
The Proteome Profile of Olfactory Ecto-Mesenchymal Stem Cells-Derived from Patients with Familial Alzheimer’s Disease Reveals New Insights for AD Study

Abstract
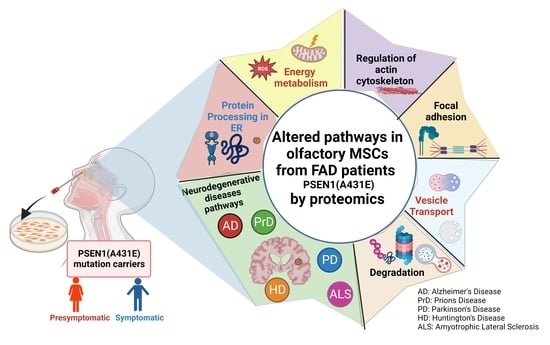
1. Introduction
2. Results
2.1. Protein Expression in Symptomatic and Presymptomatic PSEN1(A431E) Carriers (P1 and P2) and Controls (C1 and C2)
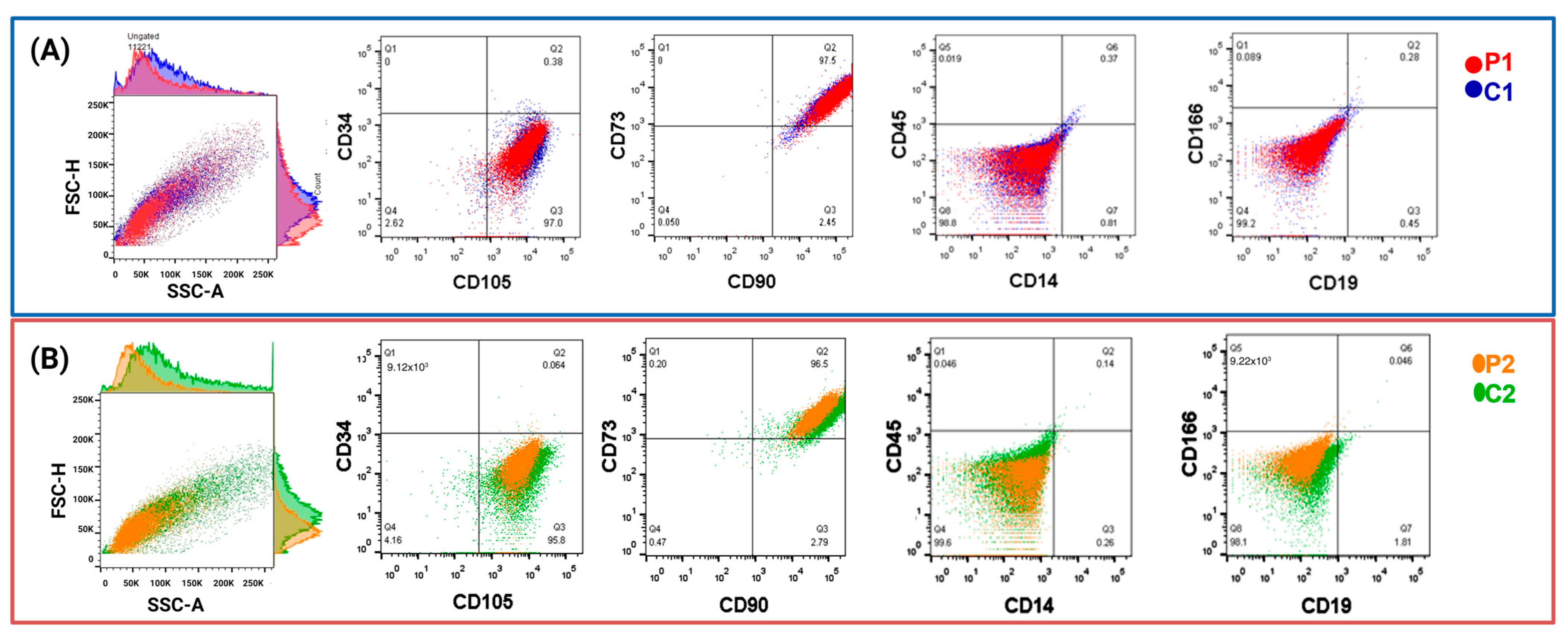
2.1.1. Isolation and Characterization of Olfactory MSCs
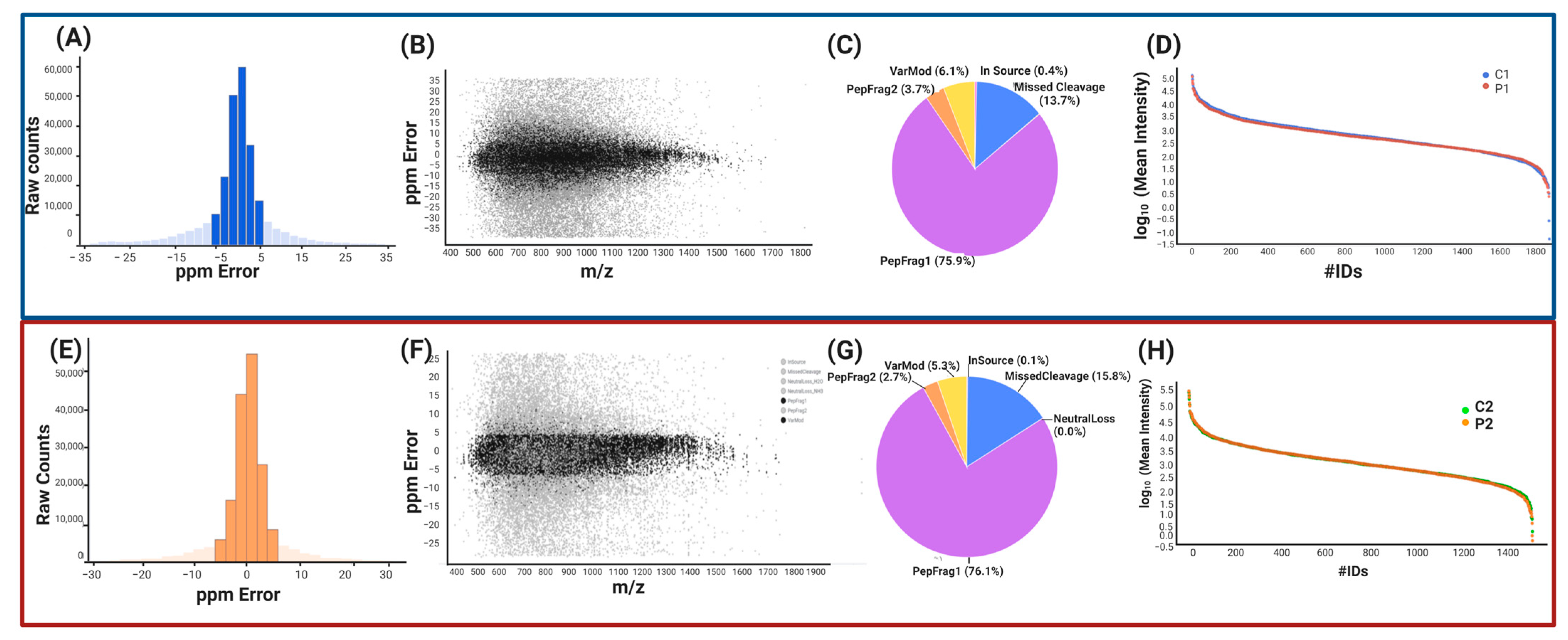
2.1.2. Label-Free UPLC-HDMSE Analysis for P1 vs. C1
2.1.3. Peptide and Protein Reliability and Confidence
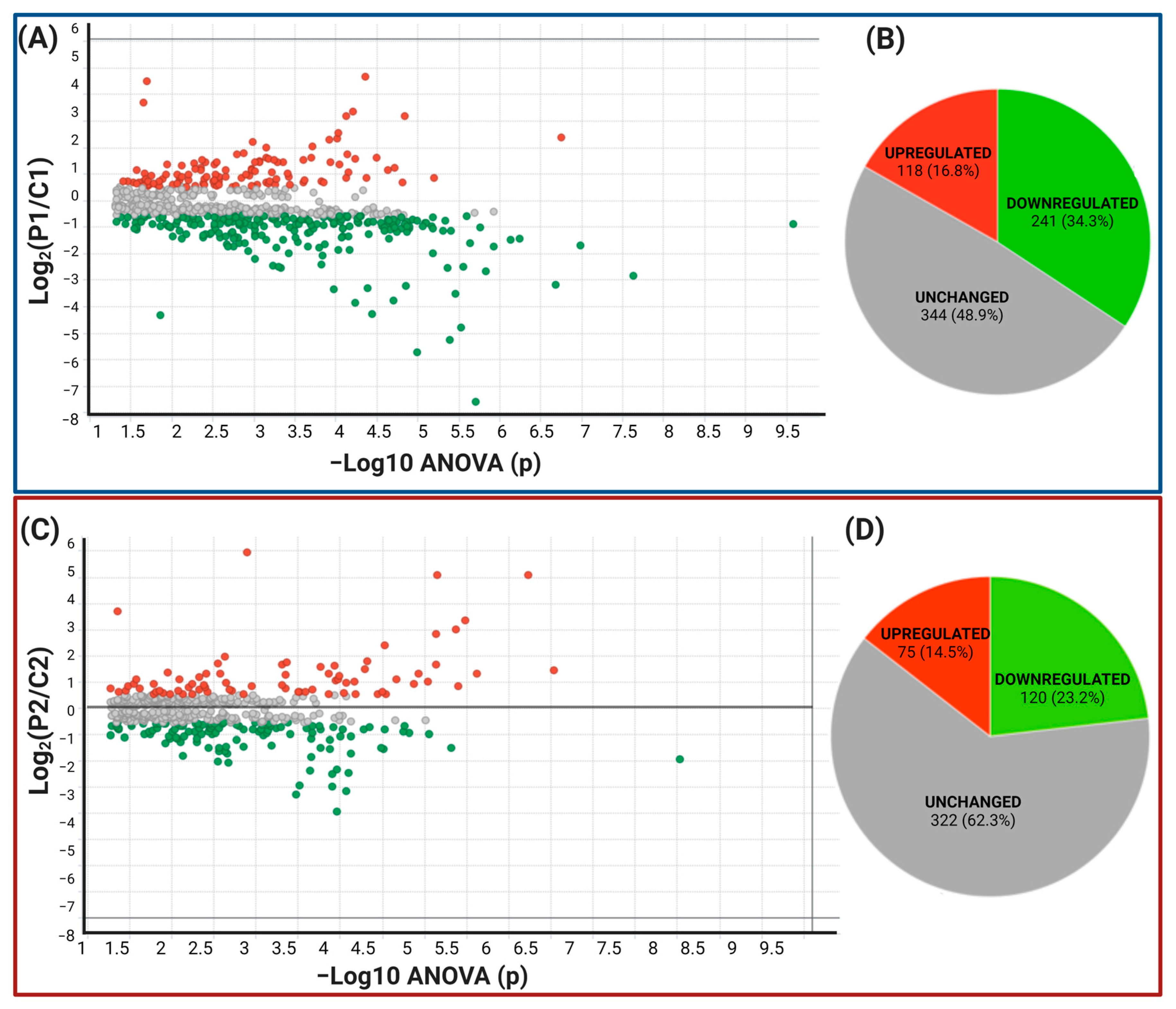
2.2. Data Filtering and Differentially Expressed Proteins in P1 and P2 MSCs
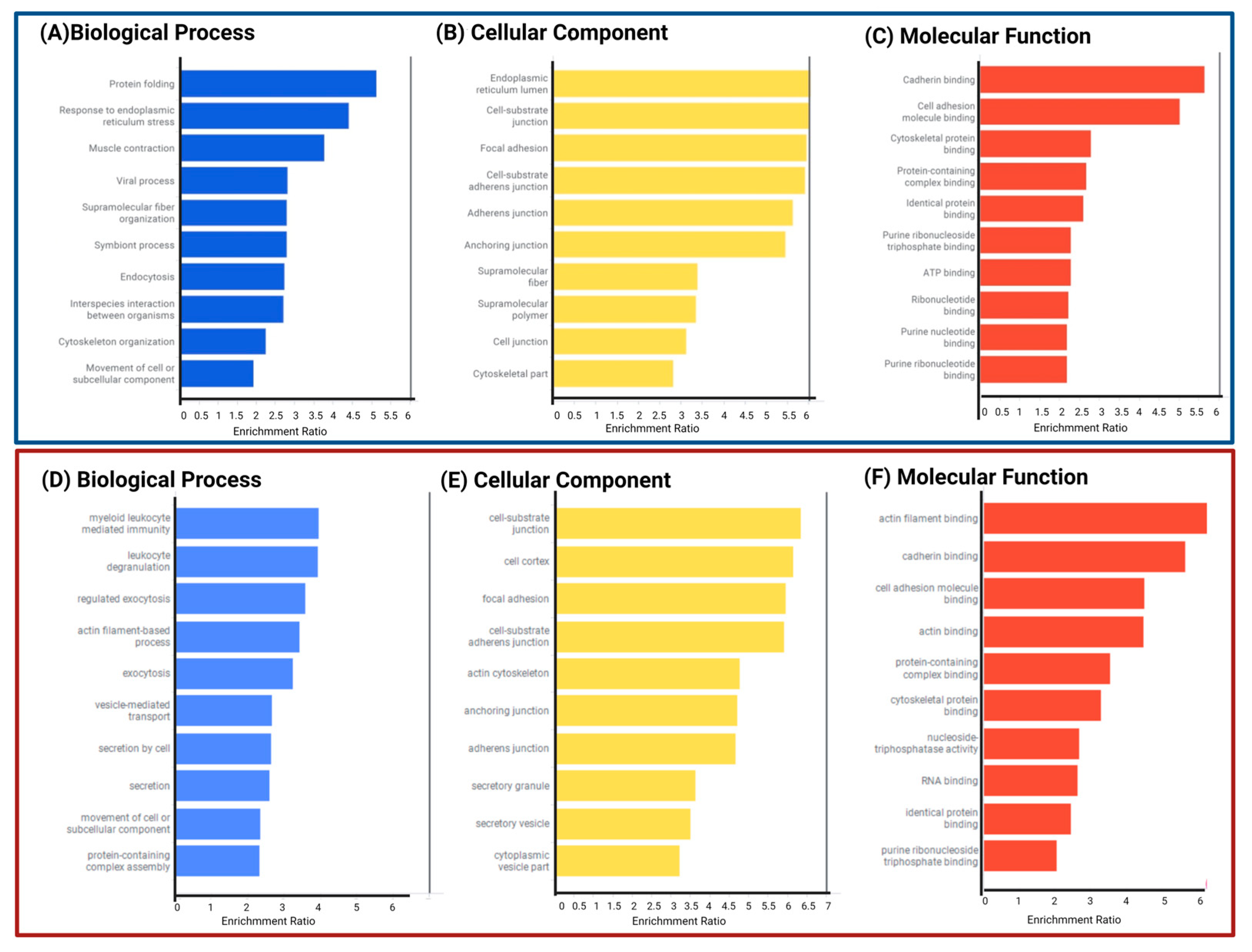
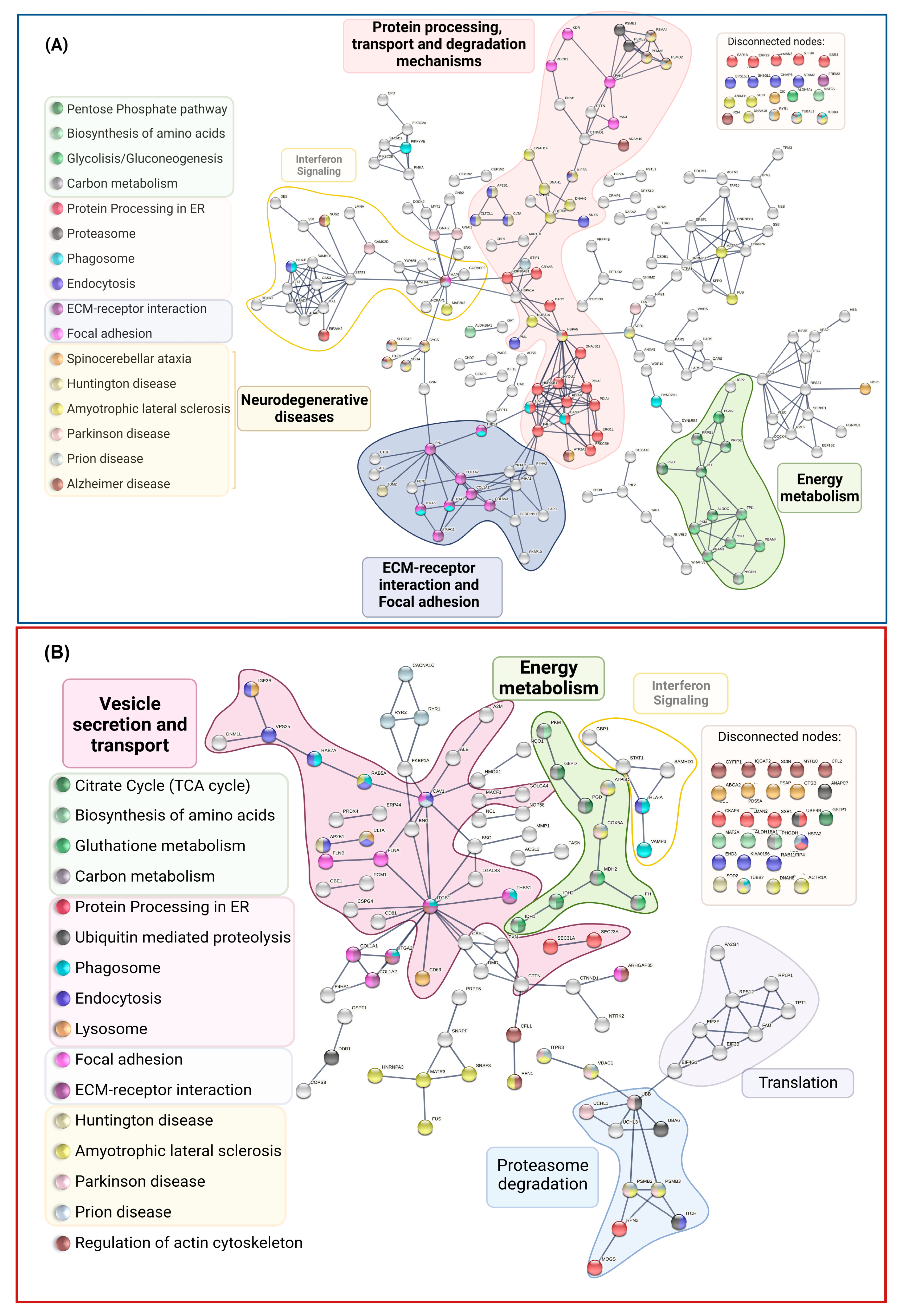
Functional and Biological Analysis of DEPs in P1/C1 and P2/C2 Analysis
2.3. Comparison of DEPs between PSEN(A431E) Mutation Carriers and with Other NDD and AD DEPs Previously Reported
Correlation between the Proteomes of Olfactory MSCs from PSEN1(A431E) Mutant Carriers with Their Clinical Histories
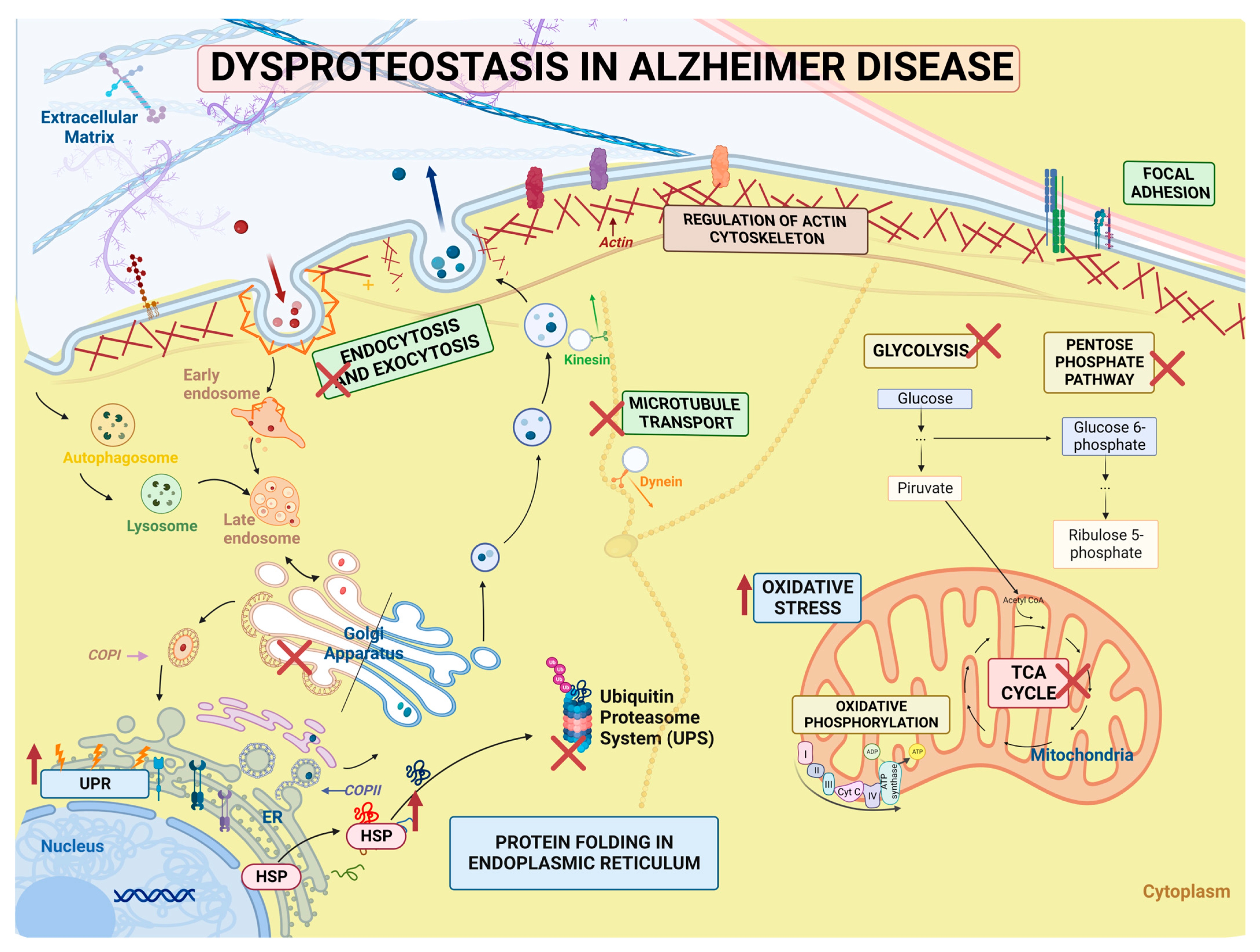
3. Discussion
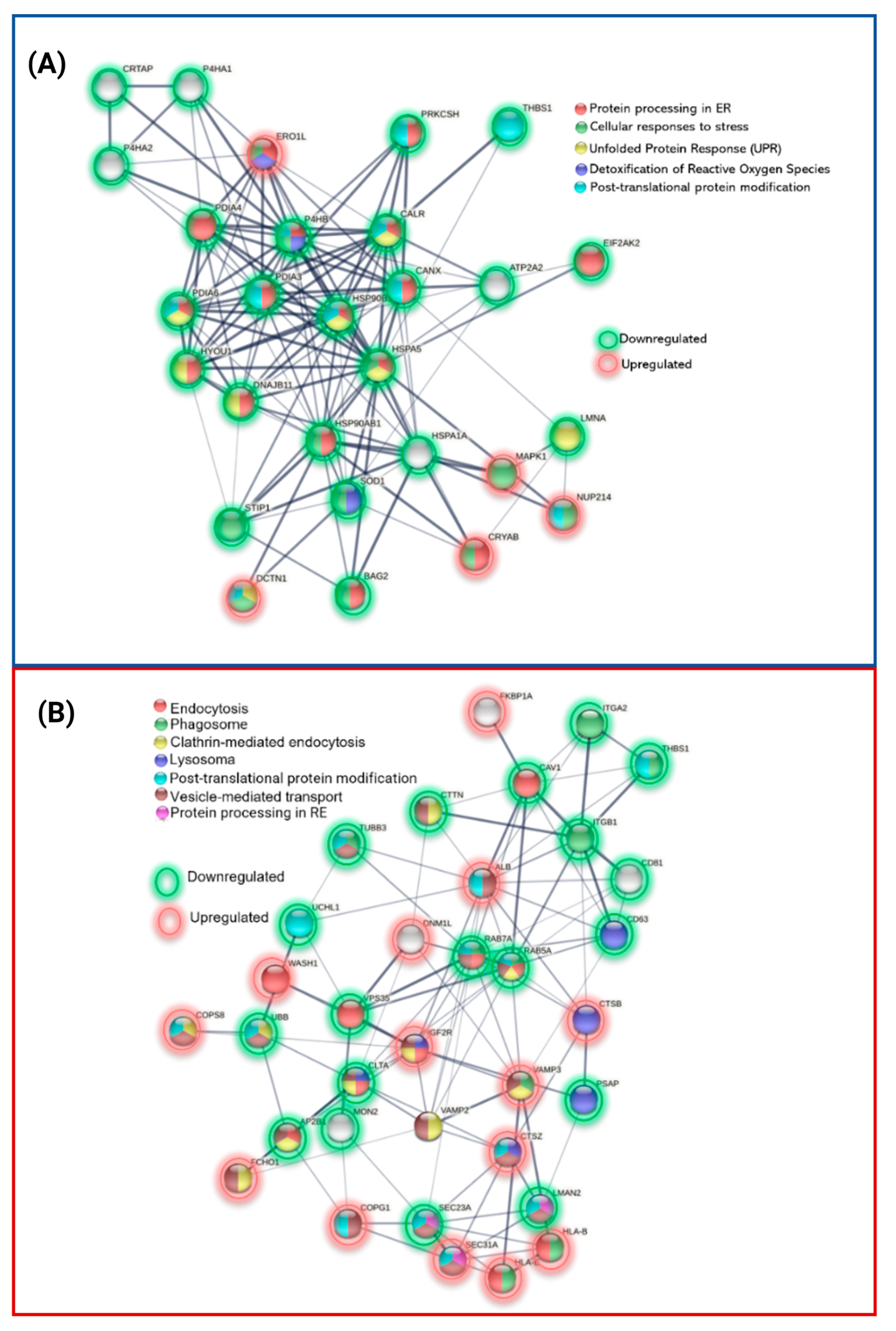
3.1. Exclusive Proteins of PSEN1(A431E) Mutation Carriers
3.2. Downregulated Proteins in PSEN1(A431E) Mutation Carriers
3.3. Upregulated Proteins in PSEN1(A431E) Mutation Carriers
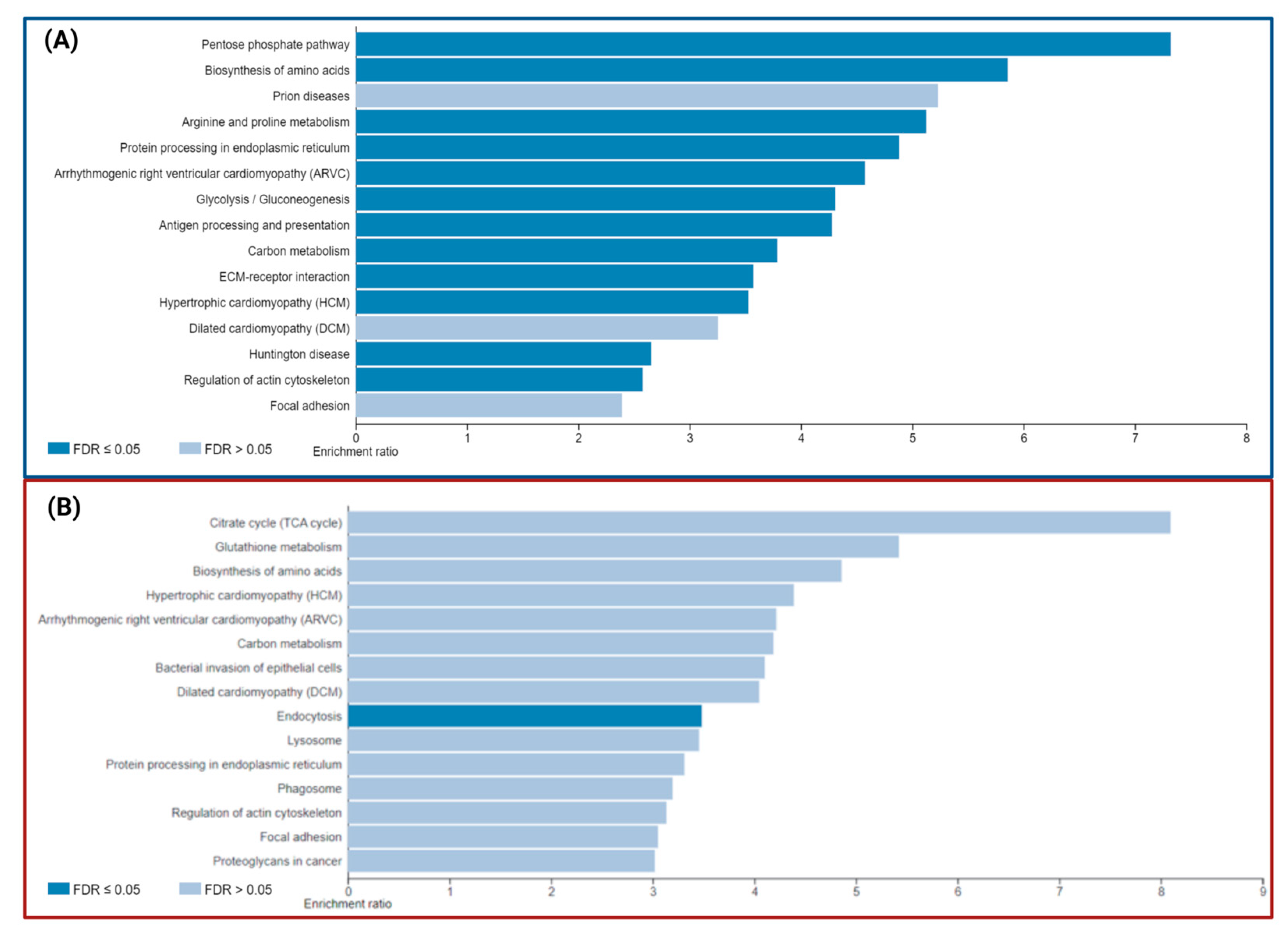
3.4. Signaling Pathways and Biological Processes Altered in PSEN1(A431E) Mutation Carriers
3.4.1. Protein Processing in Endoplasmic Reticulum
3.4.2. Regulation of Actin Cytoskeleton
3.4.3. Focal Adhesion and Extracellular Matrix–Receptor Interactions
3.4.4. Neurodegenerative Diseases Pathways
3.5. Comparison with AD Studies Previously Reported
4. Materials and Methods
4.1. Subjects
4.2. Sample Collection and Cell Culture Conditions
4.3. Flow Cytometry
4.4. Proteomic Sample
4.5. Filter-Aided Sample Preparation (FASP)
4.6. Liquid Chromatography-Tandem Mass Spectrometry
4.7. Data Analysis
4.8. Bioinformatic Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haque, R.U.; Levey, A.I. Alzheimer’s disease: A clinical perspective and future nonhuman primate research opportunities. Proc. Natl. Acad. Sci. USA 2019, 116, 26224–26229. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.J. World Alzheimer Report 2015: The Global Impact of Dementia. 2015. Available online: https://www.alz.co.uk/research/world-report-2015 (accessed on 23 July 2020).
- Nisbet, R.M.; Götz, J. Amyloid-β and Tau in Alzheimer’s Disease: Novel Pathomechanisms and Non-Pharmacological Treatment Strategies. J. Alzheimers Dis. 2018, 64, S517–S527. Available online: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi=10.3233/JAD-179907 (accessed on 23 July 2020). [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hymans, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Tellechea, P.; Pujol, N.; Esteve-Belloch, P.; Echeveste, B.; García-Eulate, M.R.; Arbizu, J.; Riverol, M. Enfermedad de Alzheimer de inicio precoz y de inicio tardío: ¿son la misma entidad? Neurología 2018, 33, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Chouraki, V.; Seshadri, S. Genetics of Alzheimer’s Disease. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2014; pp. 245–294. Available online: http://linkinghub.elsevier.com/retrieve/pii/B9780128001493000056 (accessed on 4 July 2018).
- Soosman, S.K.; Joseph-Mathurin, N.; Braskie, M.N.; Bordelon, Y.M.; Wharton, D.; Casado, M.; Coppola, G.; McCallum, H.; Nuwer, M.; Coutin-Churchman, P.; et al. Widespread white matter and conduction defects in PSEN1- related spastic paraparesis. Neurobiol. Aging 2016, 47, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Casado, M.; Van Berlo, V.; Pa, J.; Joseph-Mathurin, N.; Fagan, A.M.; Benzinger, T.; Bateman, R.J.; Morris, J.C. A novel PSEN1 (S230N) mutation causing early-onset Alzheimer’s Disease associated with prosopagnosia, hoarding, and Parkinsonism. Neurosci. Lett. 2017, 657, 11–15. [Google Scholar] [CrossRef]
- Rudzinski, L.A.; Fletcher, R.M.; Dickson, D.W.; Crook, R.; Hutton, M.L.; Adamson, J.; Graff-Radford, N.R. Early Onset Familial Alzheimer Disease With Spastic Paraparesis, Dysarthria, and Seizures and N135S Mutation in PSEN1. Alzheimer Dis. Assoc. Disord. 2008, 22, 299–307. [Google Scholar] [CrossRef]
- Yescas, P.; Huertas-Vazquez, A.; Villarreal-Molina, M.T.; Rasmussen, A.; Tusié-Luna, M.T.; López, M.; Canizales-Quinteros, S.; Alonso, M.E. Founder effect for the Ala431Glu mutation of the presenilin 1 gene causing early-onset Alzheimer’s disease in Mexican families. Neurogenetics 2006, 7, 195–200. [Google Scholar] [CrossRef]
- Orozco-Barajas, M.; Oropeza-Ruvalcaba, Y.; Canales-Aguirre, A.A.; Sánchez-González, V.J. PSEN1 c.1292C<A Variant and Early-Onset Alzheimer’s Disease: A Scoping Review. Front. Aging Neurosci. 2022, 14, 860529. [Google Scholar] [CrossRef]
- Souza, G.H.M.F.; Guest, P.C.; Martins-de-Souza, D. LC-MSE, Multiplex MS/MS, Ion Mobility, and Label-Free Quantitation in Clinical Proteomics. In Multiplex Biomarker Techniques; Guest, P.C., Ed.; Springer: New York, NY, USA, 2017; pp. 57–73. Available online: http://link.springer.com/10.1007/978-1-4939-6730-8_4 (accessed on 31 July 2022).
- Jain, A.P.; Sathe, G. Proteomics Landscape of Alzheimer’s Disease. Proteomes 2021, 9, 13. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8005944/ (accessed on 3 September 2022). [CrossRef]
- Craft, G.E.; Chen, A.; Nairn, A.C. Recent advances in quantitative neuroproteomics. Methods 2013, 61, 186–218. [Google Scholar] [CrossRef]
- Hondius, D.C.; van Nierop, P.; Li, K.W.; Hoozemans, J.J.M.; van der Schors, R.C.; van Haastert, E.S.; van der Vies, S.M.; Rozemuller, A.J.M.; Smit, A.B. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Andreev, V.P.; Petyuk, V.A.; Brewer, H.M.; Karpievitch, Y.V.; Xie, F.; Clarke, J.; Camp, D.; Smith, R.D.; Lieberman, A.P.; Albin, R.L.; et al. Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 2012, 11, 3053–3067. [Google Scholar] [CrossRef]
- Lachén-Montes, M.; Íñigo-Marco, I.; Cartas-Cejudo, P.; Fernández-Irigoyen, J.; Santamaría, E. Olfactory Bulb Proteomics Reveals Widespread Proteostatic Disturbances in Mixed Dementia and Guides for Potential Serum Biomarkers to Discriminate Alzheimer Disease and Mixed Dementia Phenotypes. J. Pers. Med. 2021, 11, 503. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8227984/ (accessed on 18 July 2021). [CrossRef]
- Dayon, L.; Núñez Galindo, A.; Wojcik, J.; Cominetti, O.; Corthésy, J.; Oikonomidi, A.; Henry, H.; Kussmann, M.; Migliavacca, E.; Severin, I.; et al. Alzheimer disease pathology and the cerebrospinal fluid proteome. Alzheimers Res. Ther. 2018, 10, 66. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6052524/ (accessed on 24 September 2019). [CrossRef]
- Khoonsari, P.E.; Häggmark, A.; Lönnberg, M.; Mikus, M.; Kilander, L.; Lannfelt, L.; Bergquist, J.; Ingelsson, M.; Nilsson, P.; Kultima, K.; et al. Analysis of the Cerebrospinal Fluid Proteome in Alzheimer’s Disease. PLoS ONE 2016, 11, e0150672. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; Wang, H.; Niu, M.; Bai, B.; Wang, X.; Li, Y.; Cho, J.-H.; Tan, H.; Mishra, A.; High, A.A.; et al. Deep undepleted human serum proteome profiling toward biomarker discovery for Alzheimer’s disease. Clin. Proteom. 2019, 16, 16. [Google Scholar] [CrossRef]
- Hye, A.; Lynham, S.; Thambisetty, M.; Causevic, M.; Campbell, J.; Byers, H.L.; Hooper, C.; Rijsdijk, F.; Tabrizi, S.J.; Banner, S. Proteome-based plasma biomarkers for Alzheimer’s disease. Brain 2006, 129, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- François, M.; Bull, C.F.; Fenech, M.F.; Leifert, W.R. Current State of Saliva Biomarkers for Aging and Alzheimer’s Disease. 2019. Available online: https://www.ingentaconnect.com/contentone/ben/car/2019/00000016/00000001/art00007 (accessed on 24 September 2019).
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Jiménez-Acosta, M.A.; Hernández, L.J.R.; Cristerna, M.L.P.; Tapia-Ramírez, J.; Meraz-Ríos, M.A. Review: Neuronal Differentiation Protocols of Mesenchymal Stem Cells. ABB 2022, 13, 15–71. Available online: https://www.scirp.org/journal/doi.aspx?doi=10.4236/abb.2022.131002 (accessed on 31 July 2022). [CrossRef]
- Benítez-King, G.; Riquelme, A.; Ortíz-López, L.; Berlanga, C.; Rodríguez-Verdugo, M.S.; Romo, F.; Calixto, E.; Solís-Chagoyán, H.; Jímenez, M.; Montaño, L.M.; et al. A non-invasive method to isolate the neuronal linage from the nasal epithelium from schizophrenic and bipolar diseases. J. Neurosci. Methods 2011, 201, 35–45. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0165027011003980 (accessed on 31 July 2022). [CrossRef] [PubMed]
- Jiménez-Acosta, M.A.; Hernández, L.J.R.; Cristerna, M.L.P.; Meraz-Ríos, M.A. Mesenchymal Stem Cells: New Alternatives for Nervous System Disorders. CSCR 2023, 18, 299–321. [Google Scholar] [CrossRef]
- Duan, D.; Lu, M. Olfactory mucosa: A rich source of cell therapy for central nervous system repair. Rev. Neurosci. 2015, 26, 281–293. Available online: https://www.degruyter.com/document/doi/10.1515/revneuro-2014-0065/html (accessed on 20 July 2023). [CrossRef]
- Delorme, B.; Nivet, E.; Gaillard, J.; Häupl, T.; Ringe, J.; Devèze, A.; Magnan, J.; Sohier, J.; Khrestchatisky, M.; Roman, F.S.; et al. The human nose harbors a niche of olfactory ectomesenchymal stem cells displaying neurogenic and osteogenic properties. Stem Cells Dev. 2010, 19, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Vaca, A.L.; Benitez-King, G.; Ruiz, V.; Ramírez-Rodríguez, G.B.; Hernández-de la Cruz, B.; Salamanca-Gómez, F.A.; González-Márquez, H.; Ramírez-Sánchez, I.; Ortíz-López, L.; Vélez-Del Valle, C.; et al. Exfoliated Human Olfactory Neuroepithelium: A Source of Neural Progenitor Cells. Mol. Neurobiol. 2018, 55, 2516–2523. [Google Scholar] [CrossRef] [PubMed]
- Murrell, W.; Féron, F.; Wetzig, A.; Cameron, N.; Splatt, K.; Bellette, B.; Bianco, J.; Perry, C.; Lee, G.; Mackay-Sim, A. Multipotent stem cells from adult olfactory mucosa. Dev. Dyn. 2005, 233, 496–515. [Google Scholar] [CrossRef]
- Murrell, W.; Sanford, E.; Anderberg, L.; Cavanagh, B.; Mackay-Sim, A. Olfactory stem cells can be induced to express chondrogenic phenotype in a rat intervertebral disc injury model. Spine J. 2009, 9, 585–594. Available online: https://linkinghub.elsevier.com/retrieve/pii/S152994300900093X (accessed on 25 July 2023). [CrossRef]
- Ge, L.; Zhuo, Y.; Duan, D.; Zhao, Z.; Teng, X.; Wang, L.; Lu, M. Biological characteristics of human olfactory mucosa mesenchymal stem cells. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2015, 40, 53–58. [Google Scholar] [CrossRef]
- Veron, A.D.; Bienboire-Frosini, C.; Feron, F.; Codecasa, E.; Deveze, A.; Royer, D.; Watelet, P.; Asproni, P.; Sadelli, K.; Chabaud, C.; et al. Isolation and characterization of olfactory ecto-mesenchymal stem cells from eight mammalian genera. BMC Vet. Res. 2018, 14, 17. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5772688/ (accessed on 20 July 2023). [CrossRef]
- Murrell, W.; Wetzig, A.; Donnellan, M.; Féron, F.; Burne, T.; Meedeniya, A.; Kesby, J.; Bianco, J.; Perry, C.; Silburn, P.; et al. Olfactory Mucosa Is a Potential Source for Autologous Stem Cell Therapy for Parkinson’s Disease. Stem Cells 2008, 26, 2183–2192. Available online: https://academic.oup.com/stmcls/article/26/8/2183-2192/6403124 (accessed on 20 July 2023). [CrossRef]
- McDonald, C.; Mackay-Sim, A.; Crane, D.; Murrell, W. Could cells from your nose fix your heart? Transplantation of olfactory stem cells in a rat model of cardiac infarction. Sci. World J. 2010, 10, 422–433. [Google Scholar] [CrossRef][Green Version]
- Stamegna, J.-C.; Sadelli, K.; Escoffier, G.; Girard, S.D.; Veron, A.D.; Bonnet, A.; Khrestchatisky, M.; Gauthier, P.; Roman, F.S. Grafts of Olfactory Stem Cells Restore Breathing and Motor Functions after Rat Spinal Cord Injury. J. Neurotrauma 2018, 35, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, A.; Valdés-Tovar, M.; Ugalde, O.; Maya-Ampudia, V.; Fernández, M.; Mendoza-Durán, L.; Rodríguez-Cárdenas, L.; Benítez-King, G. Potential Use of Exfoliated and Cultured Olfactory Neuronal Precursors for In Vivo Alzheimer’s Disease Diagnosis: A Pilot Study. Cell. Mol. Neurobiol. 2020, 40, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Dan, X.; Wechter, N.; Gray, S.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Olfactory dysfunction in aging and neurodegenerative diseases. Ageing Res. Rev. 2021, 70, 101416. Available online: https://linkinghub.elsevier.com/retrieve/pii/S156816372100163X (accessed on 31 July 2022). [CrossRef]
- Kotecha, A.; Corrêa, A.; Fisher, K.; Rushworth, J. Olfactory Dysfunction as a Global Biomarker for Sniffing out Alzheimer’s Disease: A Meta-Analysis. Biosensors 2018, 8, 41. Available online: https://www.mdpi.com/2079-6374/8/2/41 (accessed on 31 July 2022). [CrossRef]
- Talamo, B.R.; Rudel, R.; Kosik, K.S.; Lee, V.M.-Y.; Neff, S.; Adelman, L.; Kauer, J.S. Pathological changes in olfactory neurons in patients with Alzheimer’s disease. Nature 1989, 337, 736–739. Available online: http://www.nature.com/articles/337736a0 (accessed on 31 July 2022). [CrossRef] [PubMed]
- Kovács, T.; Cairns, N.J.; Lantos, P.L. Olfactory centres in Alzheimer’s disease: Olfactory bulb is involved in early Braak’s stages. Neuroreport 2001, 12, 285–288. [Google Scholar] [CrossRef]
- Scopa, C.; Marrocco, F.; Latina, V.; Ruggeri, F.; Corvaglia, V.; La Regina, F.; Ammassari-Teule, M.; Middei, S.; Amadoro, G.; Meli, G.; et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 2020, 27, 934–948. Available online: http://www.nature.com/articles/s41418-019-0409-3 (accessed on 31 July 2022). [CrossRef]
- Santos-Mandujano, R.A.; Ryan, N.S.; Chávez-Gutiérrez, L.; Sánchez-Torres, C.; Meraz-Ríos, M.A. Clinical Association of White Matter Hyperintensities Localization in a Mexican Family with Spastic Paraparesis Carrying the PSEN1 A431E Mutation. J. Alzheimers Dis. 2020, 73, 1075–1083. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1465324906708817 (accessed on 31 July 2022). [CrossRef]
- Erten-Lyons, D.; Wilmot, B.; Anur, P.; McWeeney, S.; Westaway, S.K.; Silbert, L.; Kramer, P.; Kaye, J. Microcephaly genes and risk of late-onset Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 276–282. [Google Scholar] [CrossRef]
- Miron, J.; Picard, C.; Nilsson, N.; Frappier, J.; Dea, D.; Théroux, L.; Alzheimer’s Disease Neuroimaging Initiative; United Kingdom Brain Expression Consortium; Poirier, J. CDK5RAP2 gene and tau pathophysiology in late-onset sporadic Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, T.; Katsenelson, K.; Daniliuc, S.; Hasin, T.; Choder, M.; Aronheim, A. A Novel c-Jun N-terminal Kinase (JNK)-binding Protein WDR62 Is Recruited to Stress Granules and Mediates a Nonclassical JNK Activation. Mol. Biol. Cell 2010, 21, 117–130. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2801705/ (accessed on 3 August 2022). [CrossRef]
- Duce, J.A.; Smith, D.P.; Blake, R.E.; Crouch, P.J.; Li, Q.-X.; Masters, C.L.; Trounce, I.A. Linker Histone H1 Binds to Disease Associated Amyloid-like Fibrils. J. Mol. Biol. 2006, 361, 493–505. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0022283606007595 (accessed on 1 August 2022). [CrossRef]
- Roque, A.; Sortino, R.; Ventura, S.; Ponte, I.; Suau, P. Histone H1 Favors Folding and Parallel Fibrillar Aggregation of the 1–42 Amyloid-β Peptide. Langmuir 2015, 31, 6782–6790. Available online: https://pubs.acs.org/doi/10.1021/la504089g (accessed on 1 August 2022). [CrossRef] [PubMed]
- Sepulveda-Falla, D.; Barrera-Ocampo, A.; Hagel, C.; Korwitz, A.; Vinueza-Veloz, M.F.; Zhou, K.; Schonewille, M.; Zhou, H.; Velazquez-Perez, L.; Rodriguez-Labrada, R.; et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Investig. 2014, 124, 1552–1567. [Google Scholar] [CrossRef] [PubMed]
- Guo, E.; Liu, H.; Liu, X. Overexpression of SCUBE2 Inhibits Proliferation, Migration, and Invasion in Glioma Cells. Oncol. Res. 2017, 25, 437–444. Available online: https://www.ingentaconnect.com/content/10.3727/096504016X14747335734344 (accessed on 1 August 2022). [CrossRef]
- Area-Gomez, E.; del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease: Upregulated function of MAM in AD. EMBO J. 2012, 31, 4106–4123. Available online: http://emboj.embopress.org/cgi/doi/10.1038/emboj.2012.202 (accessed on 1 August 2022). [CrossRef]
- Ihara, S.; Nakayama, S.; Murakami, Y.; Suzuki, E.; Asakawa, M.; Kinoshita, T.; Sawa, H. PIGN prevents protein aggregation in the endoplasmic reticulum independently of its function in the GPI synthesis. J. Cell Sci. 2017, 130, 602–613. [Google Scholar] [CrossRef]
- Ebanks, B.; Ingram, T.L.; Chakrabarti, L. ATP synthase and Alzheimer’s disease: Putting a spin on the mitochondrial hypothesis. Aging 2020, 12, 16647–16662. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7485717/ (accessed on 4 August 2022). [CrossRef]
- Patro, S.; Ratna, S.; Yamamoto, H.A.; Ebenezer, A.T.; Ferguson, D.S.; Kaur, A.; McIntyre, B.C.; Snow, R.; Solesio, M.E. ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11185. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8539681/ (accessed on 4 August 2022). [CrossRef] [PubMed]
- Lin, Q.; Liang, Q.; Qin, C.; Li, Y. CircANKRD36 Knockdown Suppressed Cell Viability and Migration of LPS-Stimulated RAW264.7 Cells by Sponging MiR-330. Inflammation 2021, 44, 2044–2053. Available online: https://link.springer.com/10.1007/s10753-021-01480-5 (accessed on 7 August 2022). [CrossRef] [PubMed]
- Ma, J.; Zhang, L.-Q.; He, Z.-X.; He, X.-X.; Wang, Y.-J.; Jian, Y.-L.; Wang, X.; Zhang, B.-B.; Su, C.; Lu, J.; et al. Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 2019, 17, e3000461. [Google Scholar] [CrossRef] [PubMed]
- Jordanova, A.; Irobi, J.; Thomas, F.P.; Van Dijck, P.; Meerschaert, K.; Dewil, M.; Dierick, I.; Jacobs, A.; De Vriendt, E.; Guergueltcheva, V.; et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006, 38, 197–202. [Google Scholar] [CrossRef]
- Prpar Mihevc, S.; Baralle, M.; Buratti, E.; Rogelj, B. TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 2016, 6, 33996. [Google Scholar] [CrossRef]
- Niedowicz, D.M.; Beckett, T.L.; Holler, C.J.; Weidner, A.M.; Murphy, M.P. APP(DeltaNL695) expression in murine tissue downregulates CNBP expression. Neurosci. Lett. 2010, 482, 57–61. [Google Scholar] [CrossRef][Green Version]
- Sun, C.; Van Ghelue, M.; Tranebjærg, L.; Thyssen, F.; Nilssen, Ø.; Torbergsen, T. Myotonia congenita and myotonic dystrophy in the same family: Coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin. Genet. 2011, 80, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, M.; Stamler, J.S. Hemoglobin induction in mouse macrophages. Proc. Natl. Acad. Sci. USA 1999, 96, 6643–6647. Available online: https://pnas.org/doi/full/10.1073/pnas.96.12.6643 (accessed on 1 August 2022). [CrossRef]
- Biagioli, M.; Pinto, M.; Cesselli, D.; Zaninello, M.; Lazarevic, D.; Roncaglia, P.; Simone, R.; Vlachouli, C.; Plessy, C.; Bertin, N.; et al. Unexpected expression of α- and β-globin in mesencephalic dopaminergic neurons and glial cells. Proc. Natl. Acad. Sci. USA 2009, 106, 15454–15459. Available online: https://pnas.org/doi/full/10.1073/pnas.0813216106 (accessed on 1 August 2022). [CrossRef]
- Richter, F.; Meurers, B.H.; Zhu, C.; Medvedeva, V.P.; Chesselet, M.-F. Neurons express hemoglobin α- and β-chains in rat and human brains. J. Comp. Neurol. 2009, 515, 538–547. Available online: https://onlinelibrary.wiley.com/doi/10.1002/cne.22062 (accessed on 1 August 2022). [CrossRef]
- Wu, C.; Liao, P.; Yu, L.; Wang, S.; Chen, S.; Wu, C.; Kuo, Y. Hemoglobin promotes A? oligomer formation and localizes in neurons and amyloid deposits. Neurobiol. Dis. 2004, 17, 367–377. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0969996104001925 (accessed on 3 August 2022). [CrossRef] [PubMed]
- Fernández-Irigoyen, J.; Corrales, F.J.; Santamaría, E. Proteomic atlas of the human olfactory bulb. J. Proteom. 2012, 75, 4005–4016. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1874391912002990 (accessed on 1 August 2022). [CrossRef] [PubMed]
- Ferrer, I.; Gómez, A.; Carmona, M.; Huesa, G.; Porta, S.; Riera-Codina, M.; Biagioli, M.; Gustincich, S.; Aso, E. Neuronal hemoglobin is reduced in Alzheimer’s disease, argyrophilic grain disease, Parkinson’s disease, and dementia with Lewy bodies. J. Alzheimers Dis. 2011, 23, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, M.; Chambers, I. Loss of Resf1 reduces the efficiency of embryonic stem cell self-renewal and germline entry. Life Sci. Alliance 2021, 4, e202101190. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8500223/ (accessed on 26 September 2022). [CrossRef]
- Pitzen, V.; Sander, S.; Baumann, O.; Gräf, R.; Meyer, I. Cep192, a Novel Missing Link between the Centrosomal Core and Corona in Dictyostelium Amoebae. Cells 2021, 10, 2384. [Google Scholar] [CrossRef]
- Yan, S.; Song, M.; Ping, J.; Lai, S.; Cao, X.; Bai, C.-J.; Xie, D.-F.; Guan, H.; Gao, S.; Zhou, P.-K. ZGRF1 promotes end resection of DNA homologous recombination via forming complex with BRCA1/EXO1. Cell Death Discov. 2021, 7, 260. [Google Scholar] [CrossRef]
- Tirilomi, A.; Elakad, O.; Yao, S.; Li, Y.; Hinterthaner, M.; Danner, B.C.; Ströbel, P.; Tirilomis, T.; Bohnenberger, H.; von Hammerstein-Equord, A. Expression and prognostic impact of CD49b in human lung cancer. Medicine 2022, 101, e28814. [Google Scholar] [CrossRef]
- Liu, H.; Al-aidaroos, A.Q.O.; Wang, H.; Guo, K.; Li, J.; Zhang, H.F.; Zeng, Q. PRL-3 suppresses c-Fos and integrin α2 expression in ovarian cancer cells. BMC Cancer 2013, 13, 80. [Google Scholar] [CrossRef]
- Zutter, M.M.; Santoro, S.A.; Staatz, W.D.; Tsung, Y.L. Re-expression of the alpha 2 beta 1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc. Natl. Acad. Sci. USA 1995, 92, 7411–7415. [Google Scholar] [CrossRef]
- Yamada, T.; Horisberger, M.A.; Kawaguchi, N.; Moroo, I.; Toyoda, T. Immunohistochemistry using antibodies to alpha-interferon and its induced protein, MxA, in Alzheimer’s and Parkinson’s disease brain tissues. Neurosci. Lett. 1994, 181, 61–64. [Google Scholar] [CrossRef]
- Furuyama, H.; Chiba, S.; Okabayashi, T.; Yokota, S.; Nonaka, M.; Imai, T.; Fujii, N.; Matsumoto, H. Single nucleotide polymorphisms and functional analysis of MxA promoter region in multiple sclerosis. J. Neurol. Sci. 2006, 249, 153–157. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Shi, S.-F.; Wu, W.-L.; Ho, L.-J.; Lai, J.-H. Protective roles of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) in dengue virus infection of human lung epithelial cells. PLoS ONE 2013, 8, e79518. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, E.M.; Xu, H.; Murphy, R.M.; Grounds, M.D.; Pinniger, G.J. Dysferlin-deficiency has greater impact on function of slow muscles, compared with fast, in aged BLAJ mice. PLoS ONE 2019, 14, e0214908. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu Pene, T.; Rosé, S.D.; Lejen, T.; Marcu, M.G.; Trifaró, J.-M. Expression of various scinderin domains in chromaffin cells indicates that this protein acts as a molecular switch in the control of actin filament dynamics and exocytosis. J. Neurochem. 2005, 92, 780–789. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1471-4159.2004.02907.x (accessed on 20 July 2023). [CrossRef]
- Li, X.; Jiang, H.; Huang, Y.; Gong, Q.; Wang, J.; Ling, J. Expression and Function of the Actin-severing Protein Adseverin in the Proliferation, Migration, and Differentiation of Dental Pulp Cells. J. Endod. 2015, 41, 493–500. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0099239914011698 (accessed on 13 December 2022). [CrossRef]
- Pohlkamp, T.; Wasser, C.R.; Herz, J. Functional Roles of the Interaction of APP and Lipoprotein Receptors. Front. Mol. Neurosci. 2017, 10, 54. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5331069/ (accessed on 13 December 2022). [CrossRef] [PubMed]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4033011/ (accessed on 13 December 2022). [CrossRef]
- Bu, G.; Cam, J.; Zerbinatti, C. LRP in amyloid-beta production and metabolism. In Integrated Molecular Medicine for Neuronal and Neoplastic Disorders; Sobue, G., Takahashi, M., Yoshida, J., Kaibuchi, K., Naoe, T., Lahiri, D.K., Eds.; Blackwell Publishing: Oxford, UK, 2006; pp. 35–53. Available online: http://www.webofscience.com/wos/woscc/full-record/WOS:000244112400004 (accessed on 13 December 2022).
- Benoit, M.E.; Hernandez, M.X.; Dinh, M.L.; Benavente, F.; Vasquez, O.; Tenner, A.J. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. J. Biol. Chem. 2013, 288, 654–665. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, Y.; Kim, D.; Jang, H.; Bae, J.S.; Park, M.K.; Kim, H.; Kim, S.; Lee, H. AIMP3 depletion causes genome instability and loss of stemness in mouse embryonic stem cells. Cell Death Dis. 2018, 9, 972. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6155375/ (accessed on 12 December 2022). [CrossRef]
- Kim, C.; Park, J.; Song, Y.; Kim, S.; Moon, J. HIF1α-mediated AIMP3 suppression delays stem cell aging via the induction of autophagy. Aging Cell 2019, 18, e12909. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6413650/ (accessed on 12 December 2022). [CrossRef]
- Wang, Y.; Yang, Q.; Cheng, Y.; Gao, M.; Kuang, L.; Wang, C. Myosin Heavy Chain 10 (MYH10) Gene Silencing Reduces Cell Migration and Invasion in the Glioma Cell Lines U251, T98G, and SHG44 by Inhibiting the Wnt/β-Catenin Pathway. Med. Sci. Monit. 2018, 24, 9110–9119. [Google Scholar] [CrossRef]
- Zeng, F.; Zou, H.-Q.; Zhou, H.-D.; Li, J.; Wang, L.; Cao, H.-Y.; Yi, X.; Wang, X.; Liang, C.-R.; Wang, Y.-R.; et al. The relationship between single nucleotide polymorphisms of the NTRK2 gene and sporadic Alzheimer’s disease in the Chinese Han population. Neurosci. Lett. 2013, 550, 55–59. Available online: https://www.sciencedirect.com/science/article/pii/S0304394013006083 (accessed on 12 December 2022). [CrossRef]
- Takahashi, R.; Takeshita, F.; Honma, K.; Ono, M.; Kato, K.; Ochiya, T. Ribophorin II regulates breast tumor initiation and metastasis through the functional suppression of GSK3β. Sci. Rep. 2013, 3, 2474. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3747512/ (accessed on 14 December 2022). [CrossRef]
- Sun, W.; Hua, X.; Gu, Y.; Xu, Q.; Zhu, S.; Lv, T.; Song, Y. E3 ubiquitin ligase RNF6 promotes antiviral immune responses through enhancing the expression of interferon stimulated genes in myeloid cells. Clin. Immunol. 2022, 242, 109099. Available online: https://www.sciencedirect.com/science/article/pii/S1521661622001802 (accessed on 14 December 2022). [CrossRef] [PubMed]
- Li, Q.; Wang, G.; Tao, J.; Chen, W. RNF6 promotes colorectal cancer invasion and migration via the Wnt/β-catenin pathway by inhibiting GSK3β activity. Pathol. Res. Pract. 2021, 225, 153545. Available online: https://www.sciencedirect.com/science/article/pii/S0344033821002065 (accessed on 14 December 2022). [CrossRef]
- Zhu, K.; Bai, H.; Mu, M.; Xue, Y.; Duan, Z. Knockdown of RNF6 inhibits HeLa cervical cancer cell growth via suppression of MAPK/ERK signaling. FEBS Open Bio 2021, 11, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-Y.; Diao, C.-F.; Zhang, Y.; Wu, N.; Wan, H.-Y.; Nong, X.-Y.; Liu, M.; Tang, H. miR-371-5p down-regulates pre mRNA processing factor 4 homolog B (PRPF4B) and facilitates the G1/S transition in human hepatocellular carcinoma cells. Cancer Lett. 2013, 335, 351–360. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0304383513001717 (accessed on 4 December 2022). [CrossRef] [PubMed]
- Ou, S.; Liao, Y.; Shi, J.; Tang, J.; Ye, Y.; Wu, F.; Wang, W.; Fei, J.; Xie, F.; Bai, L. S100A16 suppresses the proliferation, migration and invasion of colorectal cancer cells in part via the JNK/p38 MAPK pathway. Mol. Med. Rep. 2021, 23, 164. [Google Scholar] [CrossRef]
- Bose, A.; Banerjee, S.; Visweswariah, S.S. Mutational landscape of receptor guanylyl cyclase C: Functional analysis and disease-related mutations. IUBMB Life 2020, 72, 1145–1159. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7611479/ (accessed on 4 December 2022). [CrossRef]
- Li, P.; Schulz, S.; Bombonati, A.; Palazzo, J.P.; Hyslop, T.M.; Xu, Y.; Baran, A.A.; Siracusa, L.D.; Pitari, G.M.; Waldman, S.A. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology 2007, 133, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Basu, N.; Saha, S.; Khan, I.; Ramachandra, S.G.; Visweswariah, S.S. Intestinal Cell Proliferation and Senescence Are Regulated by Receptor Guanylyl Cyclase C and p21. J. Biol. Chem. 2014, 289, 581–593. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3879579/ (accessed on 4 December 2022). [CrossRef]
- Rescher, U.; Gerke, V. S100A10/p11: Family, friends and functions. Pflugers Arch. Eur. J. Physiol. 2007, 455, 575–582. Available online: http://link.springer.com/10.1007/s00424-007-0313-4 (accessed on 4 December 2022). [CrossRef]
- Sun, Y.; Fan, Y.; Wang, Z.; Li, M.; Su, D.; Liu, Y.; Liang, X. S100A16 promotes acute kidney injury by activating HRD1-induced ubiquitination and degradation of GSK3β and CK1α. Cell Mol. Life Sci. 2022, 79, 184. [Google Scholar] [CrossRef]
- Jin, R.; Zhao, A.; Han, S.; Zhang, D.; Sun, H.; Li, M.; Su, D.; Liang, X. The interaction of S100A16 and GRP78 actives endoplasmic reticulum stress-mediated through the IRE1α/XBP1 pathway in renal tubulointerstitial fibrosis. Cell Death Dis. 2021, 12, 942. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Tan, X.; Lv, D.-J.; Xing, R.-W.; Shu, F.-P.; Zhong, C.-F.; Li, C.; Zou, Y.-G.; Mao, X.-M. Phosphoribosyl pyrophosphate synthetases 2 knockdown inhibits prostate cancer progression by suppressing cell cycle and inducing cell apoptosis. J. Cancer 2020, 11, 1027–1037. [Google Scholar] [CrossRef]
- Miao, W.; Wang, Y. Targeted Quantitative Kinome Analysis Identifies PRPS2 as a Promoter for Colorectal Cancer Metastasis. J. Proteome Res. 2019, 18, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Sahin, M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011, 585, 973–980. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Ferrando-Miguel, R.; Rosner, M.; Freilinger, A.; Lubec, G.; Hengstschläger, M. Tuberin—A New Molecular Target in Alzheimer’s Disease? Neurochem. Res. 2005, 30, 1413–1419. Available online: http://link.springer.com/10.1007/s11064-005-8511-y (accessed on 5 December 2022). [CrossRef]
- Habib, S.L.; Michel, D.; Masliah, E.; Thomas, B.; Ko, H.S.; Dawson, T.M.; Abboud, H.; Clark, R.A.; Imam, S.Z. Role of Tuberin in Neuronal Degeneration. Neurochem. Res. 2008, 33, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Hahn, I.; Voelzmann, A.; Parkin, J.; Fülle, J.B.; Slater, P.G.; Lowery, L.A.; Sanchez-Soriano, N.; Prokop, A. Tau, XMAP215/Msps and Eb1 co-operate interdependently to regulate microtubule polymerisation and bundle formation in axons. PLoS Genet. 2021, 17, e1009647. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8284659/ (accessed on 4 December 2022). [CrossRef] [PubMed]
- Kim, M.-J.; Yun, H.S.; Hong, E.-H.; Lee, S.-J.; Baek, J.-H.; Lee, C.-W.; Yim, J.-H.; Kim, J.-S.; Park, J.K.; Um, H.-D.; et al. Depletion of end-binding protein 1 (EB1) promotes apoptosis of human non-small-cell lung cancer cells via reactive oxygen species and Bax-mediated mitochondrial dysfunction. Cancer Lett. 2013, 339, 15–24. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0304383513005478 (accessed on 4 December 2022). [CrossRef] [PubMed]
- Karbowski, M.; Jeong, S.-Y.; Youle, R.J. Endophilin B1 is required for the maintenance of mitochondrial morphology. J. Cell Biol. 2004, 166, 1027–1039. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2172012/ (accessed on 4 December 2022). [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2254521/ (accessed on 4 December 2022). [CrossRef]
- Odierna, G.L.; Kerwin, S.K.; Harris, L.E.; Shin, G.J.-E.; Lavidis, N.A.; Noakes, P.G.; Millard, S.S. Dscam2 suppresses synaptic strength through a PI3K-dependent endosomal pathway. J. Cell Biol. 2020, 219, e201909143. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Liu, J.; Huo, Y.; Luo, X.-J. The schizophrenia susceptibility gene NAGA regulates dendritic spine density: Further evidence for the dendritic spine pathology of schizophrenia. Mol. Psychiatry 2021, 26, 7102–7104. Available online: https://www.nature.com/articles/s41380-021-01261-4 (accessed on 4 December 2022). [CrossRef] [PubMed]
- Kapuralin, K.; Ćurlin, M.; Mitrečić, D.; Kosi, N.; Schwarzer, C.; Glavan, G.; Gajović, S. STAM2, a member of the endosome-associated complex ESCRT-0 is highly expressed in neurons. Mol. Cell. Neurosci. 2015, 67, 104–115. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1044743115000974 (accessed on 5 December 2022). [CrossRef]
- Furić Cunko, V.; Mitrecić, D.; Mavrić, S.; Gajović, S. Expression pattern and functional analysis of mouse Stam2 in the olfactory system. Coll. Antropol. 2008, 32 (Suppl. S1), 59–63. [Google Scholar]
- Mizuno, E.; Kawahata, K.; Kato, M.; Kitamura, N.; Komada, M. STAM Proteins Bind Ubiquitinated Proteins on the Early Endosome via the VHS Domain and Ubiquitin-interacting Motif. Mol. Biol. Cell 2003, 14, 3675–3689. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC196559/ (accessed on 5 December 2022). [CrossRef]
- Shehadeh, L.A.; Yu, K.; Wang, L.; Guevara, A.; Singer, C.; Vance, J.; Papapetropoulos, S. SRRM2, a potential blood biomarker revealing high alternative splicing in Parkinson’s disease. PLoS ONE 2010, 5, e9104. [Google Scholar] [CrossRef]
- Ilik, İ.A.; Malszycki, M.; Lübke, A.K.; Schade, C.; Meierhofer, D.; Aktaş, T. SON and SRRM2 are essential for nuclear speckle formation. elife 2020, 9, e60579. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.J.; Strovas, T.J.; Baum, M.; Mitchell, B.K.; Eck, R.J.; Hendricks, N.; Wheeler, J.M.; Latimer, C.S.; Keene, C.D.; Kraemer, B.C. Pathological tau drives ectopic nuclear speckle scaffold protein SRRM2 accumulation in neuron cytoplasm in Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 117. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8243890/ (accessed on 5 December 2022). [CrossRef] [PubMed]
- Burgener, A.-V.; Bantug, G.R.; Meyer, B.J.; Higgins, R.; Ghosh, A.; Bignucolo, O.; Ma, E.H.; Loeliger, J.; Unterstab, G.; Geigges, M.; et al. SDHA gain-of-function engages inflammatory mitochondrial retrograde signaling via KEAP1-Nrf2. Nat. Immunol. 2019, 20, 1311–1321. [Google Scholar] [CrossRef]
- Kinoshita, M.O.; Shinoda, Y.; Sakai, K.; Hashikawa, T.; Watanabe, M.; Machida, T.; Hirabayashi, Y.; Furuya, S. Selective upregulation of 3-phosphoglycerate dehydrogenase (Phgdh) expression in adult subventricular zone neurogenic niche. Neurosci. Lett. 2009, 453, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Yoshida, K.; Yang, J.H.; Suzuki, T.; Azuma, N.; Sakai, K.; Hashikawa, T.; Watanabe, M.; Yasuda, K.; Kuhara, S.; et al. Impaired neurogenesis in embryonic spinal cord of Phgdh knockout mice, a serine deficiency disorder model. Neurosci. Res. 2009, 63, 184–193. [Google Scholar] [CrossRef]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.-A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e8. Available online: https://www.sciencedirect.com/science/article/pii/S1550413120300632 (accessed on 5 December 2022). [CrossRef]
- Chen, X.; Calandrelli, R.; Girardini, J.; Yan, Z.; Tan, Z.; Xu, X.; Hiniker, A.; Zhong, S. PHGDH expression increases with progression of Alzheimer’s disease pathology and symptoms. Cell Metab. 2022, 34, 651–653. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9531314/ (accessed on 5 December 2022). [CrossRef]
- Castora, F.J.; Kerns, K.A.; Pflanzer, H.K.; Hitefield, N.L.; Gershon, B.; Shugoll, J.; Shelton, M.; Coleman, R.A. Expression Changes in Mitochondrial Genes Affecting Mitochondrial Morphology, Transmembrane Potential, Fragmentation, Amyloidosis, and Neuronal Cell Death Found in Brains of Alzheimer’s Disease Patients. J. Alzheimers Dis. 2022, 90, 119–137. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Pradeepkiran, J.A.; Morton, H.; Reddy, P.H. A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic Tau mouse model of Alzheimer disease. Hum. Mol. Genet. 2022, 31, 1788–1805. [Google Scholar] [CrossRef]
- Lei, D.; Li, F.; Su, H.; Liu, J.; Wei, N.; Wang, X. Hepatic deficiency of COP9 signalosome subunit 8 induces ubiquitin-proteasome system impairment and Bim-mediated apoptosis in murine livers. PLoS ONE 2013, 8, e67793. [Google Scholar] [CrossRef]
- Su, H.; Li, J.; Zhang, H.; Ma, W.; Wei, N.; Liu, J.; Wang, X. COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ. Res. 2015, 117, 956–966. [Google Scholar] [CrossRef]
- Akiyama, H.; Nishimura, T.; Kondo, H.; Ikeda, K.; Hayashi, Y.; McGeer, P.L. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res. 1994, 639, 171–174. [Google Scholar] [CrossRef]
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef]
- Luo, J.; Elwood, F.; Britschgi, M.; Villeda, S.; Zhang, H.; Ding, Z.; Zhu, L.; Alabsi, H.; Getachew, R.; Narasimhan, R.; et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 2013, 210, 157–172. [Google Scholar] [CrossRef]
- Liu, F.-L.; Liu, T.-Y.; Kung, F.-L. FKBP12 regulates the localization and processing of amyloid precursor protein in human cell lines. J. Biosci. 2014, 39, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Criado-Marrero, M.; Zheng, D.; Wang, X.; Kamath, S.; Nordhues, B.A.; Weeber, E.J.; Dickey, C.A. The Disease-Associated Chaperone FKBP51 Impairs Cognitive Function by Accelerating AMPA Receptor Recycling. eNeuro 2019, 6, ENEURO.0242-18.2019. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6450497/ (accessed on 24 May 2023). [CrossRef] [PubMed]
- Zhang, J.; Rubio, V.; Lieberman, M.W.; Shi, Z.-Z. OLA1, an Obg-like ATPase, suppresses antioxidant response via nontranscriptional mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 15356–15361. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2741255/ (accessed on 26 December 2022). [CrossRef] [PubMed]
- Mao, R.-F.; Rubio, V.; Chen, H.; Bai, L.; Mansour, O.C.; Shi, Z.-Z. OLA1 protects cells in heat shock by stabilizing HSP70. Cell Death Dis. 2013, 4, e491. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3734832/ (accessed on 26 December 2022). [CrossRef]
- Ding, Y.; Zhang, H.; Liu, Z.; Li, Q.; Guo, Y.; Chen, Y.; Chang, Y.; Cui, H. Carnitine palmitoyltransferase 1 (CPT1) alleviates oxidative stress and apoptosis of hippocampal neuron in response to beta-Amyloid peptide fragment Aβ25-35. Bioengineered 2021, 12, 5440–5449. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8806834/ (accessed on 24 May 2023). [CrossRef]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5495060/ (accessed on 2 September 2022). [CrossRef] [PubMed]
- Kabir, M.d.T.; Uddin, M.d.S.; Abdeen, A.; Ashraf, G.M.; Perveen, A.; Hafeez, A.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Evidence Linking Protein Misfolding to Quality Control in Progressive Neurodegenerative Diseases. CTMC 2020, 20, 2025–2043. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. Available online: https://www.mdpi.com/2227-9059/9/1/63 (accessed on 12 January 2023). [CrossRef] [PubMed]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. Available online: http://www.aginganddisease.org/EN/10.14336/AD.2015.1007 (accessed on 12 January 2023). [CrossRef] [PubMed]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. Available online: https://www.frontiersin.org/articles/10.3389/fnins.2020.530219 (accessed on 12 January 2023). [CrossRef]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain Glucose Hypometabolism and Oxidative Stress in Preclinical Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1196/annals.1427.007 (accessed on 31 October 2022). [CrossRef]
- Ou, Y.-N.; Xu, W.; Li, J.-Q.; Guo, Y.; Cui, M.; Chen, K.-L.; Huang, Y.-Y.; Dong, Q.; Tan, L.; Yu, J.-T.; et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimer’s Res. Ther. 2019, 11, 57. [Google Scholar] [CrossRef]
- Mosconi, L.; Sorbi, S.; de Leon, M.J.; Li, Y.; Nacmias, B.; Myoung, P.S.; Tsui, W.; Ginestroni, A.; Bessi, V.; Fayyazz, M.; et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J. Nucl. Med. 2006, 47, 1778–1786. [Google Scholar]
- Koh, K.; Ishiura, H.; Beppu, M.; Shimazaki, H.; Ichinose, Y.; Mitsui, J.; Kuwabara, S.; Tsuji, S.; Takiyama, Y.; Japan Spastic Paraplegia Research Consortium. Novel mutations in the ALDH18A1 gene in complicated hereditary spastic paraplegia with cerebellar ataxia and cognitive impairment. J. Hum. Genet. 2018, 63, 1009–1013. [Google Scholar] [CrossRef]
- Patel, A.; Rees, S.D.; Kelly, M.A.; Bain, S.C.; Barnett, A.H.; Thalitaya, D.; Prasher, V.P. Association of variants within APOE, SORL1, RUNX1, BACE1 and ALDH18A1 with dementia in Alzheimer’s disease in subjects with Down syndrome. Neurosci. Lett. 2011, 487, 144–148. [Google Scholar] [CrossRef]
- Palmer, A.M. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J. Neural Transm. 1999, 106, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Patel, A.B. Pyruvate Carboxylase and Pentose Phosphate Fluxes are Reduced in AβPP-PS1 Mouse Model of Alzheimer’s Disease: A 13C NMR Study. J. Alzheimer Dis. 2014, 41, 387–399. Available online: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi=10.3233/JAD-122449 (accessed on 12 January 2023). [CrossRef] [PubMed]
- Schrötter, A.; Pfeiffer, K.; El Magraoui, F.; Platta, H.W.; Erdmann, R.; Meyer, H.E.; Egensperger, R.; Marcus, K.; Müller, T. The Amyloid Precursor Protein (APP) Family Members are Key Players in S-adenosylmethionine Formation by MAT2A and Modify BACE1 and PSEN1 Gene Expression-Relevance for Alzheimer’s Disease. Mol. Cell Proteom. 2012, 11, 1274–1288. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3494178/ (accessed on 12 January 2023). [CrossRef]
- Han, J.; Hyun, J.; Park, J.; Jung, S.; Oh, Y.; Kim, Y.; Ryu, S.-H.; Kim, S.-H.; Jeong, E.I.; Jo, D.-G.; et al. Aberrant role of pyruvate kinase M2 in the regulation of gamma-secretase and memory deficits in Alzheimer’s disease. Cell Rep. 2021, 37, 110102. Available online: https://www.cell.com/cell-reports/abstract/S2211-1247(21)01596-5 (accessed on 12 January 2023). [CrossRef]
- Cao, J.; Zhong, M.B.; Toro, C.A.; Zhang, L.; Cai, D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci. Lett. 2019, 703, 68–78. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Mathews, P.M.; Boiteau, A.B.; Hassinger, L.C.; Peterhoff, C.M.; Jiang, Y.; Mullaney, K.; Neve, R.L.; Gruenberg, J.; Nixon, R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology: Accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008, 173, 370–384. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: Impact of genetic risk factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Zwart, R.; van der Sluijs, P.; Annaert, W.; Gool, W.A.; Baas, F. Alzheimer’s presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum. Mol. Genet. 2000, 9, 303–310. [Google Scholar] [CrossRef]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S.B. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. Available online: https://linkinghub.elsevier.com/retrieve/pii/S2211124716312566 (accessed on 5 July 2020). [CrossRef]
- Malm, T.; Loppi, S.; Kanninen, K.M. Exosomes in Alzheimer’s disease. Neurochem. Int. 2016, 97, 193–199. [Google Scholar] [CrossRef]
- Lee, S.; Tsai, F.T.F. Molecular chaperones in protein quality control. J. Biochem. Mol. Biol. 2005, 38, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, M.M.M.; Boelens, W.C.; Otte-Höller, I.; Kamps, B.; de Waal, R.M.W.; Verbeek, M.M. Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res. 2006, 1089, 67–78. [Google Scholar] [CrossRef]
- Gammazza, A.M.; Bavisotto, C.C.; Barone, R.; de Macario, E.C.; Macario, A.J.L. Alzheimer’s Disease and Molecular Chaperones: Current Knowledge and the Future of Chaperonotherapy. 2016, pp. 4040–4049. Available online: https://www.eurekaselect.com/142309/article (accessed on 24 July 2020).
- Singh, R.; Kaur, N.; Dhingra, N.; Kaur, T. Protein misfolding, ER stress and chaperones: An approach to develop chaperone-based therapeutics for Alzheimer’s disease. Int. J. Neurosci. 2023, 133, 714–734. [Google Scholar] [CrossRef]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. Available online: http://www.nature.com/articles/ncb1299_479 (accessed on 6 August 2020). [CrossRef] [PubMed]
- Penzes, P.; VanLeeuwen, J.-E. Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res. Rev. 2011, 67, 184–192. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0165017311000130 (accessed on 12 January 2023). [CrossRef] [PubMed]
- Knafo, S.; Alonso-Nanclares, L.; Gonzalez-Soriano, J.; Merino-Serrais, P.; Fernaud-Espinosa, I.; Ferrer, I.; DeFelipe, J. Widespread Changes in Dendritic Spines in a Model of Alzheimer’s Disease. Cereb. Cortex 2009, 19, 586–592. Available online: https://academic.oup.com/cercor/article-lookup/doi/10.1093/cercor/bhn111 (accessed on 12 January 2023). [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. Available online: https://www.science.org/doi/10.1126/science.274.5284.99 (accessed on 12 January 2023). [CrossRef]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. Available online: https://www.neurology.org/lookup/doi/10.1212/WNL.56.1.127 (accessed on 12 January 2023). [CrossRef]
- Xie, Z.; Srivastava, D.P.; Photowala, H.; Kai, L.; Cahill, M.E.; Woolfrey, K.M.; Shum, C.Y.; Surmeier, D.J.; Penzes, P. Kalirin-7 Controls Activity-Dependent Structural and Functional Plasticity of Dendritic Spines. Neuron 2007, 56, 640–656. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0896627307007611 (accessed on 12 January 2023). [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble Oligomers of Amyloid β Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron 2009, 62, 788–801. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0896627309003870 (accessed on 12 January 2023). [CrossRef]
- Ma, Q.-L.; Yang, F.; Calon, F.; Ubeda, O.J.; Hansen, J.E.; Weisbart, R.H.; Beech, W.; Frautschy, S.A.; Cole, G.M. p21-activated Kinase-aberrant Activation and Translocation in Alzheimer Disease Pathogenesis. J. Biol. Chem. 2008, 283, 14132–14143. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0021925820716467 (accessed on 12 January 2023). [CrossRef]
- Huesa, G.; Baltrons, M.A.; Gómez-Ramos, P.; Morán, A.; García, A.; Hidalgo, J.; Francés, S.; Santpere, G.; Ferrer, I.; Galea, E. Altered Distribution of RhoA in Alzheimer’s Disease and AβPP Overexpressing Mice. J. Alzheimer Dis. 2010, 19, 37–56. Available online: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi=10.3233/JAD-2010-1203 (accessed on 12 January 2023). [CrossRef]
- Liu, Y.; Xu, Y.-F.; Zhang, L.; Huang, L.; Yu, P.; Zhu, H.; Deng, W.; Qin, C. Effective expression of Drebrin in hippocampus improves cognitive function and alleviates lesions of Alzheimer’s disease in APP (swe)/PS1 (ΔE9) mice. CNS Neurosci. Ther. 2017, 23, 590–604. Available online: https://onlinelibrary.wiley.com/doi/10.1111/cns.12706 (accessed on 12 January 2023). [CrossRef]
- Barone, E.; Mosser, S.; Fraering, P.C. Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and γ-secretase. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2500–2509. Available online: https://linkinghub.elsevier.com/retrieve/pii/S092544391400310X (accessed on 12 January 2023). [CrossRef] [PubMed]
- Bao, X.; Liu, G.; Jiang, Y.; Jiang, Q.; Liao, M.; Feng, R.; Zhang, L.; Ma, G.; Zhang, S.; Chen, Z.; et al. Cell adhesion molecule pathway genes are regulated by cis-regulatory SNPs and show significantly altered expression in Alzheimer’s disease brains. Neurobiol. Aging 2015, 36, 2904.e1–2904.e7. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0197458015003176 (accessed on 12 January 2023). [CrossRef] [PubMed]
- Yew, D.T.; Li, W.P.; Webb, S.E.; Lai, H.W.L.; Zhang, L. Neurotransmitters, peptides, and neural cell adhesion molecules in the cortices of normal elderly humans and alzheimer patients: A comparison. Exp. Gerontol. 1999, 34, 117–133. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0531556598000175 (accessed on 12 January 2023). [CrossRef] [PubMed]
- Gautam, V.; D’Avanzo, C.; Hebisch, M.; Kovacs, D.M.; Kim, D.Y. BACE1 activity regulates cell surface contactin-2 levels. Mol Neurodegener. 2014, 9, 4. Available online: https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/1750-1326-9-4 (accessed on 12 January 2023). [CrossRef]
- Ando, K.; Uemura, K.; Kuzuya, A.; Maesako, M.; Asada-Utsugi, M.; Kubota, M.; Aoyagi, N.; Yoshioka, K.; Okawa, K.; Inoue, H.; et al. N-cadherin Regulates p38 MAPK Signaling via Association with JNK-associated Leucine Zipper Protein. J. Biol. Chem. 2011, 286, 7619–7628. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0021925820519546 (accessed on 12 January 2023). [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. Available online: http://academic.oup.com/hmg/article/16/1/15/2355921/The-DYRK1A-gene-encoded-in-chromosome-21-Down (accessed on 12 January 2023). [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. Available online: http://www.nature.com/articles/ng.440 (accessed on 12 January 2023). [CrossRef]
- For the AddNeuroMed consortium and for the Alzheimer’s Disease Neuroimaging Initiative; Khondoker, M.; Newhouse, S.; Westman, E.; Muehlboeck, J.-S.; Mecocci, P.; Vellas, B.; Tsolaki, M.; Kłoszewska, I.; Soininen, H.; et al. Linking Genetics of Brain Changes to Alzheimer’s Disease: Sparse Whole Genome Association Scan of Regional MRI Volumes in the ADNI and AddNeuroMed Cohorts. J. Alzheimer Dis. 2015, 45, 851–864. Available online: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi=10.3233/JAD-142214 (accessed on 12 January 2023). [CrossRef]
- Rosenthal, S.B.; Wang, H.; Shi, D.; Liu, C.; Abagyan, R.; McEvoy, L.K.; Chen, C.-H. Mapping the gene network landscape of Alzheimer’s disease through integrating genomics and transcriptomics. PLoS Comput. Biol. 2022, 18, e1009903. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. Available online: https://www.nature.com/articles/s41591-020-0815-6 (accessed on 31 July 2022). [CrossRef] [PubMed]
- Pedrero-Prieto, C.M.; García-Carpintero, S.; Frontiñán-Rubio, J.; Llanos-González, E.; Aguilera García, C.; Alcaín, F.J.; Lindberg, I.; Durán-Prado, M.; Peinado, J.R.; Rabanal-Ruiz, Y. A comprehensive systematic review of CSF proteins and peptides that define Alzheimer’s disease. Clin. Proteom. 2020, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, L.; Ping, L.; Dammer, E.B.; Duong, D.M.; Zhou, M.; Gearing, M.; Hurst, C.; Glass, J.D.; Factor, S.A.; Johnson, E.C.B.; et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci. Adv. 2020, 6, eaaz9360. [Google Scholar] [CrossRef]
- Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics. Antioxid. Redox Signal 2017, 26, 364–387. [Google Scholar] [CrossRef]
- Lopez-Toledo, G.; Silva-Lucero, M.-D.-C.; Herrera-Díaz, J.; García, D.-E.; Arias-Montaño, J.-A.; Cardenas-Aguayo, M.-D.-C. Patient-Derived Fibroblasts With Presenilin-1 Mutations, That Model Aspects of Alzheimer’s Disease Pathology, Constitute a Potential Object for Early Diagnosis. Front. Aging Neurosci. 2022, 14, 921573. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Filter-Aided Sample Preparation for Proteome Analysis. In Microbial Proteomics; Becher, D., Ed.; Springer: New York, NY, USA, 2018; pp. 3–10. Available online: http://link.springer.com/10.1007/978-1-4939-8695-8_1 (accessed on 31 July 2022).
- Ríos-Castro, E.; Souza, G.H.M.F.; Delgadillo-Álvarez, D.M.; Ramírez-Reyes, L.; Torres-Huerta, A.L.; Velasco-Suárez, A.; Cruz-Cruz, C.; Hernández-Hernández, J.M.; Tapia-Ramírez, J. Quantitative Proteomic Analysis of MARC-145 Cells Infected with a Mexican Porcine Reproductive and Respiratory Syndrome Virus Strain Using a Label-Free Based DIA approach. J. Am. Soc. Mass Spectrom. 2020, 31, 1302–1312. Available online: https://pubs.acs.org/doi/10.1021/jasms.0c00134 (accessed on 31 July 2022). [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot ID | Gene Symbol | Description | ANOVA (p) | |
|---|---|---|---|---|
| Symptomatic carrier (P1) | O60282-2 | KIF5C | Kinesin heavy chain isoform 5C | 5.92 × 10−9 |
| Q8IV33 | KIAA0825 | Uncharacterized protein KIAA0825 | 7.01 × 10−8 | |
| A6NCS7 | UTY | Histone demethylase UTY | 1.23 × 10−7 | |
| P16401 | HIST1H1B | Histone H1.5 | 3.42 × 10−7 | |
| Q5JPF3-3 | ANKRD36C | Ankyrin repeat domain-containing protein 36C | 4.84 × 10−7 | |
| Q9NQ36-3 | SCUBE2 | Signal peptide_ CUB and EGF-like domain-containing protein 2 | 8.00 × 10−7 | |
| F8WBJ0 | CDK5RAP2 | CDK5 regulatory subunit-associated protein 2 | 1.87 × 10−5 | |
| O43379-3 | WDR62 | WD repeat-containing protein 62 | 2.44 × 10−5 | |
| Q8NEM0-3 | MCPH1 | Microcephalin | 3.85 × 10−5 | |
| P18859-2 | ATP5PF | ATP synthase-coupling factor 6_ mitochondrial | 1.75 × 10−3 | |
| K7ESH9 | PIGN | GPI ethanolamine phosphate transferase 1 | 2.50 × 10−3 | |
| Presymptomatic Carrier (P2) | P62633 | CNBP | Cellular nucleic acid-binding protein | 1.6801 × 10−9 |
| Q92786 | PROX1 | Prospero Homeobox protein 1 | 2.733 × 10−9 | |
| H7BYV6 | BIN3 | Bridging integrator 3 | 3.9233 × 10−9 | |
| G3V4C6 | RTRAF | RNA transcription_ translation, and transport factor protein | 2.3644 × 10−7 | |
| O75947 | ATP5H | ATP synthase subunit d_ mitochondrial | 6.5903 × 10−7 | |
| B1AKR6 | DYNLRB1 | Dynein light chain roadblock-type 1 | 8.1222 × 10−7 | |
| Q95365 | HLA-B | HLA class I histocompatibility antigen_ B-38 alpha chain | 1.348 × 10−5 | |
| A0A0C3SFZ9 | FCHO1 | F-BAR domain only protein 1 | 1.5595 × 10−5 | |
| Q8IWT3 | CUL9 | Cullin-9 | 2.1129 × 10−5 | |
| P54577 | YARS | Tyrosine--tRNA ligase_ cytoplasmic | 3.6024 × 10−5 | |
| Q9Y5L4 | TIMM13 | Mitochondrial import inner membrane translocase subunit Tim13 | 0.0020643 | |
| Q9Y2E4 | DIP2C | Disco-interacting protein 2 homolog C | 0.00959067 |
| Uniprot ID | Gene Symbol (GS) | Description | FC | ANOVA (p) | |
|---|---|---|---|---|---|
| Symptomatic carrier (P1) | P68871 | HBB | Hemoglobin subunit beta | −189.5 | 1.99 × 10−6 |
| Q9HCM1 | KIAA1551 | Uncharacterized protein KIAA1551 | −52.0 | 1.05 × 10−5 | |
| P20591 | MX1 | Interferon-induced GTP-binding protein Mx1 | −37.1 | 4.19 × 10−6 | |
| A0A2R8Y7C0 | HBA2 | Hemoglobin subunit alpha (Fragment) | −27.6 | 3.04 × 10−6 | |
| E7EMF1 | ITGA2 | Integrin alpha-2 | −26.9 | 7.13 × 10−5 | |
| Q86YA3 | ZGRF1 | Protein ZGRF1 | −29.7 | 1.42 × 10−2 | |
| Q8TEP8 | CEP192 | Centrosomal protein of 192 kDa | −19.2 | 3.72 × 10−5 | |
| Q15283 | RASA2 | Ras GTPase-activating protein 2 | −14.3 | 5.99 × 10−5 | |
| Q9Y6K5 | OAS3 | 2′-5′-oligoadenylate synthase 3 | −13.5 | 2.02 × 10−5 | |
| O14879 | IFIT3 | Interferon-induced protein with tetratricopeptide repeats 3 | −11.2 | 3.56 × 10−6 | |
| Presymptomatic Carrier (P2) | Q9Y6U3 | SCIN | Adseverin | −14.9 | 1.10 × 10−4 |
| C9J1V9 | EEF1E1-BLOC1S5 | EEF1E1-BLOC1S5 readthrough (NMD candidate) | −9.7 | 3.39 × 10−4 | |
| Q7Z2Z1 | TICRR | Treslin | −8.7 | 8.58 × 10−5 | |
| P35580 | MYH10 | Myosin-10 | −7.8 | 1.28 × 10−4 | |
| Q16620 | NTRK2 | BDNF/NT-3 growth factors receptor | −7.6 | 3.14 × 10−4 | |
| P32455 | GBP1 | Guanylate-binding protein 1 | −5.6 | 1.28 × 10−4 | |
| Q9NZR2 | LRP1B | Low-density lipoprotein receptor-related protein 1B | −5.5 | 8.13 × 10−5 | |
| O75131 | CPNE3 | Copine-3 | −5.2 | 2.32 × 10−4 | |
| C9JD73 | PPP1R7 | Protein phosphatase 1 regulatory subunit 7 | −5.0 | 1.13 × 10−4 | |
| A0A087X0Y2 | UTY | Histone demethylase UTY | −4.1 | 2.15 × 10−3 |
| Uniprot ID | GS | Description | FC | ANOVA (p) | |
|---|---|---|---|---|---|
| Symptomatic carrier (P1) | P25092 | GUCY2C | Heat-stable enterotoxin receptor | 34.9 | 2.07 × 10−2 |
| Q13523 | PRPF4B | Serine/threonine-protein kinase PRP4 homolog | 25.9 | 4.42 × 10−5 | |
| Q15691 | MAPRE1 | Microtubule-associated protein RP/EB family member 1 | 19.8 | 2.28 × 10−2 | |
| Q8TD84 | DSCAML1 | Down syndrome cell adhesion molecule-like protein 1 | 10.4 | 6.39 × 10−5 | |
| Q4LDE5 | SVEP1 | Sushi_ von Willebrand factor type A_ EGF and pentraxin domain-containing protein 1 | 9.4 | 7.57 × 10−5 | |
| Q96FQ6 | S100A16 | Protein S100-A16 | 9.3 | 1.45 × 10−5 | |
| P11908 | PRPS2 | Ribose-phosphate pyrophosphokinase 2 | 5.9 | 9.41 × 10−5 | |
| P17050 | NAGA | Alpha-N-acetylgalactosaminidase | 5.4 | 1.82 × 10−7 | |
| O00750 | PIK3C2B | Phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit beta | 5.2 | 9.90 × 10−5 | |
| A0A2R8Y5F1 | TSC2 | Tuberin | 5.0 | 1.22 × 10−4 | |
| Presymptomatic Carrier (P2) | O00429 | DNM1L | Dynamin-1-like protein | 94.5 | 1.29 × 10−3 |
| Q99627 | COPS8 | COP9 signalosome complex subunit 8 | 35.0 | 6.07 × 10−7 | |
| P07333 | CSF1R | Macrophage colony-stimulating factor 1 receptor | 34.3 | 7.27 × 10−6 | |
| P05386 | RPLP1 | 60S acidic ribosomal protein P1 | 19.8 | 4.54 × 10−2 | |
| P62942 | FKBP1A | Peptidyl-prolyl cis-trans isomerase FKBP1A | 10.3 | 3.36 × 10−6 | |
| P02768 | ALB | Serum albumin | 8.2 | 4.31 × 10−6 | |
| J3KQ32 | OLA1 | Obg-like ATPase 1 | 7.3 | 7.48 × 10−6 | |
| Q92817 | EVPL | Envoplakin | 5.3 | 3.00 × 10−5 | |
| P50416 | CPT1A | Carnitine O-palmitoyltransferase 1_ liver isoform | 4.0 | 2.37 × 10−3 | |
| Q9Y2X3 | NOP58 | Nucleolar protein 58 | 3.5 | 4.94 × 10−5 |
| KEGG Entry | Pathway | Size | Overlapped Proteins | Enrich Ratio | p Value | FDR |
|---|---|---|---|---|---|---|
| hsa00030 | Pentose phosphate pathway | 30 | 6 (↑: PRPS1, PRPS2. ↓: ALDOC, PGD, PGM2, TKT) | 7.32 | 1.32 × 10−4 | 8.86 × 10−3 |
| hsa01230 | Biosynthesis of amino acids | 75 | 12 (↑: ALDH18A1, PRPS1, PRPS2, PHGDH. ↓: ALDOC, MAT2A, PGAM1, PGAM4, PGK1, PKM, TKT, TPI1) | 5.86 | 7.19 × 10−7 | 1.17 × 10−4 |
| hsa05020 | Prion diseases | 35 | 5 (↑: MAPK1. ↓: HSPA1A, HSPA5, SOD1, STIP1) | 5.23 | 2.41 × 10−3 | 6.04 × 10−2 |
| hsa00330 | Arginine and proline metabolism | 50 | 7 (↑: ALDH18A1, OAT,↓: ALDH7A1, LAP3, NOS2, P4HA1. P4HA2) | 5.13 | 3.77 × 10−5 | 1.76 × 10−5 |
| hsa04141 | Protein processing in the endoplasmic reticulum | 165 | 22 (↑: SAR1A, CRYAB. ERO1A. ↓: BAG2, CALR, CANX, DNAJB11, HSP90AB1, HSP90B1, HSPA1A, HSPA5, PDIA3, PDIA4, PDIA6, ERP29, P4HB, EIF2AK2, HYOU1, LMAN2, PRKCSH, SSR4, STT3A) | 4.88 | 5.29 × 10−10 | 1.73 × 10−7 |
| hsa05412 | Arrhythmogenic right ventricular cardiomyopathy | 72 | 9 (↓: ACTN2, ATP2A2, CTNNA2, DES, DSP, ITGA11, ITGA2, ITGA5, LMNA) | 4.58 | 1.36 × 10−4 | 8.86 × 10−3 |
| hsa00010 | Glycolysis/Gluconeogenesis | 68 | 8 (↓: ALDH7A1, ALDOC, PGAM1, PGAM4, PGK1, PGM2, PKM, TPI1) | 4.31 | 4.89 × 10−4 | 1.99 × 10−2 |
| hsa04612 | Antigen processing and presentation | 77 | 9 (↓: CANX, CALR, HLA-E, HSP90AB1, HSPA1A, HSPA5, PDIA3, PSME1, PSME2) | 4.28 | 2.29 × 10−4 | 1.24 × 10−2 |
| hsa01200 | Carbon metabolism | 116 | 12 (↑: PHGDH, PRPS1, PRPS2, SDHA, ↓: PKM, PGAM1, PGAM4, PGD, PGK1, TKT, ALDOC, TPI1.) | 3.79 | 7.08 × 10−5 | 7.70 × 10−3 |
| hsa04512 | ECM-receptor interaction | 82 | 8 (↑:COL6A3, ↓: COL1A2, COL1A1, FN1, ITGA11, ITGA2, ITGA5, THBS1) | 3.57 | 1.70× 10−3 | 5.00× 10−2 |
| hsa05410 | Hypertrophic cardiomyopathy (HCM) | 83 | 8 (↓: ATP2A2, DES, ITGA11, ITGA5, ITGA2, LMNA, TPM2, TPM3) | 3.53 | 1.84 × 10−3 | 5.00 × 10−2 |
| hsa05414 | Dilated cardiomyopathy (DCM) | 90 | 8 (↓: ATP2A2, DES, ITGA11, ITGA5, ITGA2, LMNA, TPM2, TPM3) | 3.25 | 3.07 × 10−3 | 6.68 × 10−2 |
| hsa05016 | Huntington disease | 193 | 14 (↑: SLC25A5, DCTN1, CLTCL1, CYCS, SDHA. ↓: AP2B1, CLTA, DNAH1, DNAH10, DNAH14, DNAH6, ITPR1, SOD1, TGM2) | 2.66 | 7.83 × 10−4 | 2.55 × 10−2 |
| hsa04810 | Regulation of actin cytoskeleton | 213 | 15 (↑: ENAH, MAPK1, NCKAP1, PAK2, ROCK1. ↓: FN1, GSN, IQGAP3, ITGA11, ITGA2, ITGA5, PAK3, PIKFYVE, RRAS, SCIN) | 2.58 | 6.95 × 10−4 | 2.52 × 10−2 |
| hsa04510 | Focal adhesion | 199 | 13 (↑: COL6A3, PAK2, MAPK1, ROCK1.↓: COL1A2, COL1A1, FN1, ITGA11, ITGA2, ITGA5, KDR, PAK3, THBS1) | 2.39 | 3.06 × 10−3 | 6.68 × 10−2 |
| KEGG Entry | Description | Size | Overlap | EnrRatio | p Value | FDR |
|---|---|---|---|---|---|---|
| hsa00020 | Citrate cycle (TCA cycle) | 30 | 4 (↑: FH, MDH2, ↓: IDH1, IDH2) | 8.10 | 1.38 × 10−3 | 6.93 × 10−2 |
| hsa00480 | Glutathione metabolism | 56 | 5 (↓: G6PD, GSTP1, IDH1, IDH2,PGD) | 5.42 | 2.18 × 10−3 | 7.73 × 10−2 |
| hsa01230 | Biosynthesis of amino acids | 75 | 6 (↓: ALDH18A1, IDH1, IDH2, MAT2A, PHGDH, PKM) | 4.86 | 1.41 × 10−3 | 6.93 × 10−2 |
| hsa05410 | Hypertrophic cardiomyopathy (HCM) | 83 | 6 (↓: CACNA1C, DMD, ITGA2, ITGB1, RYR2, TPM1) | 4.39 | 2.37 × 10−3 | 7.73 × 10−2 |
| hsa05412 | Arrhythmogenic right ventricular cardiomyopathy | 72 | 5 (↓: CACNA1C, DMD, ITGA2, ITGB1, RYR2) | 4.22 | 6.48 × 10−3 | 1.51 × 10−1 |
| hsa01200 | Carbon metabolism | 116 | 8 (↑: FH, MDH2, ↓: G6PD, IDH1, IDH2, PGD, PHGDH, PKM) | 4.19 | 6.20 × 10−4 | 6.93 × 10−2 |
| hsa05100 | Bacterial invasion of epithelial cells | 74 | 5 (↓: CAV1, CLTA, CTTN, ITGB1, PXN) | 4.10 | 7.27 × 10−3 | 1.58 × 10−1 |
| hsa05414 | Dilated cardiomyopathy (DCM) | 90 | 6 (↓: CACNA1C, DMD, ITGA2, ITGB1, RYR2, TPM1) | 4.05 | 3.56 × 10−3 | 9.67 × 10−2 |
| hsa04144 | Endocytosis | 244 | 14 (↑: IGF2R, WASHC5 ↓: AP2B1, CAV1, CLTA, EHD3, HLA-A, HSPA2, ITCH, RAB11FIP4, RAB5A, RAB7A, UBB, VPS35) | 3.48 | 4.38 × 10−5 | 1.43 × 10−2 |
| hsa04142 | Lysosome | 123 | 7 (↑: ABCA2, CTSB, CTSZ, IGF2R ↓: CD63, CLTA, PSAP) | 3.46 | 4.06 × 10−3 | 1.02 × 10−1 |
| hsa04141 | Protein processing in the endoplasmic reticulum | 165 | 9 (↑: SEC31A, SSR1, UBE4B ↓: CKAP4, HSPA2, LMAN2, MOGS, RPN2, SEC23A) | 3.31 | 1.54 × 10−3 | 6.93 × 10−2 |
| hsa04145 | Phagosome | 152 | 8 (↑: VAMP3. ↓: HLA-A, ITGA2, ITGB1, RAB5A, RAB7A, THBS1, TUBB3) | 3.20 | 3.50 × 10−3 | 9.67 × 10−2 |
| hsa04810 | Regulation of actin cytoskeleton | 213 | 11 (↑: ARHGAP35, CFL1, CFL2, CYFIP1, IQGAP2, PFN1, ↓: ITGA2, ITGB1, MYH10, PXN, SCIN) | 3.14 | 7.35 × 10−4 | 6.93 × 10−2 |
| hsa04510 | Focal adhesion | 199 | 10 (↑: ARHGAP35, COL1A1, COL1A2, FLNA, FLNB, ↓: CAV1, ITGA2, ITGB1, PXN, THBS1) | 3.05 | 1.58 × 10−3 | 6.93 × 10−2 |
| hsa05205 | Proteoglycans in cancer | 201 | 10 (↑: FLNA, FLNB, ↓: CAV1, CD63, CTTN, ITGA2, ITGB1, ITPR3, PXN, THBS1) | 3.02 | 1.70 × 10−3 | 6.93 × 10−2 |
| Pathways | DEPs in Symptomatic Carrier | DEPs in Presymptomatic Carrier |
|---|---|---|
| ENERGY METABOLISM | ↑: PRPS1 1,2,3, PRPS2 1,2,3, ALDH18A1 2,6, PHGDH 2,3, SDHA 3,5,8, OAT 6, CYCS 8. ↓: ALDOC 1,2,3,4, PGD 1,3,7, PGM2 1,4, TKT 1,2,3, MAT2A 2, PGAM1 2,3,4, PGAM4 2,3,4, PGK1 2,3,4, PKM 2,3,4, TPI1 2,3,4, ALDH7A1 4,6, LAP3 6,7, P4HA1 6, P4HA2 6, NOS2 6, GSTO1 7, PRDX6 7. | ↑: FH 3,5, MDH2 3,5, GSTP1 7, COX5A 8. ↓: G6PD 1,3,7, PGD 1,3,7, PGM1 1,4, ALDH18A1 2,6, IDH1 2,3,5,7, IDH2 2,3,5,7, MAT2A 2, PHGDH 2,3, PKM 2,3,4, P4HA1 6, ATP5PO 8. |
| VESICLE TRANSPORT AND DEGRADATION | ↑: KIF5B, STAM2 a, CLTCL1 a,b, NAGA b, TUBB3 c, DYNC2H1 c, TUBAL3 c, PSMD2 d, UBRS e, MAPK1 f,g,TSC2 g, IRS4 g, ATG4C g ↓: AP2B1 a, CLTA a,b, HLA-E a,c, HSPA1A a, KIF5C a, CHMPS a, PML a, EPS15L1 a, SNX6 a, SH3GL1 a, CTSD b,g,, SCARB2 b, PIKFYVE c, ITGA2 c, ITGA5 c, THBS1 c, CALR c, CANX c, PSMA4 d, PSMA6 d, PSME1 d, PSME2 d, PML e, HERC1 e, GSN f, SCIN f, ITPR1 g, RRAS g | ↑: IGF2R a,b,, WASHC5 a,, ABCA2 b, CTSB b, CTSZ b, IGF2R a,b, VAMP3 c, UBE4B e, DDB1 e, CFL1 f, CFL2 f, PP2CB g ↓: AP2B1 a, CAV1 a,, CLTA a,b, EHD3 a,, HLA-A a,c, HSPA2 a,, ITCH a,e,, RAB11FIP4 a, RAB5A a,c, RAB7A a,c,g, UBB a,e, VPS35 a,, CD63 b, PSAP b, ITGA2 c, ITGB1 c, THBS1 c, TUBB3 c, PSMB2 d, PSMB3 d, ANAPC7 e, UBA6 e, MARCKS f, SCIN f |
| PROTEIN PROCESSING IN THE ENDOPLASMIC RETICULUM | 22 (↑: CRYAB, SAR1A ↓: BAG2, CALR, CANX, DNAJB11, HSP90AB1, HSP90B1, HSPA1A, HSPA5, PDIA3, PDIA4, PDIA6, ERP29, ERO1A, P4HB, EIF2AK2, HYOU1, LMAN2, PRKCSH, SSR4, STT3A) | 9 (↑: SEC31A, SSR1, UBE4B ↓: CKAP4, HSPA2, LMAN2, MOGS, RPN2, SEC23A) |
| REGULATION OF ACTIN CYTOSKELETON | 15 (↑:ENAH, MAPK1, NCKAP1, PAK2, ROCK1 ↓:FN1, GSN, IQGAP3, ITGA11, ITGA2, ITGA5, PAK3, PIKFYVE, RRAS, SCIN) | 11 (↑: ARHGAP35, CFL1. CFL2, CYFIP1, IQGAP2, PFN1, ↓: ITGA2, ITGB1, MYH10, PXN, SCIN) |
| FOCAL ADHESION | 13 (↑: COL6A3, MAPK1, PAK2, ROCK1, ↓: COL1A1, COL1A2, FN1, ITGA11, ITGA2, ITGA5, KDR, PAK3, THBS1, FREM2) | 10 (↑: ARHGAP35, COL1A1, COL1A2, FLNA, FLNB, ↓: CAV1, ITGA2, ITGB1, PXN, THBS1) |
| ARRHYTHMOGENIC CARDIOMYOPATHY | ↓: ACTN2, ATP2A2, CTNNA2, DES, DSP, ITGA11, ITGA2, ITGA5, LMNA TPM2, TPM3 | ↓: DMD, ITGA2, ITGB1, RYR2, TPM1, CACNA1C |
| NEURODEGENERATIVE DISEASES (NDDs) | ↑: TUBB3 A,B,C,D,E, TUBAL3 A,B,C,D,E, KIF5B A,B,C,D,E, SLC25A5 A,B,C,D, CYCS A,B,C,D,E, PSMD2 A,B,C,D,E, MAPK1 A, SDHA A,B,C,D,E, IRS4 A, CAMK2D B RYR1 C, DCTN1 D,E, CLTCL1 D, NUP214 E ↓: ITPR1 A,B,C,D, PSMA4 A,B,C,D,E, PSMA6 A,B,C,D,E, ADAM10 A, NOS2 A,E, ATP2A2 A, EIF2AK2 A HSPA5 B, SOD1 B,C,D,E, TXN B, GNAI1 B, GNAI2 B, HSPA5 C,E, HSPA1A C, STIP1 C, CLTA H, DNAH14 D,E, AP2B1 D, DNAH6 D,E, DNAH10 D,E, DNAH1 D,E, TGM2 D, U:KIF5C A,B,D,E, ATP5PF A,B,D,E, SETX E, FUS E, ANXA11 E, MAP2K3 E, MATR3 E | ↑: VDAC1 A,B,C,D,E, COX5A A,B,C,D,E, MME A, RYR1 C, PFN1 E, SRSF3 E, HNRNPA3 E ↓: TUBB3 A,B,C,D,E, ITPR3 A,B,C,E, ATP5PO A,B,C,D,E, PSMB2 A,B,C,D,E, PSMB3 A,B,C,D,E, CACNA1C A,C, UBB B, UCHL1 B CAV1 C, RYR2 C, HSPA2 C, ACTR1A D,E, CLTA D, AP2B1 D, DNAH6 D,E, SOD2 D, FUS E, MATR3 E, RAB5A E |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochín-Hernández, L.J.; Jiménez-Acosta, M.A.; Ramírez-Reyes, L.; Figueroa-Corona, M.d.P.; Sánchez-González, V.J.; Orozco-Barajas, M.; Meraz-Ríos, M.A. The Proteome Profile of Olfactory Ecto-Mesenchymal Stem Cells-Derived from Patients with Familial Alzheimer’s Disease Reveals New Insights for AD Study. Int. J. Mol. Sci. 2023, 24, 12606. https://doi.org/10.3390/ijms241612606
Rochín-Hernández LJ, Jiménez-Acosta MA, Ramírez-Reyes L, Figueroa-Corona MdP, Sánchez-González VJ, Orozco-Barajas M, Meraz-Ríos MA. The Proteome Profile of Olfactory Ecto-Mesenchymal Stem Cells-Derived from Patients with Familial Alzheimer’s Disease Reveals New Insights for AD Study. International Journal of Molecular Sciences. 2023; 24(16):12606. https://doi.org/10.3390/ijms241612606
Chicago/Turabian StyleRochín-Hernández, Lory J., Miguel A. Jiménez-Acosta, Lorena Ramírez-Reyes, María del Pilar Figueroa-Corona, Víctor J. Sánchez-González, Maribel Orozco-Barajas, and Marco A. Meraz-Ríos. 2023. "The Proteome Profile of Olfactory Ecto-Mesenchymal Stem Cells-Derived from Patients with Familial Alzheimer’s Disease Reveals New Insights for AD Study" International Journal of Molecular Sciences 24, no. 16: 12606. https://doi.org/10.3390/ijms241612606
APA StyleRochín-Hernández, L. J., Jiménez-Acosta, M. A., Ramírez-Reyes, L., Figueroa-Corona, M. d. P., Sánchez-González, V. J., Orozco-Barajas, M., & Meraz-Ríos, M. A. (2023). The Proteome Profile of Olfactory Ecto-Mesenchymal Stem Cells-Derived from Patients with Familial Alzheimer’s Disease Reveals New Insights for AD Study. International Journal of Molecular Sciences, 24(16), 12606. https://doi.org/10.3390/ijms241612606