens, H.L.; Guarnieri, A.; Brumana, G.; Zanconato, F.; Battilana, G.; Xiccato, R.L.; Panciera, T.; Forcato, M.; Bicciato, S.; Guzzardo, V.; et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature 2022, 607, 790–798. [Google Scholar] [CrossRef]
- Santinon, G.; Brian, I.; Pocaterra, A.; Romani, P.; Franzolin, E.; Rampazzo, C.; Bicciato, S.; Dupont, S. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. EMBO J. 2018, 37, e97780. [Google Scholar] [CrossRef] [PubMed]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.A.; Tan, N.Y.; Thiam, C.H.; Khatoo, M.; MacAry, P.A.; Angeli, V.; Gasser, S.; Zhang, Y.L. cGAS-STING cytosolic DNA sensing pathway is suppressed by JAK2-STAT3 in tumor cells. Sci. Rep. 2021, 11, 7243. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, C.; Wang, Z. The induction of inflammation by the cGAS-STING pathway in human dental pulp cells: A laboratory investigation. Int. Endod. J. 2022, 55, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Tigges, J.; Krutmann, J.; Fritsche, E.; Haendeler, J.; Schaal, H.; Fischer, J.W.; Kalfalah, F.; Reinke, H.; Reifenberger, G.; Stuhler, K.; et al. The hallmarks of fibroblast ageing. Mech. Ageing Dev. 2014, 138, 26–44. [Google Scholar] [CrossRef] [PubMed]
- Heckenbach, I.; Mkrtchyan, G.V.; Ezra, M.B.; Bakula, D.; Madsen, J.S.; Nielsen, M.H.; Oro, D.; Osborne, B.; Covarrubias, A.J.; Idda, M.L.; et al. Nuclear morphology is a deep learning biomarker of cellular senescence. Nat. Aging 2022, 2, 742–755. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Smith, M.A.; Jensen, C.C.; Yoshigi, M.; Blankman, E.; Ullman, K.S.; Beckerle, M.C. Mechanical stress triggers nuclear remodeling and the formation of transmembrane actin nuclear lines with associated nuclear pore complexes. Mol. Biol. Cell 2020, 31, 1774–1787. [Google Scholar] [CrossRef]
- Kim, J.K.; Louhghalam, A.; Lee, G.; Schafer, B.W.; Wirtz, D.; Kim, D.H. Nuclear lamin A/C harnesses the perinuclear apical actin cables to protect nuclear morphology. Nat. Commun. 2017, 8, 2123. [Google Scholar] [CrossRef]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef]
- Hatch, E.M.; Hetzer, M.W. Nuclear envelope rupture is induced by actin-based nucleus confinement. J. Cell Biol. 2016, 215, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Wolf, K. Nuclear envelope rupture: Actin fibers are putting the squeeze on the nucleus. J. Cell Biol. 2016, 215, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, T.; Zhao, R.; Gao, D.; Cui, Y.; Wang, K.; Guo, Y. Caveolin-1 Inhibits Proliferation, Migration, and Invasion of Human Colorectal Cancer Cells by Suppressing Phosphorylation of Epidermal Growth Factor Receptor. Med. Sci. Monit. 2018, 24, 332–341. [Google Scholar] [CrossRef]
- Nishio, K.; Inoue, A. Senescence-associated alterations of cytoskeleton: Extraordinary production of vimentin that anchors cytoplasmic p53 in senescent human fibroblasts. Histochem. Cell Biol. 2005, 123, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Seawright, J.W.; Sreenivasappa, H.; Gibbs, H.C.; Padgham, S.; Shin, S.Y.; Chaponnier, C.; Yeh, A.T.; Trzeciakowski, J.P.; Woodman, C.R.; Trache, A. Vascular Smooth Muscle Contractile Function Declines with Age in Skeletal Muscle Feed Arteries. Front. Physiol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Sine, C.C.; Jeganathan, K.B.; Zhang, C.; Fierro Velasco, R.O.; Baker, D.J.; Li, H.; van Deursen, J.M. Senescent cells limit p53 activity via multiple mechanisms to remain viable. Nat. Commun. 2022, 13, 3722. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, L.; Ke, Y.; Lei, J.; Shen, S.; Shao, S.; Zhang, C.; Zhu, Z.; Dang, E.; Wang, G. EZH2-dependent epigenetic modulation of histone H3 lysine-27 contributes to psoriasis by promoting keratinocyte proliferation. Cell Death Dis. 2020, 11, 826. [Google Scholar] [CrossRef]
- Hiroyasu, S.; Stimac, G.P.; Hopkinson, S.B.; Jones, J.C.R. Loss of beta-PIX inhibits focal adhesion disassembly and promotes keratinocyte motility via myosin light chain activation. J. Cell Sci. 2017, 130, 2329–2343. [Google Scholar] [CrossRef]
- Alimirah, F.; Pulido, T.; Valdovinos, A.; Alptekin, S.; Chang, E.; Jones, E.; Diaz, D.A.; Flores, J.; Velarde, M.C.; Demaria, M.; et al. Cellular Senescence Promotes Skin Carcinogenesis through p38MAPK and p44/42MAPK Signaling. Cancer Res. 2020, 80, 3606–3619. [Google Scholar] [CrossRef]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
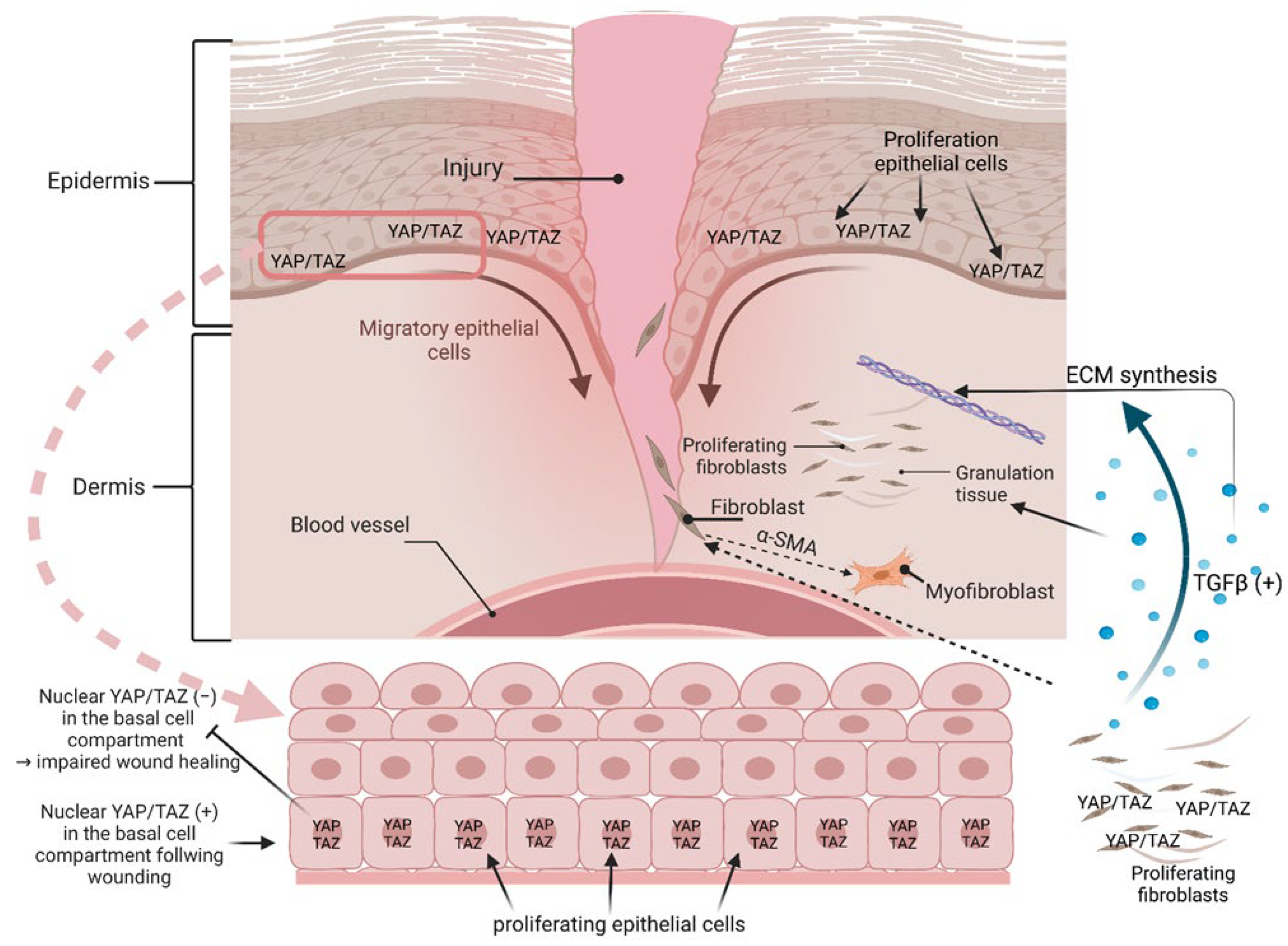
- Lee, M.J.; Byun, M.R.; Furutani-Seiki, M.; Hong, J.H.; Jung, H.S. YAP and TAZ regulate skin wound healing. J. Investig. Dermatol. 2014, 134, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Pandya, U.M.; Manzanares, M.A.; Tellechea, A.; Egbuta, C.; Daubriac, J.; Jimenez-Jaramillo, C.; Samra, F.; Fredston-Hermann, A.; Saadipour, K.; Gold, L.I. Calreticulin exploits TGF-beta for extracellular matrix induction engineering a tissue regenerative process. FASEB J. 2020, 34, 15849–15874. [Google Scholar] [CrossRef]
- Wang, L.; Qin, W.; Zhou, Y.; Chen, B.; Zhao, X.; Zhao, H.; Mi, E.; Mi, E.; Wang, Q.; Ning, J. Transforming growth factor beta plays an important role in enhancing wound healing by topical application of Povidone-iodine. Sci. Rep. 2017, 7, 991. [Google Scholar] [CrossRef]
- Jiang, X.; Hu, J.; Wu, Z.; Cafarello, S.T.; Di Matteo, M.; Shen, Y.; Dong, X.; Adler, H.; Mazzone, M.; Ruiz de Almodovar, C.; et al. Protein Phosphatase 2A Mediates YAP Activation in Endothelial Cells Upon VEGF Stimulation and Matrix Stiffness. Front. Cell Dev. Biol. 2021, 9, 675562. [Google Scholar] [CrossRef]
- Brewer, C.M.; Nelson, B.R.; Wakenight, P.; Collins, S.J.; Okamura, D.M.; Dong, X.R.; Mahoney, W.M., Jr.; McKenna, A.; Shendure, J.; Timms, A.; et al. Adaptations in Hippo-Yap signaling and myofibroblast fate underlie scar-free ear appendage wound healing in spiny mice. Dev. Cell 2021, 56, 2722–2740.e6. [Google Scholar] [CrossRef]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef]
- Kegelman, C.D.; Nijsure, M.P.; Moharrer, Y.; Pearson, H.B.; Dawahare, J.H.; Jordan, K.M.; Qin, L.; Boerckel, J.D. YAP and TAZ Promote Periosteal Osteoblast Precursor Expansion and Differentiation for Fracture Repair. J. Bone Miner. Res. 2021, 36, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Yui, S.; Azzolin, L.; Maimets, M.; Pedersen, M.T.; Fordham, R.P.; Hansen, S.L.; Larsen, H.L.; Guiu, J.; Alves, M.R.P.; Rundsten, C.F.; et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018, 22, 35–49.e7. [Google Scholar] [CrossRef]
- Ranes, M.; Zaleska, M.; Sakalas, S.; Knight, R.; Guettler, S. Reconstitution of the destruction complex defines roles of AXIN polymers and APC in beta-catenin capture, phosphorylation, and ubiquitylation. Mol. Cell 2021, 81, 3246–3261.e11. [Google Scholar] [CrossRef]
- Upadhyay, G. Emerging Role of Lymphocyte Antigen-6 Family of Genes in Cancer and Immune Cells. Front. Immunol. 2019, 10, 819. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; You, H.; Li, Y.; Xu, Y.; He, Q.; Harris, R.C. EGF Receptor-Dependent YAP Activation Is Important for Renal Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 2372–2385. [Google Scholar] [CrossRef]
- Murphy, J.M.; Jeong, K.; Rodriguez, Y.A.R.; Kim, J.H.; Ahn, E.E.; Lim, S.S. FAK and Pyk2 activity promote TNF-alpha and IL-1beta-mediated pro-inflammatory gene expression and vascular inflammation. Sci. Rep. 2019, 9, 7617. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 2011, 18, 148–152. [Google Scholar] [CrossRef]
- Liu, X.; Fang, S.; Liu, H.; Wang, X.; Dai, X.; Yin, Q.; Yun, T.; Wang, W.; Zhang, Y.; Liao, H.; et al. Role of human pulmonary fibroblast-derived MCP-1 in cell activation and migration in experimental silicosis. Toxicol. Appl. Pharmacol. 2015, 288, 152–160. [Google Scholar] [CrossRef]
- Chen, K.; Kwon, S.H.; Henn, D.; Kuehlmann, B.A.; Tevlin, R.; Bonham, C.A.; Griffin, M.; Trotsyuk, A.A.; Borrelli, M.R.; Noishiki, C.; et al. Disrupting biological sensors of force promotes tissue regeneration in large organisms. Nat. Commun. 2021, 12, 5256. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.G.; Cormier, A.; Ito, S.; Seed, R.I.; Bondesson, A.J.; Lou, J.; Marks, J.D.; Baron, J.L.; Cheng, Y.; Nishimura, S.L. Cryo-EM Reveals Integrin-Mediated TGF-beta Activation without Release from Latent TGF-beta. Cell 2020, 180, 490–501.e16. [Google Scholar] [CrossRef]
- Abe, M.; Harpel, J.G.; Metz, C.N.; Nunes, I.; Loskutoff, D.J.; Rifkin, D.B. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 1994, 216, 276–284. [Google Scholar] [CrossRef]
- Weng, Y.; Lieberthal, T.J.; Zhou, V.X.; Lopez-Ichikawa, M.; Armas-Phan, M.; Bond, T.K.; Yoshida, M.C.; Choi, W.T.; Chang, T.T. Liver epithelial focal adhesion kinase modulates fibrogenesis and hedgehog signaling. JCI Insight 2020, 5, e141217. [Google Scholar] [CrossRef]
- Patel, R.B.; Colangelo, L.A.; Reiner, A.P.; Gross, M.D.; Jacobs, D.R., Jr.; Launer, L.J.; Lima, J.A.C.; Lloyd-Jones, D.M.; Shah, S.J. Cellular Adhesion Molecules in Young Adulthood and Cardiac Function in Later Life. J. Am. Coll. Cardiol. 2020, 75, 2156–2165. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure with Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef] [PubMed]
- Dagouassat, M.; Suffee, N.; Hlawaty, H.; Haddad, O.; Charni, F.; Laguillier, C.; Vassy, R.; Martin, L.; Schischmanoff, P.O.; Gattegno, L.; et al. Monocyte chemoattractant protein-1 (MCP-1)/CCL2 secreted by hepatic myofibroblasts promotes migration and invasion of human hepatoma cells. Int. J. Cancer 2010, 126, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Chen, P.C.; Chang, T.M.; Hou, C.H. Monocyte Chemoattractant Protein-1 promotes cancer cell migration via c-Raf/MAPK/AP-1 pathway and MMP-9 production in osteosarcoma. J. Exp. Clin. Cancer Res. 2020, 39, 254. [Google Scholar] [CrossRef] [PubMed]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef]
- Agren, M.S.; Litman, T.; Eriksen, J.O.; Schjerling, P.; Bzorek, M.; Gjerdrum, L.M.R. Gene Expression Linked to Reepithelialization of Human Skin Wounds. Int. J. Mol. Sci. 2022, 23, 15746. [Google Scholar] [CrossRef] [PubMed]
- Caley, M.P.; Martins, V.L.; O’Toole, E.A. Metalloproteinases and Wound Healing. Adv. Wound Care 2015, 4, 225–234. [Google Scholar] [CrossRef]
- Koshikawa, N.; Giannelli, G.; Cirulli, V.; Miyazaki, K.; Quaranta, V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J. Cell Biol. 2000, 148, 615–624. [Google Scholar] [CrossRef]
- Yang, C.Q.; Li, W.; Li, S.Q.; Li, J.; Li, Y.W.; Kong, S.X.; Liu, R.M.; Wang, S.M.; Lv, W.M. MCP-1 stimulates MMP-9 expression via ERK 1/2 and p38 MAPK signaling pathways in human aortic smooth muscle cells. Cell. Physiol. Biochem. 2014, 34, 266–276. [Google Scholar] [CrossRef]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef]
- Khandoga, A.; Kessler, J.S.; Hanschen, M.; Khandoga, A.G.; Burggraf, D.; Reichel, C.; Hamann, G.F.; Enders, G.; Krombach, F. Matrix metalloproteinase-9 promotes neutrophil and T cell recruitment and migration in the postischemic liver. J. Leukoc. Biol. 2006, 79, 1295–1305. [Google Scholar] [CrossRef]
- Giambelluca, M.S.; Bertheau-Mailhot, G.; Laflamme, C.; Rollet-Labelle, E.; Servant, M.J.; Pouliot, M. TNF-alpha expression in neutrophils and its regulation by glycogen synthase kinase-3: A potentiating role for lithium. FASEB J. 2014, 28, 3679–3690. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Garg, R.K.; Sorkin, M.; Rustad, K.C.; Akaishi, S.; Levi, K.; Nelson, E.R.; Tran, M.; Rennert, R.; Liu, W.; et al. Loss of keratinocyte focal adhesion kinase stimulates dermal proteolysis through upregulation of MMP9 in wound healing. Ann. Surg. 2014, 260, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, B.; Lin, S.; Chang, P.; Tao, K. Apigenin inhibits growth and migration of fibroblasts by suppressing FAK signaling. Aging 2019, 11, 3668–3678. [Google Scholar] [CrossRef]
- Wang, Q.; More, S.K.; Vomhof-DeKrey, E.E.; Golovko, M.Y.; Basson, M.D. Small molecule FAK activator promotes human intestinal epithelial monolayer wound closure and mouse ulcer healing. Sci. Rep. 2019, 9, 14669. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Kwon, S.H.; Padmanabhan, J.; Duscher, D.; Trotsyuk, A.A.; Dong, Y.; Inayathullah, M.; Rajadas, J.; Gurtner, G.C. Controlled Delivery of a Focal Adhesion Kinase Inhibitor Results in Accelerated Wound Closure with Decreased Scar Formation. J. Investig. Dermatol. 2018, 138, 2452–2460. [Google Scholar] [CrossRef]
- Zhao, X.; Kong, Y.; Liang, B.; Xu, J.; Lin, Y.; Zhou, N.; Li, J.; Jiang, B.; Cheng, J.; Li, C.; et al. Mechanosensitive Piezo1 channels mediate renal fibrosis. JCI Insight 2022, 7, e152330. [Google Scholar] [CrossRef]
- Sun, S.; Wu, H.J.; Guan, J.L. Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci. Rep. 2018, 8, 2550. [Google Scholar] [CrossRef]
- Lan, B.; Zhang, L.; Yang, L.; Wu, J.; Li, N.; Pan, C.; Wang, X.; Zeng, L.; Yan, L.; Yang, C.; et al. Sustained delivery of MMP-9 siRNA via thermosensitive hydrogel accelerates diabetic wound healing. J. Nanobiotechnol. 2021, 19, 130. [Google Scholar] [CrossRef]
- Cunningham, R.; Hansen, C.G. The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin. Sci. 2022, 136, 197–222. [Google Scholar] [CrossRef]
- Rausch, V.; Hansen, C.G. The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends Cell Biol. 2020, 30, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, Y.; Ma, J. Interaction of non-coding RNAs and Hippo signaling: Implications for tumorigenesis. Cancer Lett. 2020, 493, 207–216. [Google Scholar] [CrossRef]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef]
- Yuan, M.; Tomlinson, V.; Lara, R.; Holliday, D.; Chelala, C.; Harada, T.; Gangeswaran, R.; Manson-Bishop, C.; Smith, P.; Danovi, S.A.; et al. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 2008, 15, 1752–1759. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, D.; Li, H.; Wang, L.; Tian, G.; Dong, Y. YAP overexpression promotes the epithelial-mesenchymal transition and chemoresistance in pancreatic cancer cells. Mol. Med. Rep. 2016, 13, 237–242. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. Increased expression of TAZ and associated upregulation of PD-L1 in cervical cancer. Cancer Cell Int. 2021, 21, 592. [Google Scholar] [CrossRef]
- Fuller, A.M.; Eisinger-Mathason, T.S.K. Context Matters: Response Heterogeneity to Collagen-Targeting Approaches in Desmoplastic Cancers. Cancers 2022, 14, 3132. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.; Zhou, W.; Chen, T.; Wu, Q.; Chutturghoon, V.K.; Lin, B.; Geng, L.; Yang, Z.; Zhou, L.; et al. YAP promotes multi-drug resistance and inhibits autophagy-related cell death in hepatocellular carcinoma via the RAC1-ROS-mTOR pathway. Cancer Cell Int. 2019, 19, 179. [Google Scholar] [CrossRef]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef]
- Panciera, T.; Citron, A.; Di Biagio, D.; Battilana, G.; Gandin, A.; Giulitti, S.; Forcato, M.; Bicciato, S.; Panzetta, V.; Fusco, S.; et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat. Mater. 2020, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Jin, L.; Chen, Y.; Xi, X.; Guo, Y. YAP promotes epithelial mesenchymal transition by upregulating Slug expression in human colorectal cancer cells. Int. J. Clin. Exp. Pathol. 2020, 13, 701–710. [Google Scholar] [PubMed]
- Liu, X.; Yu, J.; Song, S.; Yue, X.; Li, Q. Protease-activated receptor-1 (PAR-1): A promising molecular target for cancer. Oncotarget 2017, 8, 107334–107345. [Google Scholar] [CrossRef]
- Fujimoto, D.; Ueda, Y.; Hirono, Y.; Goi, T.; Yamaguchi, A. PAR1 participates in the ability of multidrug resistance and tumorigenesis by controlling Hippo-YAP pathway. Oncotarget 2015, 6, 34788–34799. [Google Scholar] [CrossRef]
- Song, S.; Ajani, J.A.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.R.; Xiao, L.; Hofstetter, W.L.; Weston, B.; et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef]
- Aldaz, P.; Otaegi-Ugartemendia, M.; Saenz-Antonanzas, A.; Garcia-Puga, M.; Moreno-Valladares, M.; Flores, J.M.; Gerovska, D.; Arauzo-Bravo, M.J.; Sampron, N.; Matheu, A.; et al. SOX9 promotes tumor progression through the axis BMI1-p21(CIP). Sci. Rep. 2020, 10, 357. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dai, X.; Cao, X.; Yan, H.; Ji, X.; Zhang, H.; Shen, S.; Si, Y.; Zhang, H.; Chen, J.; et al. PRDM4 mediates YAP-induced cell invasion by activating leukocyte-specific integrin beta2 expression. EMBO Rep. 2018, 19, e45180. [Google Scholar] [CrossRef]
- Pomella, S.; Cassandri, M.; Braghini, M.R.; Marampon, F.; Alisi, A.; Rota, R. New Insights on the Nuclear Functions and Targeting of FAK in Cancer. Int. J. Mol. Sci. 2022, 23, 1998. [Google Scholar] [CrossRef]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Cirillo, F.; Talia, M.; Muglia, L.; Gutkind, J.S.; Maggiolini, M.; Lappano, R. Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression. Cancers 2021, 13, 645. [Google Scholar] [CrossRef]
- Lietha, D. Forcing FAK into Transcriptional Activity. Structure 2016, 24, 1223–1225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef]
- Shiau, J.P.; Wu, C.C.; Chang, S.J.; Pan, M.R.; Liu, W.; Ou-Yang, F.; Chen, F.M.; Hou, M.F.; Shih, S.L.; Luo, C.W. FAK Regulates VEGFR2 Expression and Promotes Angiogenesis in Triple-Negative Breast Cancer. Biomedicines 2021, 9, 1789. [Google Scholar] [CrossRef] [PubMed]
- Pitulescu, M.E.; Schmidt, I.; Benedito, R.; Adams, R.H. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 2010, 5, 1518–1534. [Google Scholar] [CrossRef]
- Hwang-Bo, J.; Yoo, K.H.; Park, J.H.; Jeong, H.S.; Chung, I.S. Recombinant canstatin inhibits angiopoietin-1-induced angiogenesis and lymphangiogenesis. Int. J. Cancer 2012, 131, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Hwang-Bo, J.; Bae, M.G.; Park, J.H.; Chung, I.S. 3-O-Acetyloleanolic acid inhibits VEGF-A-induced lymphangiogenesis and lymph node metastasis in an oral cancer sentinel lymph node animal model. BMC Cancer 2018, 18, 714. [Google Scholar] [CrossRef]
- Pedrosa, A.R.; Bodrug, N.; Gomez-Escudero, J.; Carter, E.P.; Reynolds, L.E.; Georgiou, P.N.; Fernandez, I.; Lees, D.M.; Kostourou, V.; Alexopoulou, A.N.; et al. Tumor Angiogenesis Is Differentially Regulated by Phosphorylation of Endothelial Cell Focal Adhesion Kinase Tyrosines-397 and -861. Cancer Res. 2019, 79, 4371–4386. [Google Scholar] [CrossRef]
- Kurenova, E.; Xu, L.H.; Yang, X.; Baldwin, A.S., Jr.; Craven, R.J.; Hanks, S.K.; Liu, Z.G.; Cance, W.G. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 2004, 24, 4361–4371. [Google Scholar] [CrossRef]
- Fan, Z.; Duan, J.; Wang, L.; Xiao, S.; Li, L.; Yan, X.; Yao, W.; Wu, L.; Zhang, S.; Zhang, Y.; et al. PTK2 promotes cancer stem cell traits in hepatocellular carcinoma by activating Wnt/beta-catenin signaling. Cancer Lett. 2019, 450, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zhang, J.; Gelman, I.H.; Qu, J.; Hochwald, S.N. Phosphohistidine signaling promotes FAK-RB1 interaction and growth factor-independent proliferation of esophageal squamous cell carcinoma. Oncogene 2023, 42, 449–460. [Google Scholar] [CrossRef]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Li, Y.; Lin, S.; Wang, Z.; Xu, Y. Inhibitors Targeting YAP in Gastric Cancer: Current Status and Future Perspectives. Drug Des. Dev. Ther. 2021, 15, 2445–2456. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, X.; Feng, W.; Yu, Y.; Jeong, K.; Guo, W.; Lu, Y.; Mills, G.B. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 2016, 6, 27–37. [Google Scholar]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Mae, Y.; Kanda, T.; Sugihara, T.; Takata, T.; Kinoshita, H.; Sakaguchi, T.; Hasegawa, T.; Tarumoto, R.; Edano, M.; Kurumi, H.; et al. Verteporfin-photodynamic therapy is effective on gastric cancer cells. Mol. Clin. Oncol. 2020, 13, 10. [Google Scholar] [CrossRef]
- Song, S.; Xie, M.; Scott, A.W.; Jin, J.; Ma, L.; Dong, X.; Skinner, H.D.; Johnson, R.L.; Ding, S.; Ajani, J.A. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 443–454. [Google Scholar] [CrossRef]
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945. [Google Scholar] [CrossRef]
- Kishi, T.; Mayanagi, T.; Iwabuchi, S.; Akasaka, T.; Sobue, K. Myocardin-related transcription factor A (MRTF-A) activity-dependent cell adhesion is correlated to focal adhesion kinase (FAK) activity. Oncotarget 2016, 7, 72113–72130. [Google Scholar] [CrossRef]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef]
- Maziarz, M.; Federico, A.; Zhao, J.; Dujmusic, L.; Zhao, Z.; Monti, S.; Varelas, X.; Garcia-Marcos, M. Naturally occurring hotspot cancer mutations in Galpha(13) promote oncogenic signaling. J. Biol. Chem. 2020, 295, 16897–16904. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Z.; Wu, Y.; Wang, Y.; Wang, D.; Zhang, W.; Yuan, H.; Ye, J.; Song, X.; Yang, J.; et al. The Hippo effector TAZ promotes cancer stemness by transcriptional activation of SOX2 in head neck squamous cell carcinoma. Cell Death Dis. 2019, 10, 603. [Google Scholar] [CrossRef]
- Zhao, N.; Zhou, L.; Lu, Q.; Wang, S.; Sun, Y.; Ding, Y.; Liu, M.; He, H.; Lang, T. SOX2 maintains the stemness of retinoblastoma stem-like cells through Hippo/YAP signaling pathway. Exp. Eye Res. 2022, 214, 108887. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 1304–1317.e5. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Zhang, W.; Shams, A.; Mohammed, K.; Befeler, A.S.; Kang, N.; Lai, J. Yes-associated protein-1 may serve as a diagnostic marker and therapeutic target for residual/recurrent hepatocellular carcinoma post-transarterial chemoembolization. Liver Res. 2020, 4, 212–217. [Google Scholar] [CrossRef]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef]
- Xue, X.; Tian, X.; Zhang, C.; Miao, Y.; Wang, Y.; Peng, Y.; Qiu, S.; Wang, H.; Cui, J.; Cao, L.; et al. YAP ISGylation increases its stability and promotes its positive regulation on PPP by stimulating 6PGL transcription. Cell Death Discov. 2022, 8, 59. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, E.T.; Kim, Y.E.; Lee, M.K.; Kwon, K.M.; Kim, K.I.; Stamminger, T.; Ahn, J.H. Consecutive Inhibition of ISG15 Expression and ISGylation by Cytomegalovirus Regulators. PLoS Pathog. 2016, 12, e1005850. [Google Scholar] [CrossRef]
- Devaud, C.; Tilkin-Mariame, A.F.; Vignolle-Vidoni, A.; Souleres, P.; Denadai-Souza, A.; Rolland, C.; Duthoit, C.; Blanpied, C.; Chabot, S.; Bouille, P.; et al. FAK alternative splice mRNA variants expression pattern in colorectal cancer. Int. J. Cancer 2019, 145, 494–502. [Google Scholar] [CrossRef]
- Begum, A.; Ewachiw, T.; Jung, C.; Huang, A.; Norberg, K.J.; Marchionni, L.; McMillan, R.; Penchev, V.; Rajeshkumar, N.V.; Maitra, A.; et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0180181. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gomez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.J.; Liu, X.J.; Liu, Y.; Liu, W.B.; Li, Y.R.; Yu, G.X.; Tian, X.Y.; Zhang, Y.B.; Song, J.; Jin, C.Y.; et al. Drug Discovery Targeting Focal Adhesion Kinase (FAK) as a Promising Cancer Therapy. Molecules 2021, 26, 4250. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Spallarossa, A.; Tasso, B.; Russo, E.; Villa, C.; Brullo, C. The Development of FAK Inhibitors: A Five-Year Update. Int. J. Mol. Sci. 2022, 23, 6381. [Google Scholar] [CrossRef]
- Schultze, A.; Fiedler, W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 777–788. [Google Scholar] [CrossRef]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.P.; Alexopoulou, A.; Elia, G.; et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef]
- Mohanty, A.; Pharaon, R.R.; Nam, A.; Salgia, S.; Kulkarni, P.; Massarelli, E. FAK-targeted and combination therapies for the treatment of cancer: An overview of phase I and II clinical trials. Expert Opin. Investig. Drugs 2020, 29, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Zhang, W.; Zhao, G.; Chen, Z.; Dong, P.; Watari, H.; Narayanan, R.; Tillmanns, T.D.; Pfeffer, L.M.; Yue, J. FAK PROTAC Inhibits Ovarian Tumor Growth and Metastasis by Disrupting Kinase Dependent and Independent Pathways. Front. Oncol. 2022, 12, 851065. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Li, R.; Liu, M.; Yang, Z.; Li, J.; Gao, Y.; Tan, R. Proteolysis-Targeting Chimeras (PROTACs) in Cancer Therapy: Present and Future. Molecules 2022, 27, 8828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, H.Y.; Xi, X.X.; Liu, Y.J.; Xin, M.; Mao, S.; Zhang, J.J.; Lu, A.X.; Zhang, S.Q. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 189, 112061. [Google Scholar] [CrossRef]
- Aifuwa, I.; Kim, B.C.; Kamat, P.; Starich, B.; Agrawal, A.; Tanrioven, D.; Luperchio, T.R.; Valencia, A.M.J.; Perestrelo, T.; Reddy, K.; et al. Senescent stroma induces nuclear deformations in cancer cells via the inhibition of RhoA/ROCK/myosin II-based cytoskeletal tension. Proc. Natl. Acad. Sci. USA Nexus 2023, 2, pgac270. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinberg, T.; Dieterle, M.P.; Ramminger, I.; Klein, C.; Brossette, J.; Husari, A.; Tomakidi, P. On the Value of In Vitro Cell Systems for Mechanobiology from the Perspective of Yes-Associated Protein/Transcriptional Co-Activator with a PDZ-Binding Motif and Focal Adhesion Kinase and Their Involvement in Wound Healing, Cancer, Aging, and Senescence. Int. J. Mol. Sci. 2023, 24, 12677. https://doi.org/10.3390/ijms241612677
Steinberg T, Dieterle MP, Ramminger I, Klein C, Brossette J, Husari A, Tomakidi P. On the Value of In Vitro Cell Systems for Mechanobiology from the Perspective of Yes-Associated Protein/Transcriptional Co-Activator with a PDZ-Binding Motif and Focal Adhesion Kinase and Their Involvement in Wound Healing, Cancer, Aging, and Senescence. International Journal of Molecular Sciences. 2023; 24(16):12677. https://doi.org/10.3390/ijms241612677
Chicago/Turabian StyleSteinberg, Thorsten, Martin Philipp Dieterle, Imke Ramminger, Charlotte Klein, Julie Brossette, Ayman Husari, and Pascal Tomakidi. 2023. "On the Value of In Vitro Cell Systems for Mechanobiology from the Perspective of Yes-Associated Protein/Transcriptional Co-Activator with a PDZ-Binding Motif and Focal Adhesion Kinase and Their Involvement in Wound Healing, Cancer, Aging, and Senescence" International Journal of Molecular Sciences 24, no. 16: 12677. https://doi.org/10.3390/ijms241612677
APA StyleSteinberg, T., Dieterle, M. P., Ramminger, I., Klein, C., Brossette, J., Husari, A., & Tomakidi, P. (2023). On the Value of In Vitro Cell Systems for Mechanobiology from the Perspective of Yes-Associated Protein/Transcriptional Co-Activator with a PDZ-Binding Motif and Focal Adhesion Kinase and Their Involvement in Wound Healing, Cancer, Aging, and Senescence. International Journal of Molecular Sciences, 24(16), 12677. https://doi.org/10.3390/ijms241612677