
YAP at the Crossroads of Biomechanics and Drug Resistance in Human Cancer

 ,
, 
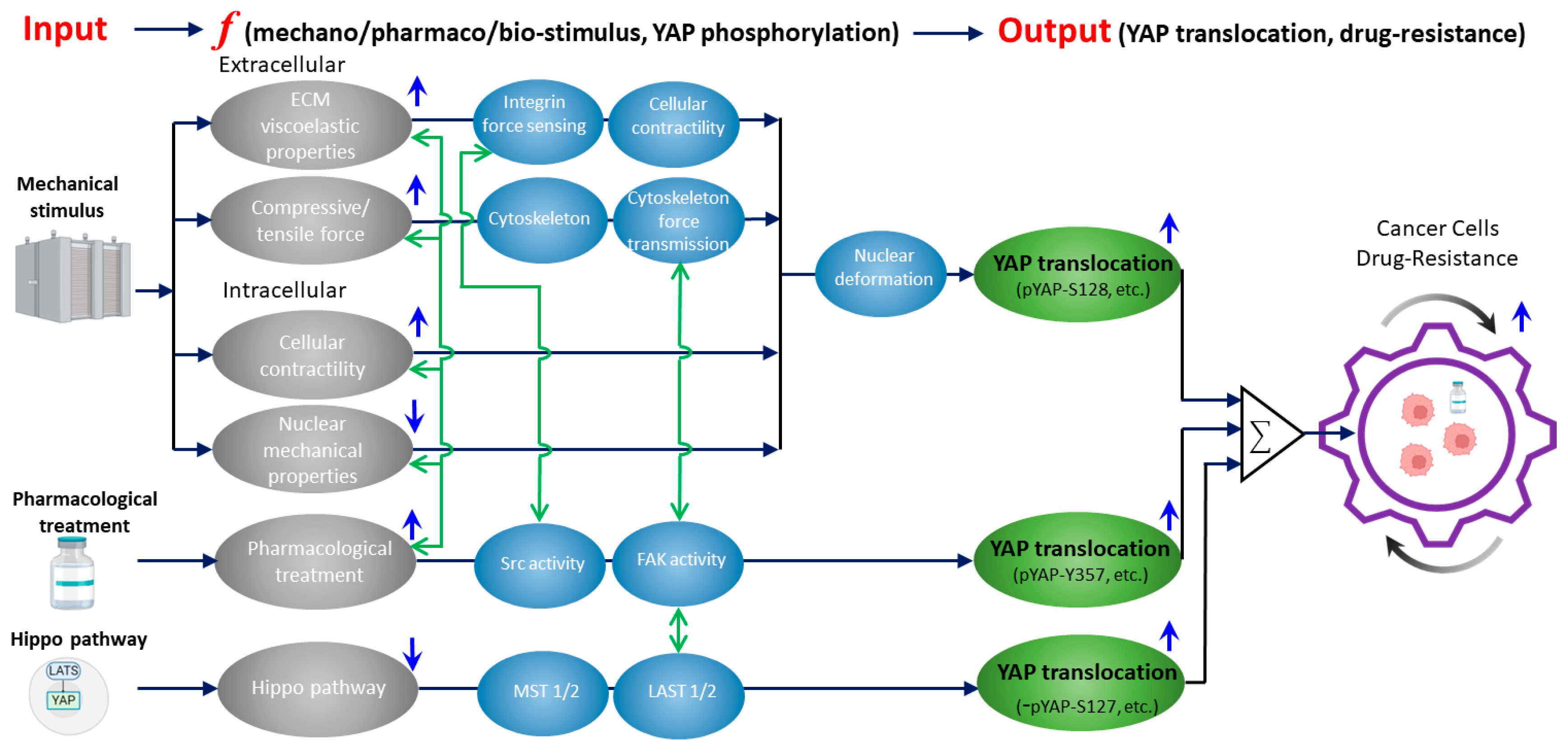{kind=link}
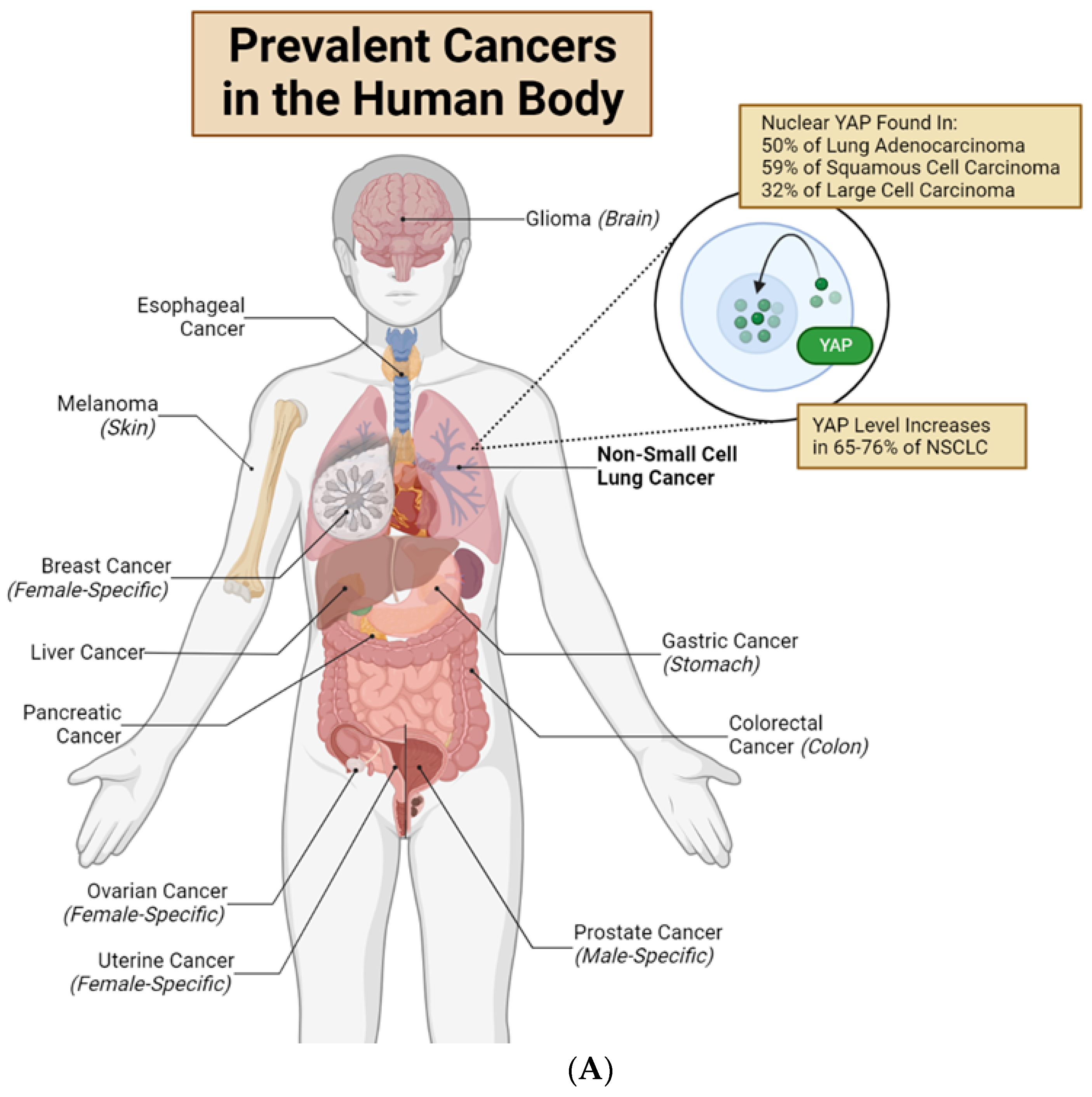{kind=link}
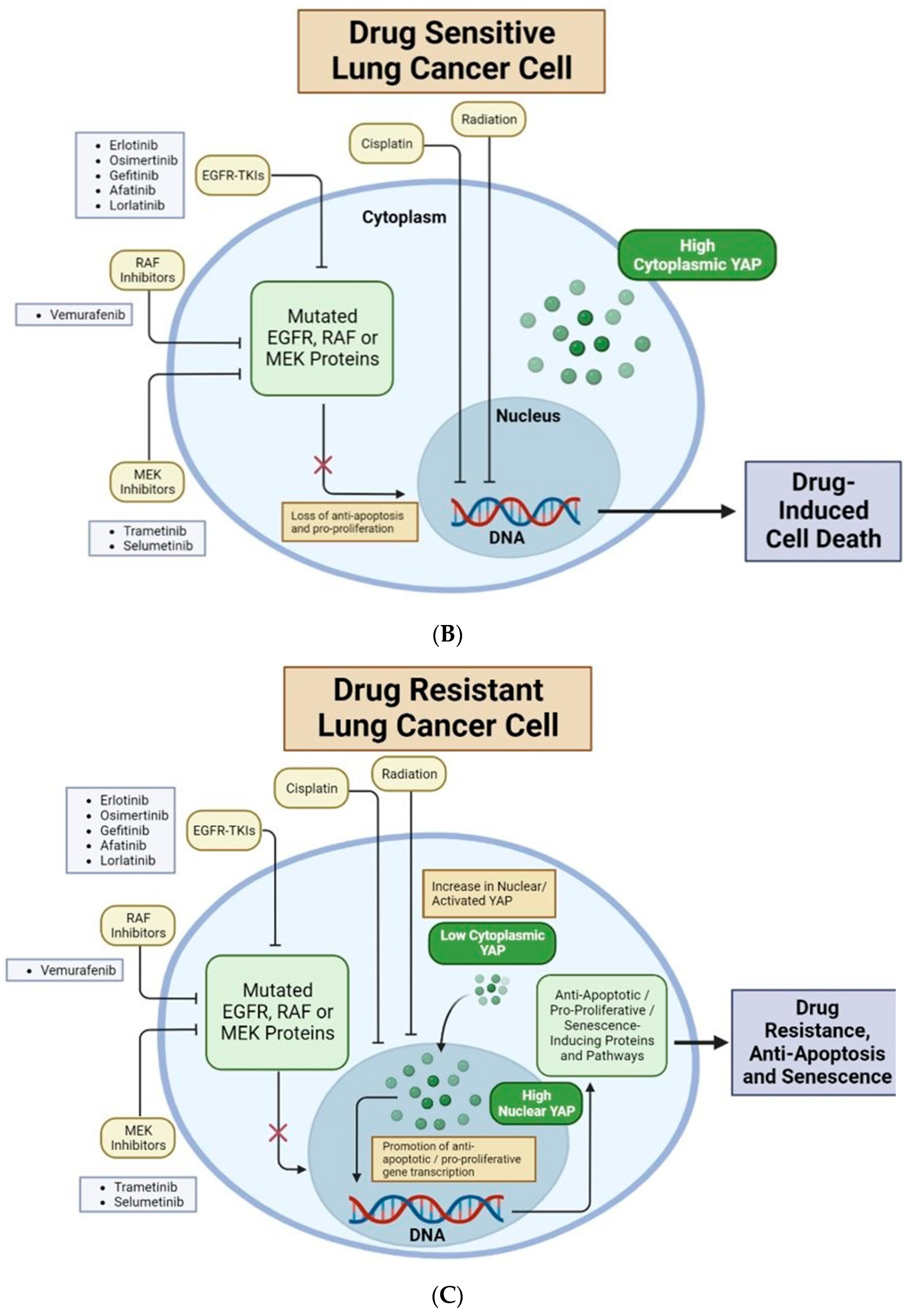{kind=link}
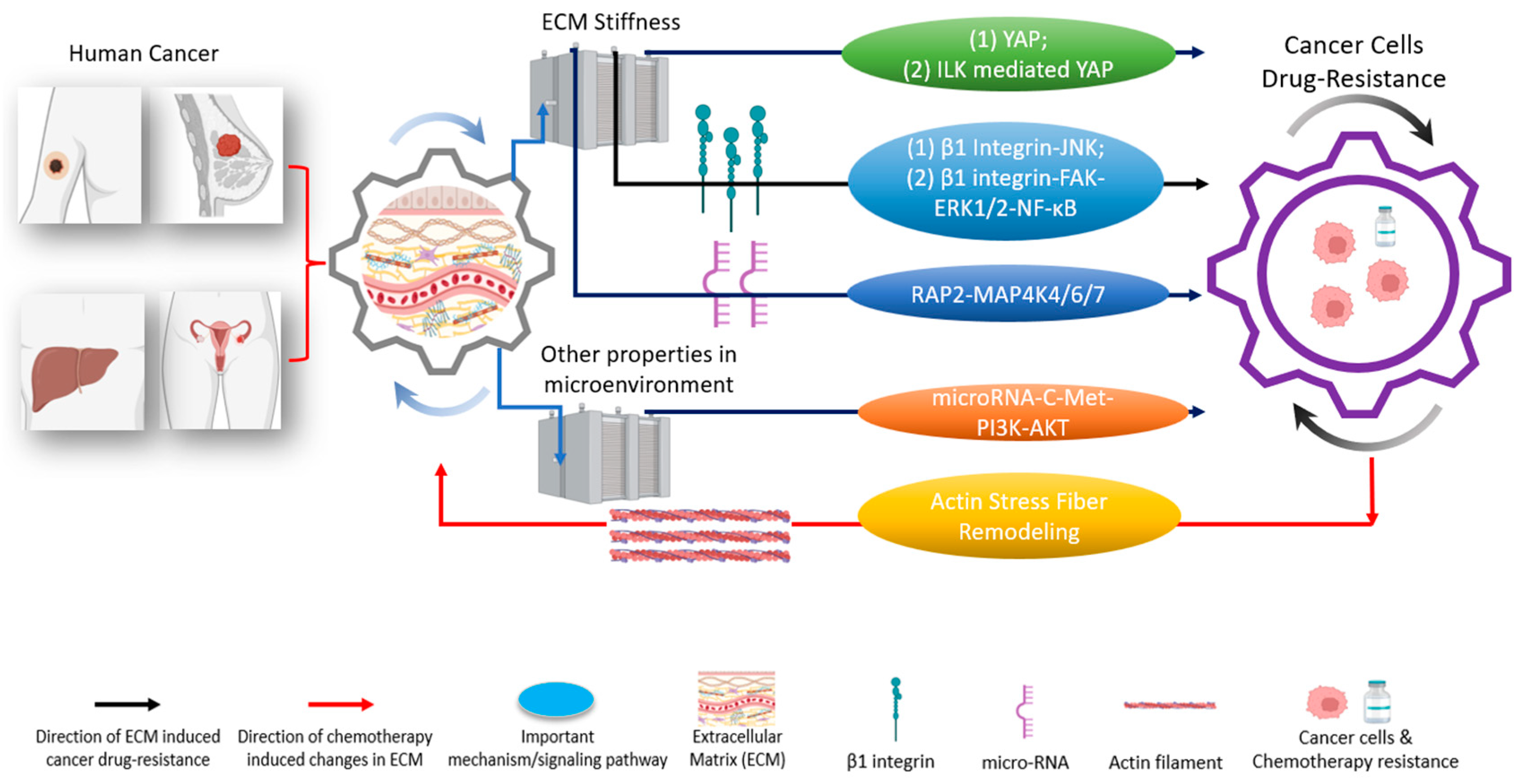{kind=link}
Abstract
1. Introduction
2. Mechanical Regulation of YAP Dynamics
2.1. Viscoelastic Mechanics of ECM Regulates YAP
2.2. Geometric Attributes and Contractility of the Cell Regulate YAP
2.3. Actively Applied Extracellular Forces Regulate YAP
2.4. Mechanism of Mechanically Regulated YAP Nuclear Translocation
3. YAP and Drug Resistance
3.1. YAP Is More Activated in Drug-Resistant Cancer Cells
3.2. Artificially Changed YAP Expression Directly Influences Drug Resistance
3.3. Mechanisms of YAP-Mediated Drug Resistance
3.4. YAP and Immunotherapy
4. Roles of ECM Played in Regulating YAP and Drug Resistance in Cancer Cells
4.1. ECM Induces YAP Nuclear Translocation and Influences the Resistance/Sensitivity of Cancer Cells to Chemotherapies
4.2. ECM Influences Resistance/Sensitivity of Cancer Cells to Chemotherapies in a YAP-Independent Manner
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| ACADL | Long-chain Acyl-CoA dehydrogenase |
| AFM | Atomic force microscopy |
| BCL-xL | B-cell Lymphoma-Extra-Large |
| BMF | Bcl2 Modifying Factor |
| BRAF | B-raf protein (v-Raf murine sarcoma viral oncogene homolog B1) |
| C-Met | Mesenchymal-epithelial transition factor |
| CTGF | Connective tissue growth factor |
| DCIS | Ductal carcinoma in situ |
| DSBs | DNA double-strand breaks |
| ECM | Extracellular matrix |
| EGFR | Epithelial Growth Factor |
| ERK | Extracellular signal-regulated kinases |
| EMT | Epithelial-Mesenchymal Transition |
| GPCR | G-protein coupled receptors |
| Her2 | Human Epidermal Growth Factor Receptor 2 |
| ILK | Integrin-linked kinase |
| IFN-γ | Interferon-γ |
| JNK | c-Jun N-terminal kinases |
| KRAS | Kirsten Rat Sarcoma |
| LATS1 | Large Tumor Suppressor Kinase 1 |
| MAP4K4/6/7 | Mitogen-activated protein kinase-4/6/7 |
| MEK | Mitogen-Activated Protein Kinase |
| MST1/2 | Macrophage-stimulating protein |
| MPM | Malignant Pleural Mesothelioma |
| NF2 | Neurofibromatosis type 2 |
| NF-κB | Nuclear factor kappa B |
| NSCLC | Non-Small Cell Lung Cancer |
| OCC | Ovarian Cancer Cell |
| P38 | P38 mitogen-activated protein kinase |
| PD-L1 | Programmed Death-Ligand 1 |
| PI3k | Phosphatidylinositol 3-kinase |
| p-YAP | Phosphorylated Yes-Associated Protein |
| Rho-ROCK | Rho/Rho-associated coiled-coil containing protein kinase |
| ROS1 | C-ROS Oncogene 1 Receptor Tyrosine Kinase |
| siRNA | Small Interfering Ribonucleic Acid |
| SLUG | Transcription Factor Encoded by the SNAI2 Gene |
| TEAD | Transcriptional Enhanced Associate Domain |
| TCP | Tissue culture plastics |
| TKI | Tyrosine Kinase Inhibitor |
| TRP | Transient Receptor Potential |
| YAP | Yes-associated protein |
| YAP N/C ratio | YAP concentrations in nucleus vs. cytoplasm |
References
- Discher, D.E.; Janmey, P.; Wang, Y. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth Factors, Matrices, and Forces Combine and Control Stem Cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.; Sheetz, M. Local Force and Geometry Sensing Regulate Cell Functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 265–275. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Chen, C.S. Mechanotransduction in Development: A Growing Role for Contractility. Nat. Rev. Mol. Cell Biol. 2009, 10, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a Distance: Mechanically Coupling the Extracellular Matrix with the Nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Huang, M.; Li, T.; Li, L.; Sussman, H.; Dai, Y.; Siemann, D.W.; Xie, M.; Tang, X. Towards an Integrative Understanding of Cancer Mechanobiology: Calcium, YAP, and MicroRNA under Biophysical Forces. Soft Matter 2022, 18, 1112–1148. [Google Scholar] [CrossRef]
- Carvalho, E.M.; Kumar, S. Lose the Stress: Viscoelastic Materials for Cell Engineering. Acta Biomater. 2023, 163, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Kumar, S. Biophysical Regulation of Tumor Cell Invasion: Moving beyond Matrix Stiffness. Integr. Biol. 2011, 3, 267. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Engler, A.J.; Kamm, R. Mechanobiology of the Cell Nucleus. APL Bioeng. 2022, 6, 040401. [Google Scholar] [CrossRef]
- Tang, X.; Bajaj, P.; Bashir, R.; Saif, T.A. How Far Cardiac Cells Can See Each Other Mechanically. Soft Matter 2011, 7, 6151. [Google Scholar] [CrossRef]
- Tang, X.; Yakut Ali, M.; Saif, M.T.A. A Novel Technique for Micro-Patterning Proteins and Cells on Polyacrylamide Gels. Soft Matter 2012, 8, 7197. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as Master Regulators in Cancer: Modulation, Function and Therapeutic Approaches. Nat. Cancer 2022, 4, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M. Yes-Associated Protein (YAP65) Is a Proline-Rich Phosphoprotein That Binds to the SH3 Domain of the Yes Proto-Oncogene Product. Oncogene 1994, 9, 2145–2152. [Google Scholar]
- Hong, W.; Guan, K.-L. The YAP and TAZ Transcription Co-Activators: Key Downstream Effectors of the Mammalian Hippo Pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD Mediates YAP-Dependent Gene Induction and Growth Control. Genes. Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef]
- Haderk, F.; Fernández-Méndez, C.; Čech, L.; Yu, J.; Meraz, I.M.; Olivas, V.; Rabago, D.B.; Lucas Kerr, D.; Gomez, C.; Allegakoen, D.V.; et al. A Focal Adhesion Kinase-YAP Signaling Axis Drives Drug Tolerant Persister Cells and Residual Disease in Lung Cancer. Cancer Biol. 2021; preprint. [Google Scholar] [CrossRef]
- Low, B.C.; Pan, C.Q.; Shivashankar, G.V.; Bershadsky, A.; Sudol, M.; Sheetz, M. YAP/TAZ as Mechanosensors and Mechanotransducers in Regulating Organ Size and Tumor Growth. FEBS Lett. 2014, 588, 2663–2670. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes. Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, Z.; Rodriguez-Barrueco, R.; Borczuk, A.; Liu, H.; Yu, J.; Silva, J.M.; Cheng, S.K.; Perez-Soler, R.; Halmos, B. Functional Genomics Screen Identifies YAP1 as a Key Determinant to Enhance Treatment Sensitivity in Lung Cancer Cells. Oncotarget 2016, 7, 28976–28988. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.K.-H.; Du, W.; Shelton, S.J.; Oldham, M.C.; DiPersio, C.M.; Klein, O.D. An FAK-YAP-MTOR Signaling Axis Regulates Stem Cell-Based Tissue Renewal in Mice. Cell Stem Cell 2017, 21, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Arang, N.; Rigiracciolo, D.C.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; König, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals That the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 2019, 35, 457–472. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Del Río Hernández, A.E. FAK Controls the Mechanical Activation of YAP, a Transcriptional Regulator Required for Durotaxis. FASEB J. 2018, 32, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gong, Y.; Guo, T.; Li, M.; Fang, L.; Yin, S.; Kamran, M.; Liu, Y.; Xu, J.; Xu, L.; et al. Activation of Aurora A Kinase Increases YAP Stability via Blockage of Autophagy. Cell Death Dis. 2019, 10, 432. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Schwarz, U.S.; Riveline, D.; Goichberg, P.; Tzur, G.; Sabanay, I.; Mahalu, D.; Safran, S.; Bershadsky, A.; Addadi, L.; et al. Force and Focal Adhesion Assembly: A Close Relationship Studied Using Elastic Micropatterned Substrates. Nat. Cell Biol. 2001, 3, 466–472. [Google Scholar] [CrossRef]
- Del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science 2009, 323, 638–641. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force Fluctuations within Focal Adhesions Mediate ECM-Rigidity Sensing to Guide Directed Cell Migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical Regulation of a Molecular Clutch Defines Force Transmission and Transduction in Response to Matrix Rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef]
- Zhou, D.W.; Fernández-Yagüe, M.A.; Holland, E.N.; García, A.F.; Castro, N.S.; O’Neill, E.B.; Eyckmans, J.; Chen, C.S.; Fu, J.; Schlaepfer, D.D.; et al. Force-FAK Signaling Coupling at Individual Focal Adhesions Coordinates Mechanosensing and Microtissue Repair. Nat. Commun. 2021, 12, 2359. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, H.; Wang, J.; Liu, Y.; Luo, T.; Hua, H. Targeting Extracellular Matrix Stiffness and Mechanotransducers to Improve Cancer Therapy. J. Hematol. Oncol. 2022, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Gargalionis, A.N.; Papavassiliou, K.A.; Papavassiliou, A.G. Mechanobiology of Solid Tumors. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2022, 1868, 166555. [Google Scholar] [CrossRef]
- Qin, X.; Lv, X.; Li, P.; Yang, R.; Xia, Q.; Chen, Y.; Peng, Y.; Li, L.; Li, S.; Li, T.; et al. Matrix Stiffness Modulates ILK-Mediated YAP Activation to Control the Drug Resistance of Breast Cancer Cells. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165625. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Sun, Q.; Li, X.; Feng, J.; Ao, Z.; Li, X.; Wang, J. Substrate Stiffness Modulates the Growth, Phenotype, and Chemoresistance of Ovarian Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 718834. [Google Scholar] [CrossRef]
- Gao, J.; Rong, Y.; Huang, Y.; Shi, P.; Wang, X.; Meng, X.; Dong, J.; Wu, C. Cirrhotic Stiffness Affects the Migration of Hepatocellular Carcinoma Cells and Induces Sorafenib Resistance through YAP. J. Cell. Physiol. 2019, 234, 2639–2648. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.; Lee, J.; Jung, E.; Kim, J. Actin Remodeling Confers BRAF Inhibitor Resistance to Melanoma Cells through YAP / TAZ Activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Yamazoe, M.; Ozasa, H.; Tsuji, T.; Funazo, T.; Yoshida, H.; Hashimoto, K.; Hosoya, K.; Ogimoto, T.; Ajimizu, H.; Yoshida, H.; et al. Yes-associated protein 1 Mediates Initial Cell Survival during Lorlatinib Treatment through AKT Signaling in ROS1 -rearranged Lung Cancer. Cancer Sci. 2023, 114, 546–560. [Google Scholar] [CrossRef]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital Imaging Reveals How BRAF Inhibition Generates Drug-Tolerant Microenvironments with High Integrin Β1/FAK Signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Liang, C.; Huang, M.; Tanaka, M.; Lightsey, S.; Temples, M.; Lepler, S.E.; Sheng, P.; Mann, W.P.; Widener, A.E.; Siemann, D.W.; et al. Functional Interrogation of Ca2+ Signals in Human Cancer Cells In Vitro and Ex Vivo by Fluorescent Microscopy and Molecular Tools. In Microfluidic Systems for Cancer Diagnosis; Methods in Molecular Biology; Garcia-Cordero, J.L., Revzin, A., Eds.; Springer: New York, NY, USA, 2023; Volume 2679, pp. 95–125. [Google Scholar] [CrossRef]
- Gonzalez-Molina, J.; Moyano-Galceran, L.; Single, A.; Gultekin, O.; Alsalhi, S.; Lehti, K. Chemotherapy as a Regulator of Extracellular Matrix-Cell Communication: Implications in Therapy Resistance. Semin. Cancer Biol. 2022, 86, 224–236. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD–YAP Complex Suppresses the Oncogenic Activity of YAP. Genes. Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, L.; Tao, Z.; Jarugumilli, G.K.; Erb, H.; Singh, A.; Li, Q.; Cotton, J.L.; Greninger, P.; Egan, R.K.; et al. Pharmacological Blockade of TEAD–YAP Reveals Its Therapeutic Limitation in Cancer Cells. Nat. Commun. 2022, 13, 6744. [Google Scholar] [CrossRef]
- Lee, J.E.; Park, H.S.; Lee, D.; Yoo, G.; Kim, T.; Jeon, H.; Yeo, M.-K.; Lee, C.-S.; Moon, J.Y.; Jung, S.S.; et al. Hippo Pathway Effector YAP Inhibition Restores the Sensitivity of EGFR-TKI in Lung Adenocarcinoma Having Primary or Acquired EGFR-TKI Resistance. Biochem. Biophys. Res. Commun. 2016, 474, 154–160. [Google Scholar] [CrossRef]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP Function Overcomes BRAF Inhibitor Resistance in Melanoma Cancer Stem Cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef]
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Diaz Espinosa, A.M.; et al. Selective YAP/TAZ Inhibition in Fibroblasts via Dopamine Receptor D1 Agonism Reverses Fibrosis. Sci. Transl. Med. 2019, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Jain, R.K. Tumor Microenvironment Abnormalities: Causes, Consequences, and Strategies to Normalize. J. Cell. Biochem. 2007, 101, 937–949. [Google Scholar] [CrossRef]
- Pfeifer, C.R.; Alvey, C.M.; Irianto, J.; Discher, D.E. Genome Variation across Cancers Scales with Tissue Stiffness—An Invasion-Mutation Mechanism and Implications for Immune Cell Infiltration. Curr. Opin. Syst. Biol. 2017, 2, 103–114. [Google Scholar] [CrossRef]
- Chin, L.; Xia, Y.; Discher, D.E.; Janmey, P.A. Mechanotransduction in Cancer. Curr. Opin. Chem. Eng. 2016, 11, 77–84. [Google Scholar] [CrossRef]
- Poh, Y.-C.; Shevtsov, S.P.; Chowdhury, F.; Wu, D.C.; Na, S.; Dundr, M.; Wang, N. Dynamic Force-Induced Direct Dissociation of Protein Complexes in a Nuclear Body in Living Cells. Nat. Commun. 2012, 3, 866. [Google Scholar] [CrossRef]
- Poh, Y.-C.; Chen, J.; Hong, Y.; Yi, H.; Zhang, S.; Chen, J.; Wu, D.C.; Wang, L.; Jia, Q.; Singh, R.; et al. Generation of Organized Germ Layers from a Single Mouse Embryonic Stem Cell. Nat. Commun. 2014, 5, 4000. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wen, Q.; Kuhlenschmidt, T.B.; Kuhlenschmidt, M.S.; Janmey, P.A.; Saif, T.A. Attenuation of Cell Mechanosensitivity in Colon Cancer Cells during In Vitro Metastasis. PLoS ONE 2012, 7, e50443. [Google Scholar] [CrossRef] [PubMed]
- Wolfenson, H.; Yang, B.; Sheetz, M.P. Steps in Mechanotransduction Pathways That Control Cell Morphology. Annu. Rev. Physiol. 2019, 81, 585–605. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; He, L.; Zhou, L.; Jing, Y.; Wang, F.; Shi, Y.; Cai, M.; Sun, J.; Xu, H.; Jiang, J.; et al. Mechanical Force Regulation of YAP by F-Actin and GPCR Revealed by Super-Resolution Imaging. Nanoscale 2020, 12, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, Q.; Chen, X.; Liu, J.; Tanaka, M.; Wang, S.; Lepler, S.E.; Jin, Z.; Siemann, D.W.; Zeng, B.; et al. Human Cancer Cells Generate Spontaneous Calcium Transients and Intercellular Waves That Modulate Tumor Growth. Biomaterials 2022, 290, 121823. [Google Scholar] [CrossRef]
- Tang, X.; Kuhlenschmidt, T.B.; Li, Q.; Ali, S.; Lezmi, S.; Chen, H.; Pires-Alves, M.; Laegreid, W.W.; Saif, T.A.; Kuhlenschmidt, M.S. A Mechanically-Induced Colon Cancer Cell Population Shows Increased Metastatic Potential. Mol. Cancer 2014, 13, 131. [Google Scholar] [CrossRef]
- Tang, X.; Kuhlenschmidt, T.B.; Zhou, J.; Bell, P.; Wang, F.; Kuhlenschmidt, M.S.; Saif, T.A. Mechanical Force Affects Expression of an In Vitro Metastasis-Like Phenotype in HCT-8 Cells. Biophys. J. 2010, 99, 2460–2469. [Google Scholar] [CrossRef]
- Tang, X.; Tofangchi, A.; Anand, S.V.; Saif, T.A. A Novel Cell Traction Force Microscopy to Study Multi-Cellular System. PLoS Comput. Biol. 2014, 10, e1003631. [Google Scholar] [CrossRef]
- Luo, Q.; Zhang, J.; Huang, M.; Lin, G.; Tanaka, M.; Lepler, S.; Guan, J.; Siemann, D.; Tang, X. Automatic Multi-Functional Integration Program (AMFIP) towards All-Optical Mechano-Electrophysiology Interrogation. PLoS ONE 2022, 17, e0266098. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Huang, M.; Liang, C.; Zhang, J.; Lin, G.; Yu, S.; Tanaka, M.; Lepler, S.; Guan, J.; Siemann, D.; et al. All-Optical Mechanobiology Interrogation of Yes-Associated Protein in Human Cancer and Normal Cells Using a Multi-Functional System. JoVE 2021, 178, 62934. [Google Scholar] [CrossRef]
- Huang, M.; Wang, H.; Delgado, A.A.; Reid, T.A.; Long, J.; Wang, S.; Sussman, H.; Guan, J.; Yamaguchi, H.; Tang, X. Combining 3D Magnetic Force Actuator and Multi-Functional Fluorescence Imaging to Study Nucleus Mechanobiology. JoVE 2022, 185, 64098. [Google Scholar] [CrossRef]
- Nardone, G.; Oliver-De La Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skládal, P.; Pešl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP Regulates Cell Mechanics by Controlling Focal Adhesion Assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Gupta, A.; Najibi, A.J.; Seo, B.R.; Garry, R.; Tringides, C.M.; De Lázaro, I.; Darnell, M.; Gu, W.; Zhou, Q.; et al. Matrix Viscoelasticity Controls Spatiotemporal Tissue Organization. Nat. Mater. 2023, 22, 117–127. [Google Scholar] [CrossRef]
- Lee, J.; Abdeen, A.A.; Tang, X.; Saif, T.A.; Kilian, K.A. Matrix Directed Adipogenesis and Neurogenesis of Mesenchymal Stem Cells Derived from Adipose Tissue and Bone Marrow. Acta Biomater. 2016, 42, 46–55. [Google Scholar] [CrossRef]
- Lee, J.; Abdeen, A.A.; Tang, X.; Saif, T.A.; Kilian, K.A. Geometric Guidance of Integrin Mediated Traction Stress during Stem Cell Differentiation. Biomaterials 2015, 69, 174–183. [Google Scholar] [CrossRef]
- Tang, X.; Saif, T.A. Adhesivity of Colon Cancer Cells During In Vitro Metastasis. Int. J. Appl. Mech. 2013, 5, 1350025. [Google Scholar] [CrossRef]
- Califano, J.P.; Reinhart-King, C.A. Substrate Stiffness and Cell Area Predict Cellular Traction Stresses in Single Cells and Cells in Contact. Cel. Mol. Bioeng. 2010, 3, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Koushki, N.; Ghagre, A.; Srivastava, L.K.; Sitaras, C.; Yoshie, H.; Molter, C.; Ehrlicher, A.J. Lamin A Redistribution Mediated by Nuclear Deformation Determines Dynamic Localization of YAP. Biophysics, 2020; preprint. [Google Scholar] [CrossRef]
- Cui, Y.; Hameed, F.M.; Yang, B.; Lee, K.; Pan, C.Q.; Park, S.; Sheetz, M. Cyclic Stretching of Soft Substrates Induces Spreading and Growth. Nat. Commun. 2015, 6, 6333. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, T.P.; Cosgrove, B.D.; Heo, S.-J.; Shurden, Z.E.; Mauck, R.L. Cytoskeletal to Nuclear Strain Transfer Regulates YAP Signaling in Mesenchymal Stem Cells. Biophys. J. 2015, 108, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.W.; Meng, Z.; Yuan, H.; Plouffe, S.W.; Moon, S.; Kim, W.; Jho, E.; Guan, K. Osmotic Stress-induced Phosphorylation by NLK at Ser128 Activates YAP. EMBO Rep. 2017, 18, 72–86. [Google Scholar] [CrossRef]
- Song, X.; Xu, H.; Wang, P.; Wang, J.; Affo, S.; Wang, H.; Xu, M.; Liang, B.; Che, L.; Qiu, W.; et al. Focal Adhesion Kinase (FAK) Promotes Cholangiocarcinoma Development and Progression via YAP Activation. J. Hepatol. 2021, 75, 888–899. [Google Scholar] [CrossRef]
- Li, B.; He, J.; Lv, H.; Liu, Y.; Lv, X.; Zhang, C.; Zhu, Y.; Ai, D. C-Abl Regulates YAPY357 Phosphorylation to Activate Endothelial Atherogenic Responses to Disturbed Flow. J. Clin. Investig. 2019, 129, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Andreu, I.; Granero-Moya, I.; Chahare, N.R.; Clein, K.; Molina-Jordán, M.; Beedle, A.E.M.; Elosegui-Artola, A.; Abenza, J.F.; Rossetti, L.; Trepat, X.; et al. Mechanical Force Application to the Nucleus Regulates Nucleocytoplasmic Transport. Nat. Cell Biol. 2022, 24, 896–905. [Google Scholar] [CrossRef]
- Tyner, J.W.; Haderk, F.; Kumaraswamy, A.; Baughn, L.B.; Van Ness, B.; Liu, S.; Marathe, H.; Alumkal, J.J.; Bivona, T.G.; Chan, K.S.; et al. Understanding Drug Sensitivity and Tackling Resistance in Cancer. Cancer Res. 2022, 82, 1448–1460. [Google Scholar] [CrossRef]
- Zargar, A.; Chang, S.; Kothari, A.; Snijders, A.M.; Mao, J.-H.; Wang, J.; Hernández, A.C.; Keasling, J.D.; Bivona, T.G. Overcoming the Challenges of Cancer Drug Resistance through Bacterial-Mediated Therapy. Chronic Dis. Transl. Med. 2019, 5, 258–266. [Google Scholar] [CrossRef]
- Chatterjee, N.; Bivona, T.G. Polytherapy and Targeted Cancer Drug Resistance. Trends Cancer 2019, 5, 170–182. [Google Scholar] [CrossRef]
- Li, J.W.; Zheng, G.; Kaye, F.J.; Wu, L. PROTAC Therapy as a New Targeted Therapy for Lung Cancer. Mol. Ther. 2023, 31, 647–656. [Google Scholar] [CrossRef]
- Wei, Y.; Au, J.L.-S. Role of Tumour Microenvironment in Chemoresistance. In Integration/Interaction of Oncologic Growth; Meadows, G.G., Ed.; Cancer Growth and Progression; Springer: Berlin/Heidelberg, Germany, 2005; Volume 15, pp. 285–321. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-F.; Tseng, Y.-C.; Nguyen, P.A.; Li, Y.-C.; Ho, C.-C.; Wu, C.-W. Enhanced YAP Expression Leads to EGFR TKI Resistance in Lung Adenocarcinomas. Sci. Rep. 2018, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- McGowan, M.; Kleinberg, L.; Halvorsen, A.R.; Hell, Å.; Brustugun, O.T. NSCLC Depend upon YAP Expression and Nuclear Localization after Acquiring Resistance to EGFR Inhibitors. Genes Cancer 2017, 8, 497–504. [Google Scholar] [CrossRef]
- Song, J.; Xie, L.; Zhang, X.; Hu, P.; Long, M.; Xiong, F.; Huang, J.; Ye, X. Role of YAP in Lung Cancer Resistance to Cisplatin. Oncol. Lett. 2018, 16, 3949–3954. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo Effector YAP Promotes Resistance to RAF- and MEK-Targeted Cancer Therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Varma, S.; Sousa, F.; Sunshine, M.; Abaan, O.D.; Davis, S.R.; Reinhold, S.W.; Kohn, K.W.; Morris, J.; Meltzer, P.S.; et al. NCI-60 Whole Exome Sequencing and Pharmacological CellMiner Analyses. PLoS ONE 2014, 9, e101670. [Google Scholar] [CrossRef]
- Morrison, H.; Sherman, L.S.; Legg, J.; Banine, F.; Isacke, C.; Haipek, C.A.; Gutmann, D.H.; Ponta, H.; Herrlich, P. The NF2 Tumor Suppressor Gene Product, Merlin, Mediates Contact Inhibition of Growth through Interactions with CD44. Genes Dev. 2001, 15, 968–980. [Google Scholar] [CrossRef]
- Gobbi, G.; Donati, B.; Do Valle, I.F.; Reggiani, F.; Torricelli, F.; Remondini, D.; Castellani, G.; Ambrosetti, D.C.; Ciarrocchi, A.; Sancisi, V. The Hippo Pathway Modulates Resistance to BET Proteins Inhibitors in Lung Cancer Cells. Oncogene 2019, 38, 6801–6817. [Google Scholar] [CrossRef]
- Rozengurt, E.; Eibl, G. Crosstalk between KRAS, SRC and YAP Signaling in Pancreatic Cancer: Interactions Leading to Aggressive Disease and Drug Resistance. Cancers 2021, 13, 5126. [Google Scholar] [CrossRef]
- Girotti, M.R.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-Breaking RAF Inhibitors That Also Target SRC Are Effective in Drug-Resistant BRAF Mutant Melanoma. Cancer Cell 2015, 27, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.-C.; Yang, C.-T.; Jablons, D.M.; You, L. The Crosstalk between Src and Hippo/YAP Signaling Pathways in Non-Small Cell Lung Cancer (NSCLC). Cancers 2020, 12, 1361. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-Y.; Chen, P.-S.; Prakash, E.; Hsu, H.-C.; Huang, H.-Y.; Lin, M.-T.; Chang, K.-J.; Kuo, M.-L. Connective Tissue Growth Factor Confers Drug Resistance in Breast Cancer through Concomitant Up-Regulation of Bcl-XL and CIAP1. Cancer Res. 2009, 69, 3482–3491. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-C.; Huang, C.-Y.; Su, H.-L.; Tang, C.-H. CTGF Increases Drug Resistance to Paclitaxel by Upregulating Survivin Expression in Human Osteosarcoma Cells. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 846–854. [Google Scholar] [CrossRef]
- Lin, M.-T.; Chang, C.-C.; Chen, S.-T.; Chang, H.-L.; Su, J.-L.; Chau, Y.-P.; Kuo, M.-L. Cyr61 Expression Confers Resistance to Apoptosis in Breast Cancer MCF-7 Cells by a Mechanism of NF-ΚB-Dependent XIAP Up-Regulation. J. Biol. Chem. 2004, 279, 24015–24023. [Google Scholar] [CrossRef]
- Hsu, P.; Miao, J.; Wang, Y.; Zhang, W.; Yang, Y.; Wang, C.; Yang, C.; Huang, Z.; You, J.; Xu, Z.; et al. Inhibition of Yes-associated Protein Down-regulates PD-L1 (CD274) Expression in Human Malignant Pleural Mesothelioma. J. Cell. Mol. Med. 2018, 22, 3139–3148. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.-L.; Wang, T.-L.; Zheng, F.-M. ACADL Suppresses PD-L1 Expression to Prevent Cancer Immune Evasion by Targeting Hippo/YAP Signaling in Lung Adenocarcinoma. Med. Oncol. 2023, 40, 118. [Google Scholar] [CrossRef]
- Yu, M.; Peng, Z.; Qin, M.; Liu, Y.; Wang, J.; Zhang, C.; Lin, J.; Dong, T.; Wang, L.; Li, S.; et al. Interferon-γ Induces Tumor Resistance to Anti-PD-1 Immunotherapy by Promoting YAP Phase Separation. Mol. Cell 2021, 81, 1216–1230.e9. [Google Scholar] [CrossRef]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular Matrix: A Dynamic Microenvironment for Stem Cell Niche. Biochim. Biophys. Acta (BBA)—General. Subj. 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Yue, B. Biology of the Extracellular Matrix: An Overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef]
- Yahyazadeh Shourabi, A.; Kashaninejad, N.; Saidi, M.S. An Integrated Microfluidic Concentration Gradient Generator for Mechanical Stimulation and Drug Delivery. J. Sci. Adv. Mater. Devices 2021, 6, 280–290. [Google Scholar] [CrossRef]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 Integration of Vascular Architecture with Physiological Force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Li, K.; Yang, M.; Tan, Y. Fluid Shear Stress Induces EMT of Circulating Tumor Cells via JNK Signaling in Favor of Their Survival during Hematogenous Dissemination. Int. J. Mol. Sci. 2020, 21, 8115. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Pelissier, F.A.; Zhang, H.; Lakins, J.; Weaver, V.M.; Park, C.; LaBarge, M.A. Microenvironment Rigidity Modulates Responses to the HER2 Receptor Tyrosine Kinase Inhibitor Lapatinib via YAP and TAZ Transcription Factors. MBoC 2015, 26, 3946–3953. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Lin, J.; Nowsheen, S.; Liu, T.; Zhao, Y.; Villalta, P.W.; Sicard, D.; Tschumperlin, D.J.; Lee, S.; Kim, J.; et al. Extracellular Matrix Stiffness Determines DNA Repair Efficiency and Cellular Sensitivity to Genotoxic Agents. Sci. Adv. 2020, 6, eabb2630. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Irnaten, M.; Hopkins, A.; O’Callaghan, J.; Stamer, W.D.; Clark, A.F.; Wallace, D.; O’Brien, C.J. Matrix Mechanotransduction via Yes-Associated Protein in Human Lamina Cribrosa Cells in Glaucoma. Investig. Ophthalmol. Vis. Sci. 2022, 63, 16. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Sleiman, M.; Moriarty, T.; Herrick, W.G.; Peyton, S.R. Sorafenib Resistance and JNK Signaling in Carcinoma during Extracellular Matrix Stiffening. Biomaterials 2014, 35, 5749–5759. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, X.; Huang, B.; Zhou, W.; Cui, X.; Zheng, C.; Liu, F.; Bi, J.; Zhang, Y.; Luo, H.; et al. Inhibition of Matrix Stiffness Relating Integrin Β1 Signaling Pathway Inhibits Tumor Growth in Vitro and in Hepatocellular Cancer Xenografts. BMC Cancer 2021, 21, 1276. [Google Scholar] [CrossRef]
- Hassan, A.A.; Artemenko, M.; Tang, M.K.S.; Shi, Z.; Chen, L.-Y.; Lai, H.-C.; Yang, Z.; Shum, H.-C.; Wong, A.S.T. Ascitic Fluid Shear Stress in Concert with Hepatocyte Growth Factor Drive Stemness and Chemoresistance of Ovarian Cancer Cells via the C-Met-PI3K/Akt-MiR-199a-3p Signaling Pathway. Cell Death Dis. 2022, 13, 537. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chang, J.K.; Dominguez, A.A.; Lee, H.; Nam, S.; Chang, J.; Varma, S.; Qi, L.S.; West, R.B.; Chaudhuri, O. YAP-Independent Mechanotransduction Drives Breast Cancer Progression. Nat. Commun. 2019, 10, 1848. [Google Scholar] [CrossRef]
- Cao, S.; Tang, J.; Huang, Y.; Li, G.; Li, Z.; Cai, W.; Yuan, Y.; Liu, J.; Huang, X.; Zhang, H. The Road of Solid Tumor Survival: From Drug-Induced Endoplasmic Reticulum Stress to Drug Resistance. Front. Mol. Biosci. 2021, 8, 620514. [Google Scholar] [CrossRef]
- Sharma, S.; Santiskulvong, C.; Bentolila, L.A.; Rao, J.; Dorigo, O.; Gimzewski, J.K. Correlative Nanomechanical Profiling with Super-Resolution F-Actin Imaging Reveals Novel Insights into Mechanisms of Cisplatin Resistance in Ovarian Cancer Cells. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 757–766. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Kieu, Q.M.N.; Dawson, M.R. The Malignancy of Metastatic Ovarian Cancer Cells Is Increased on Soft Matrices Through a Mechanosensitive Rho–ROCK Pathway. J. Cell Sci. 2014, 127, 2621–2626. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Barai, A.; Thakur, B.; Das, A.; Patwardhan, S.R.; Monteiro, M.; Gaikwad, S.; Bukhari, A.B.; Mogha, P.; Majumder, A.; et al. Soft Drug-Resistant Ovarian Cancer Cells Migrate via Two Distinct Mechanisms Utilizing Myosin II-Based Contractility. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, Á.; Cook, A.W.; Gough, R.E.; Schilling, M.; Olszok, N.A.; Brown, I.; Wang, L.; Aaron, J.; Martin-Fernandez, M.L.; Rehfeldt, F.; et al. DNA Damage Alters Nuclear Mechanics through Chromatin Reorganization. Nucleic Acids Res. 2021, 49, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.S.; Yoon, C.W.; Wang, Q.; Moon, S.; Koo, K.M.; Jung, H.; Chen, R.; Jiang, L.; Lu, G.; Fernandez, A.; et al. Focused Ultrasound Stimulates ER Localized Mechanosensitive PANNEXIN-1 to Mediate Intracellular Calcium Release in Invasive Cancer Cells. Front. Cell Dev. Biol. 2020, 8, 504. [Google Scholar] [CrossRef]
- Huo, S.; Liao, Z.; Zhao, P.; Zhou, Y.; Göstl, R.; Herrmann, A. Mechano-Nanoswitches for Ultrasound-Controlled Drug Activation. Adv. Sci. 2022, 9, 2104696. [Google Scholar] [CrossRef]
- Tijore, A.; Yao, M.; Wang, Y.-H.; Hariharan, A.; Nematbakhsh, Y.; Lee Doss, B.; Lim, C.T.; Sheetz, M. Selective Killing of Transformed Cells by Mechanical Stretch. Biomaterials 2021, 275, 120866. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Wang, H.; Mackey, C.; Chung, M.C.; Guan, J.; Zheng, G.; Roy, A.; Xie, M.; Vulpe, C.; Tang, X. YAP at the Crossroads of Biomechanics and Drug Resistance in Human Cancer. Int. J. Mol. Sci. 2023, 24, 12491. https://doi.org/10.3390/ijms241512491
Huang M, Wang H, Mackey C, Chung MC, Guan J, Zheng G, Roy A, Xie M, Vulpe C, Tang X. YAP at the Crossroads of Biomechanics and Drug Resistance in Human Cancer. International Journal of Molecular Sciences. 2023; 24(15):12491. https://doi.org/10.3390/ijms241512491
Chicago/Turabian StyleHuang, Miao, Heyang Wang, Cole Mackey, Michael C. Chung, Juan Guan, Guangrong Zheng, Arkaprava Roy, Mingyi Xie, Christopher Vulpe, and Xin Tang. 2023. "YAP at the Crossroads of Biomechanics and Drug Resistance in Human Cancer" International Journal of Molecular Sciences 24, no. 15: 12491. https://doi.org/10.3390/ijms241512491
APA StyleHuang, M., Wang, H., Mackey, C., Chung, M. C., Guan, J., Zheng, G., Roy, A., Xie, M., Vulpe, C., & Tang, X. (2023). YAP at the Crossroads of Biomechanics and Drug Resistance in Human Cancer. International Journal of Molecular Sciences, 24(15), 12491. https://doi.org/10.3390/ijms241512491