
New Clinical and Immunofluorescence Data of Collagen VI-Related Myopathy: A Single Center Cohort of 69 Patients

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Characteristics of the Study Population Stratified by Phenotype
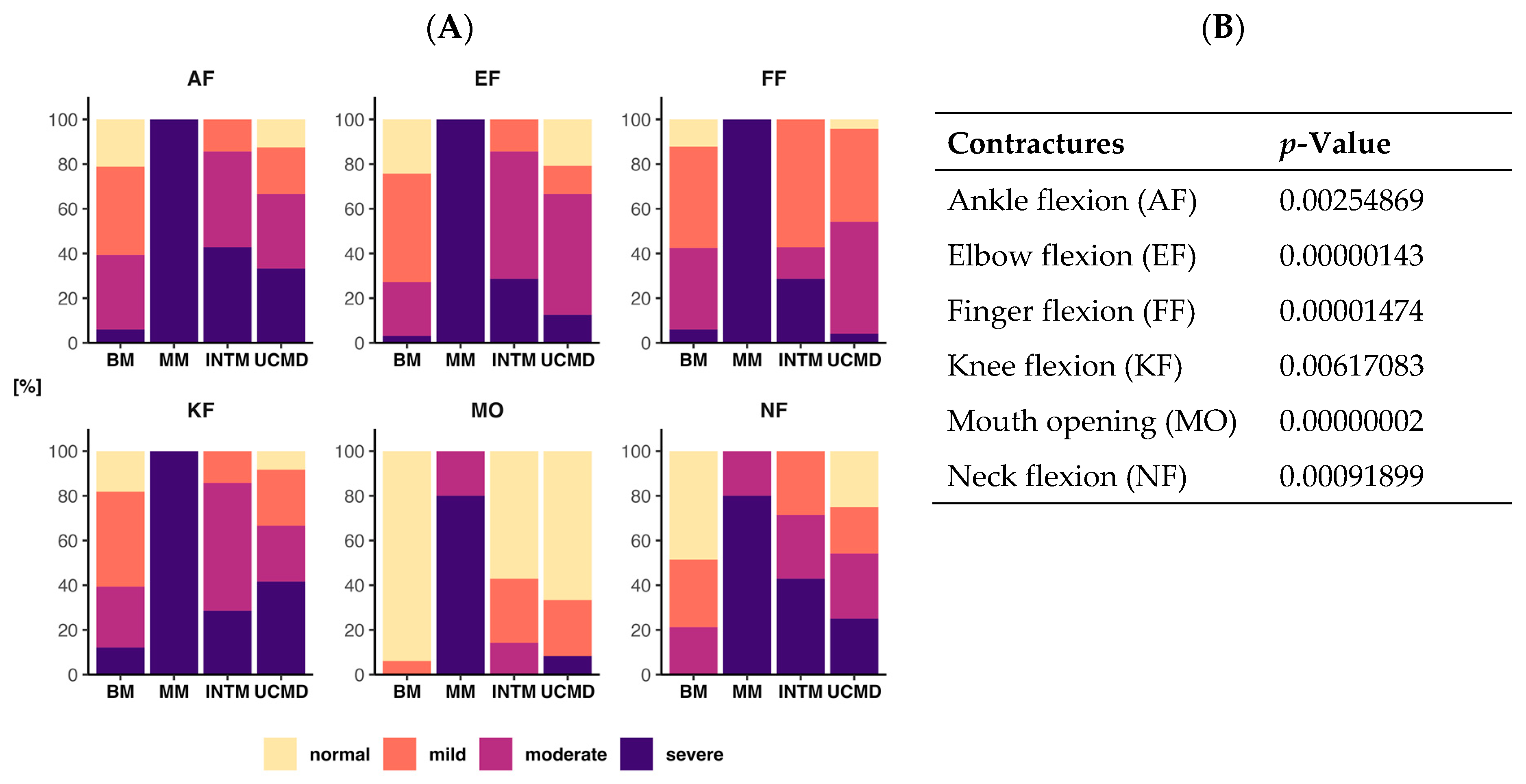
2.2. Degree of Contracture
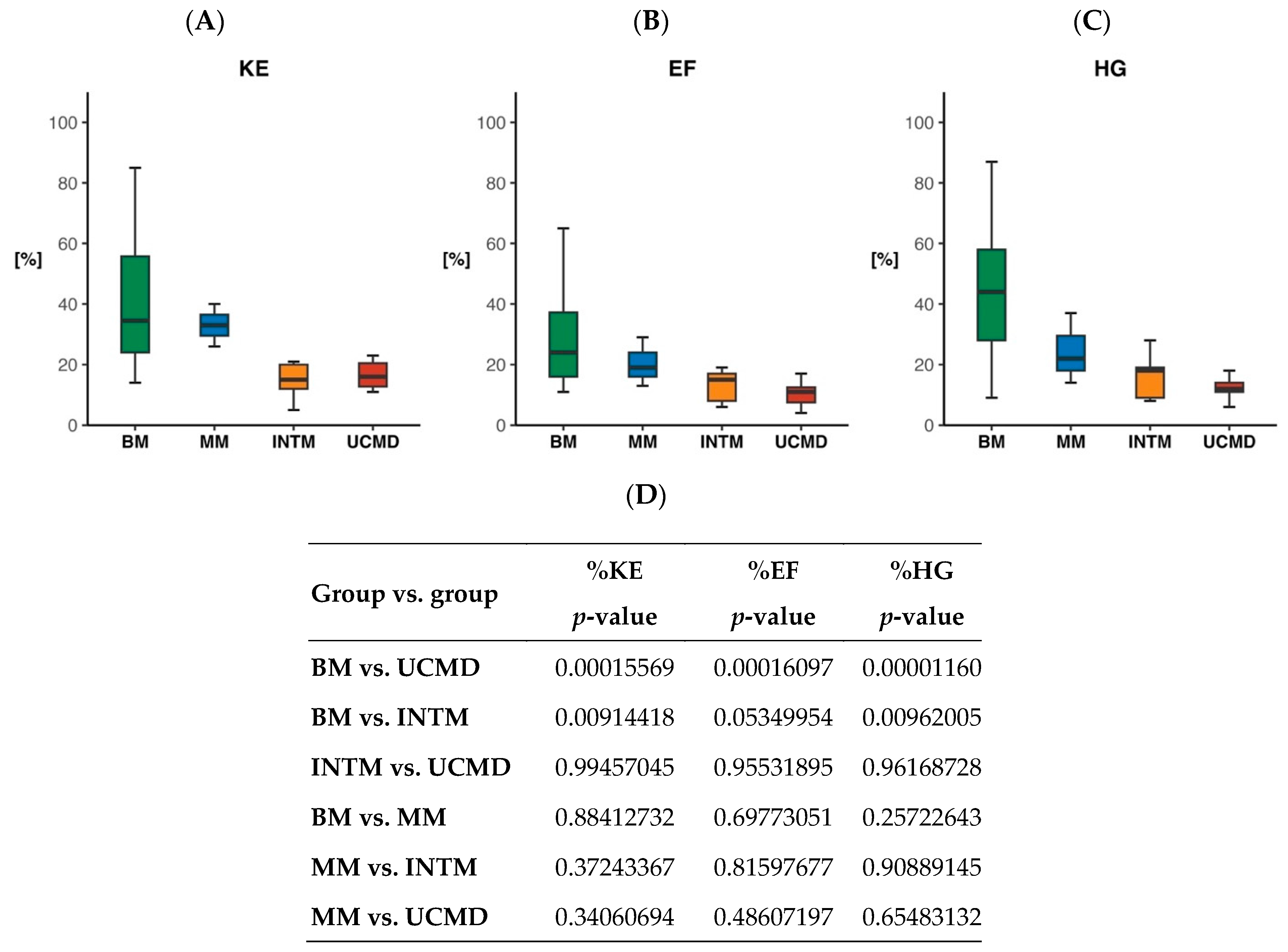
2.3. Quantitative Measure of Muscle Strength
2.4. Pulmonary Function
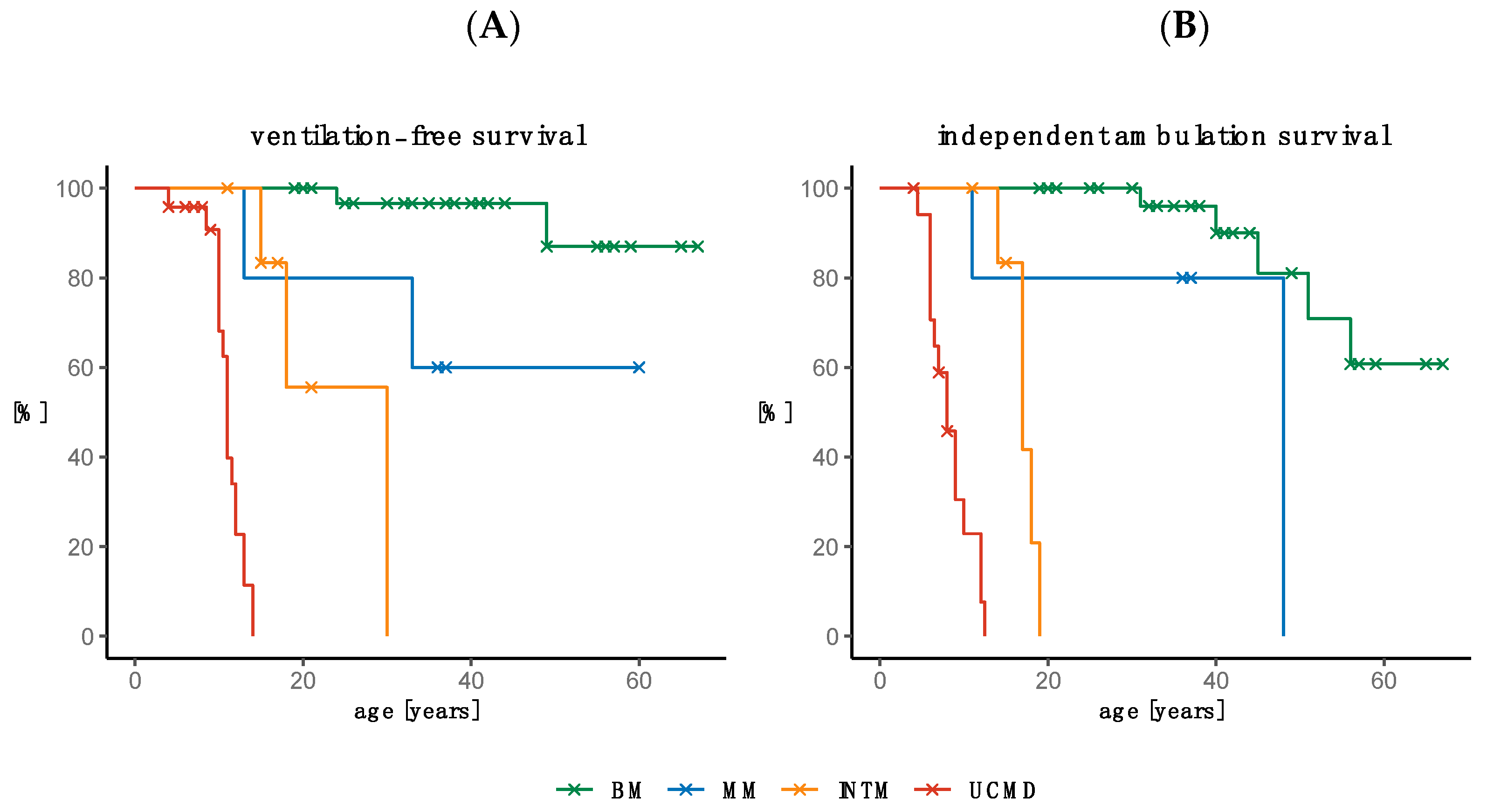
2.5. Need for Ventilation and Loss of Ambulation
2.5.1. Need for Ventilation
2.5.2. Loss of Ambulation
2.6. Survival
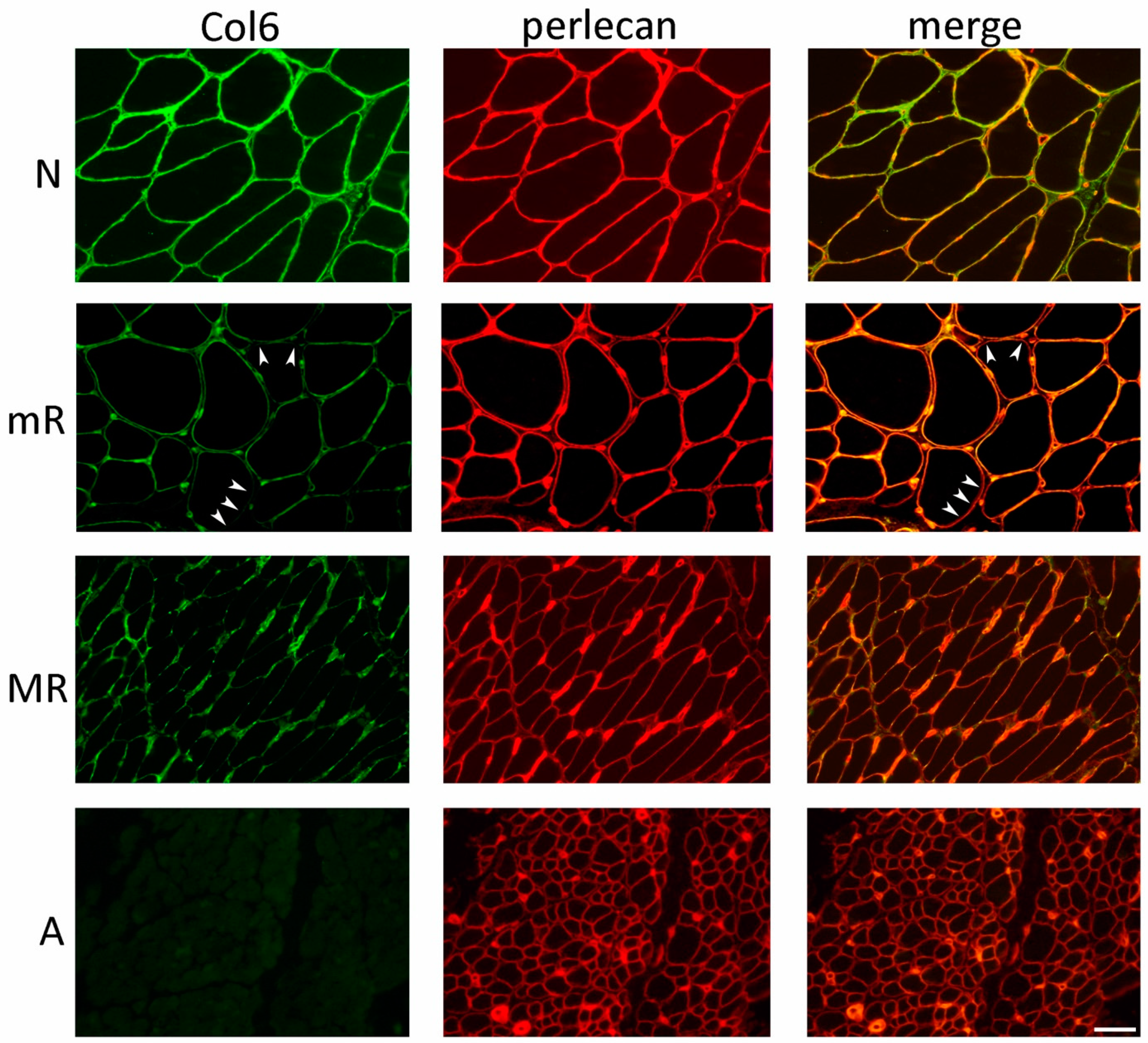
2.7. Collagen VI Immunofluorescence Analysis in Muscle Biopsy
2.8. Genetic Data
3. Discussion
4. Materials and Methods
4.1. Patients and Phenotypes Included in the Study
4.2. Data Collection
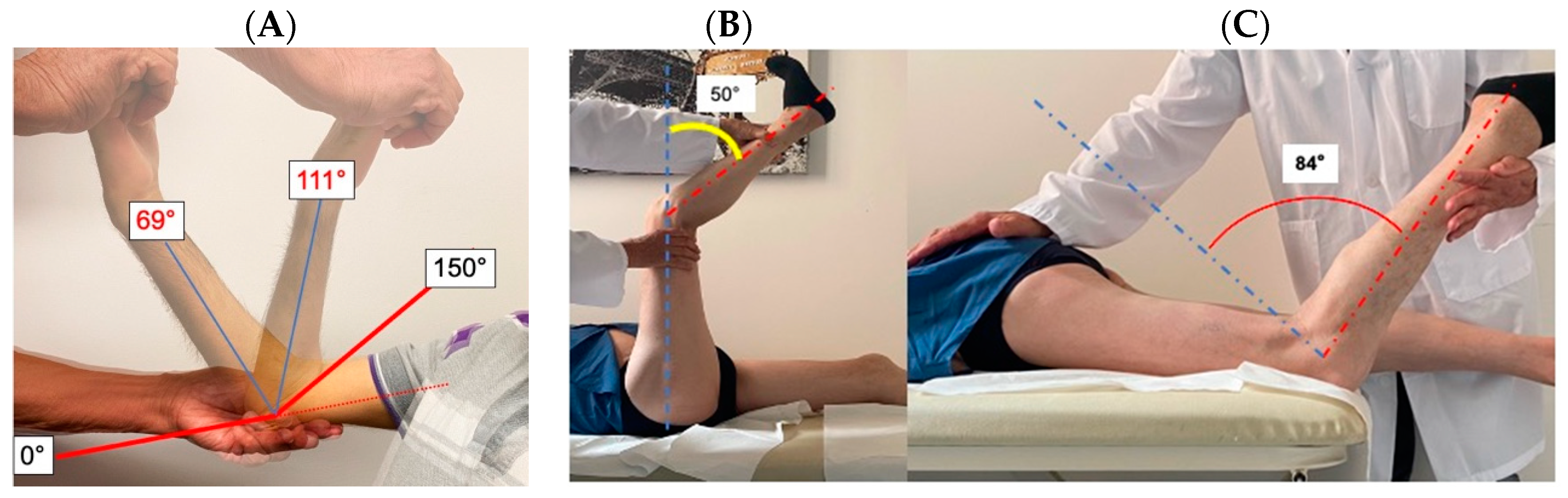
4.3. Evaluation of Contractures
4.4. Muscle Strength Measures
4.5. Immunohistochemical Analysis
4.6. Genetic Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fitzgerald, J.; Holden, P.; Hansen, U. The expanded collagen VI family: New chains and new questions. Connect. Tissue Res. 2013, 54, 345–350. [Google Scholar] [CrossRef]
- Sabatelli, P.; Gara, S.K.; Grumati, P.; Urciuolo, A.; Gualandi, F.; Curci, R.; Squarzoni, S.; Zamparelli, A.; Martoni, E.; Merlini, L.; et al. Expression of the collagen VI alpha5 and alpha6 chains in normal human skin and in skin of patients with collagen VI-related myopathies. J. Investig. Dermatol. 2011, 131, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Bethlem, J.; Wijngaarden, G.K. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain J. Neurol. 1976, 99, 91–100. [Google Scholar] [CrossRef]
- Jobsis, G.J.; Keizers, H.; Vreijling, J.P.; de Visser, M.; Speer, M.C.; Wolterman, R.A.; Baas, F.; Bolhuis, P.A. Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat. Genet. 1996, 14, 113–115. [Google Scholar] [CrossRef]
- Foley, A.R.; Hu, Y.; Zou, Y.; Columbus, A.; Shoffner, J.; Dunn, D.M.; Weiss, R.B.; Bonnemann, C.G. Autosomal recessive inheritance of classic Bethlem myopathy. Neuromuscul. Disord. NMD 2009, 19, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Bernardi, P. Therapy of collagen VI-related myopathies (Bethlem and Ullrich). Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2008, 5, 613–618. [Google Scholar] [CrossRef]
- Ullrich, O. Kongenitale atonisch-sklerotische Musckeldystrophie, ein weiterer Typus der heredodegeneration Erkrankungen des neuromuskularen Systems. Z. Ges. Neurol. Psychiat. 1930, 126, 171–201. [Google Scholar] [CrossRef]
- Camacho Vanegas, O.; Bertini, E.; Zhang, R.Z.; Petrini, S.; Minosse, C.; Sabatelli, P.; Giusti, B.; Chu, M.L.; Pepe, G. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc. Natl. Acad. Sci. USA 2001, 98, 7516–7521. [Google Scholar] [CrossRef] [PubMed]
- Demir, E.; Sabatelli, P.; Allamand, V.; Ferreiro, A.; Moghadaszadeh, B.; Makrelouf, M.; Topaloglu, H.; Echenne, B.; Merlini, L.; Guicheney, P. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am. J. Hum. Genet. 2002, 70, 1446–1458. [Google Scholar] [CrossRef]
- Pan, T.C.; Zhang, R.Z.; Sudano, D.G.; Marie, S.K.; Bonnemann, C.G.; Chu, M.L. New molecular mechanism for Ullrich congenital muscular dystrophy: A heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am. J. Hum. Genet. 2003, 73, 355–369. [Google Scholar] [CrossRef]
- Bradley, W.G.; Hudgson, P.; Gardner-Medwin, D.; Walton, J.N. The syndrome of myosclerosis. J. Neurol. Neurosurg. Psychiatry 1973, 36, 651–660. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lowenthal, A. A new heredodegenerative group: Heredofamilial myoscleroses. Acta Neurol. Psychiatr. Belg. 1954, 54, 155–165. [Google Scholar] [PubMed]
- Merlini, L.; Martoni, E.; Grumati, P.; Sabatelli, P.; Squarzoni, S.; Urciuolo, A.; Ferlini, A.; Gualandi, F.; Bonaldo, P. Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 2008, 71, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Allamand, V.; Merlini, L.; Bushby, K. 166th ENMC International Workshop on Collagen type VI-related Myopathies, 22–24 May 2009, Naarden, The Netherlands. Neuromuscul. Disord. NMD 2010, 20, 346–354. [Google Scholar] [CrossRef]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef]
- Merlini, L.; Angelin, A.; Tiepolo, T.; Braghetta, P.; Sabatelli, P.; Zamparelli, A.; Ferlini, A.; Maraldi, N.M.; Bonaldo, P.; Bernardi, P. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc. Natl. Acad. Sci. USA 2008, 105, 5225–5229. [Google Scholar] [CrossRef]
- Merlini, L.; Sabatelli, P.; Armaroli, A.; Gnudi, S.; Angelin, A.; Grumati, P.; Michelini, M.E.; Franchella, A.; Gualandi, F.; Bertini, E.; et al. Cyclosporine A in Ullrich congenital muscular dystrophy: Long-term results. Oxidative Med. Cell. Longev. 2011, 2011, 139194. [Google Scholar] [CrossRef]
- Castagnaro, S.; Pellegrini, C.; Pellegrini, M.; Chrisam, M.; Sabatelli, P.; Toni, S.; Grumati, P.; Ripamonti, C.; Pratelli, L.; Maraldi, N.M.; et al. Autophagy activation in COL6 myopathic patients by a low-protein-diet pilot trial. Autophagy 2016, 12, 2484–2495. [Google Scholar] [CrossRef]
- Tagliavini, F.; Sardone, F.; Squarzoni, S.; Maraldi, N.M.; Merlini, L.; Faldini, C.; Sabatelli, P. Ultrastructural changes in muscle cells of patients with collagen VI-related myopathies. Muscles Ligaments Tendons J. 2013, 3, 281–286. [Google Scholar] [CrossRef]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. NMD 2010, 20, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Zulian, A.; Gualandi, F.; Manzati, E.; Merlini, L.; Michelini, M.E.; Benassi, L.; Marmiroli, S.; Ferlini, A.; Sabatelli, P.; et al. Melanocytes--a novel tool to study mitochondrial dysfunction in Duchenne muscular dystrophy. J. Cell. Physiol. 2013, 228, 1323–1331. [Google Scholar] [CrossRef]
- Zulian, A.; Tagliavini, F.; Rizzo, E.; Pellegrini, C.; Sardone, F.; Zini, N.; Maraldi, N.M.; Santi, S.; Faldini, C.; Merlini, L.; et al. Melanocytes from Patients Affected by Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy have Dysfunctional Mitochondria That Can be Rescued with Cyclophilin Inhibitors. Front. Aging Neurosci. 2014, 6, 324. [Google Scholar] [CrossRef]
- Tiepolo, T.; Angelin, A.; Palma, E.; Sabatelli, P.; Merlini, L.; Nicolosi, L.; Finetti, F.; Braghetta, P.; Vuagniaux, G.; Dumont, J.M.; et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1-/- myopathic mice. Br. J. Pharmacol. 2009, 157, 1045–1052. [Google Scholar] [CrossRef]
- Angelin, A.; Tiepolo, T.; Sabatelli, P.; Grumati, P.; Bergamin, N.; Golfieri, C.; Mattioli, E.; Gualandi, F.; Ferlini, A.; Merlini, L.; et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc. Natl. Acad. Sci. USA 2007, 104, 991–996. [Google Scholar] [CrossRef]
- Merlini, L.; Nishino, I.; Consortium for Autophagy in Muscular, D. 201st ENMC International Workshop: Autophagy in muscular dystrophies—Translational approach, 1–3 November 2013, Bussum, The Netherlands. Neuromuscul. Disord. NMD 2014, 24, 546–561. [Google Scholar] [CrossRef]
- Maraldi, N.M.; Sabatelli, P.; Columbaro, M.; Zamparelli, A.; Manzoli, F.A.; Bernardi, P.; Bonaldo, P.; Merlini, L. Collagen VI myopathies: From the animal model to the clinical trial. Adv. Enzym. Regul. 2009, 49, 197–211. [Google Scholar] [CrossRef]
- Brinas, L.; Richard, P.; Quijano-Roy, S.; Gartioux, C.; Ledeuil, C.; Lacene, E.; Makri, S.; Ferreiro, A.; Maugenre, S.; Topaloglu, H.; et al. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann. Neurol. 2010, 68, 511–520. [Google Scholar] [CrossRef]
- Foley, A.R.; Quijano-Roy, S.; Collins, J.; Straub, V.; McCallum, M.; Deconinck, N.; Mercuri, E.; Pane, M.; D’Amico, A.; Bertini, E.; et al. Natural history of pulmonary function in collagen VI-related myopathies. Brain J. Neurol. 2013, 136, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Natera-de Benito, D.; Foley, A.R.; Dominguez-Gonzalez, C.; Ortez, C.; Jain, M.; Mebrahtu, A.; Donkervoort, S.; Hu, Y.; Fink, M.; Yun, P.; et al. Association of Initial Maximal Motor Ability with Long-term Functional Outcome in Patients with COL6-Related Dystrophies. Neurology 2021, 96, e1413–e1424. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liu, A.; Wei, C.; Yang, H.; Chang, X.; Wang, S.; Yuan, Y.; Bonnemann, C.; Wu, Q.; Wu, X.; et al. Genetic and clinical findings in a Chinese cohort of patients with collagen VI-related myopathies. Clin. Genet. 2018, 93, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, T.; Komaki, H.; Okada, M.; Hayashi, Y.K.; Nonaka, I.; Sugai, K.; Sasaki, M.; Nishino, I. Rapidly progressive scoliosis and respiratory deterioration in Ullrich congenital muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 982–988. [Google Scholar] [CrossRef]
- Panades-de Oliveira, L.; Rodriguez-Lopez, C.; Cantero Montenegro, D.; Marcos Toledano, M.D.M.; Fernandez-Marmiesse, A.; Esteban Perez, J.; Hernandez Lain, A.; Dominguez-Gonzalez, C. Bethlem myopathy: A series of 16 patients and description of seven new associated mutations. J. Neurol. 2019, 266, 934–941. [Google Scholar] [CrossRef]
- Zanoteli, E.; Soares, P.S.; Silva, A.; Camelo, C.G.; Fonseca, A.; Albuquerque, M.A.V.; Moreno, C.A.M.; Lopes Abath Neto, O.; Novo Filho, G.M.; Kulikowski, L.D.; et al. Clinical features of collagen VI-related dystrophies: A large Brazilian cohort. Clin. Neurol. Neurosurg. 2020, 192, 105734. [Google Scholar] [CrossRef]
- Meilleur, K.G.; Jain, M.S.; Hynan, L.S.; Shieh, C.Y.; Kim, E.; Waite, M.; McGuire, M.; Fiorini, C.; Glanzman, A.M.; Main, M.; et al. Results of a two-year pilot study of clinical outcome measures in collagen VI- and laminin alpha2-related congenital muscular dystrophies. Neuromuscul. Disord. NMD 2015, 25, 43–54. [Google Scholar] [CrossRef]
- Nadeau, A.; Kinali, M.; Main, M.; Jimenez-Mallebrera, C.; Aloysius, A.; Clement, E.; North, B.; Manzur, A.Y.; Robb, S.A.; Mercuri, E.; et al. Natural history of Ullrich congenital muscular dystrophy. Neurology 2009, 73, 25–31. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, H.Y.; Park, H.J.; Kim, S.H.; Kim, S.M.; Choi, Y.C. Clinical, Pathologic, and Genetic Features of Collagen VI-Related Myopathy in Korea. J. Clin. Neurol. 2017, 13, 331–339. [Google Scholar] [CrossRef]
- Cook, J.D.; Glass, D.S. Strength evaluation in neuromuscular disease. Neurol. Clin. 1987, 5, 101–123. [Google Scholar] [CrossRef]
- Scott, O.M.; Hyde, S.A.; Goddard, C.; Dubowitz, V. Quantitation of muscle function in children: A prospective study in Duchenne muscular dystrophy. Muscle Nerve 1982, 5, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.M.; Schierbecker, J.; Renna, R.; Florence, J. High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. NMD 2002, 12, 917–925. [Google Scholar] [CrossRef]
- Griggs, R.C.; Moxley, R.T., 3rd; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch. Neurol. 1991, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Moxley, R.T.; Griggs, R.C.; Brooke, M.H.; Fenichel, G.M.; Miller, J.P.; King, W.; Signore, L.; Pandya, S.; Florence, J.; et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N. Engl. J. Med. 1989, 320, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Cecconi, I.; Parmeggiani, A.; Cordelli, D.M.; Dormi, A. Quadriceps muscle strength in Duchenne muscular dystrophy and effect of corticosteroid treatment. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2020, 39, 200–206. [Google Scholar] [CrossRef]
- Merlini, L.; Morandi, L.; Granata, C.; Ballestrazzi, A. Bethlem myopathy: Early-onset benign autosomal dominant myopathy with contractures. Description of two new families. Neuromuscul. Disord. NMD 1994, 4, 503–511. [Google Scholar] [CrossRef]
- Martoni, E.; Urciuolo, A.; Sabatelli, P.; Fabris, M.; Bovolenta, M.; Neri, M.; Grumati, P.; D’Amico, A.; Pane, M.; Mercuri, E.; et al. Identification and characterization of novel collagen VI non-canonical splicing mutations causing Ullrich congenital muscular dystrophy. Hum. Mutat. 2009, 30, E662–E672. [Google Scholar] [CrossRef]
- Tagliavini, F.; Pellegrini, C.; Sardone, F.; Squarzoni, S.; Paulsson, M.; Wagener, R.; Gualandi, F.; Trabanelli, C.; Ferlini, A.; Merlini, L.; et al. Defective collagen VI alpha6 chain expression in the skeletal muscle of patients with collagen VI-related myopathies. Biochim. Biophys. Acta 2014, 1842, 1604–1612. [Google Scholar] [CrossRef]
- Bovolenta, M.; Neri, M.; Martoni, E.; Urciuolo, A.; Sabatelli, P.; Fabris, M.; Grumati, P.; Mercuri, E.; Bertini, E.; Merlini, L.; et al. Identification of a deep intronic mutation in the COL6A2 gene by a novel custom oligonucleotide CGH array designed to explore allelic and genetic heterogeneity in collagen VI-related myopathies. BMC Med. Genet. 2010, 11, 44. [Google Scholar] [CrossRef]
- Lucioli, S.; Giusti, B.; Mercuri, E.; Vanegas, O.C.; Lucarini, L.; Pietroni, V.; Urtizberea, A.; Ben Yaou, R.; de Visser, M.; van der Kooi, A.J.; et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology 2005, 64, 1931–1937. [Google Scholar] [CrossRef]
- Pepe, G.; Bertini, E.; Giusti, B.; Brunelli, T.; Comeglio, P.; Saitta, B.; Merlini, L.; Chu, M.L.; Federici, G.; Abbate, R. A novel de novo mutation in the triple helix of the COL6A3 gene in a two-generation Italian family affected by Bethlem myopathy. A diagnostic approach in the mutations’ screening of type VI collagen. Neuromuscul. Disord. NMD 1999, 9, 264–271. [Google Scholar] [CrossRef]
- Camacho Vanegas, O.; Zhang, R.Z.; Sabatelli, P.; Lattanzi, G.; Bencivenga, P.; Giusti, B.; Columbaro, M.; Chu, M.L.; Merlini, L.; Pepe, G. Novel COL6A1 splicing mutation in a family affected by mild Bethlem myopathy. Muscle Nerve 2002, 25, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Gualandi, F.; Urciuolo, A.; Martoni, E.; Sabatelli, P.; Squarzoni, S.; Bovolenta, M.; Messina, S.; Mercuri, E.; Franchella, A.; Ferlini, A.; et al. Autosomal recessive Bethlem myopathy. Neurology 2009, 73, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, A.; Trabanelli, C.; Scotton, C.; Venturoli, A.; Selvatici, R.; Brisca, G.; Merlini, L.; Bruno, C.; Ferlini, A.; Gualandi, F. Paternal germline mosaicism in collagen VI related myopathies. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2015, 19, 533–536. [Google Scholar] [CrossRef]
- Bolduc, V.; Foley, A.R.; Solomon-Degefa, H.; Sarathy, A.; Donkervoort, S.; Hu, Y.; Chen, G.S.; Sizov, K.; Nalls, M.; Zhou, H.; et al. A recurrent COL6A1 pseudoexon insertion causes muscular dystrophy and is effectively targeted by splice-correction therapies. JCI Insight 2019, 4, e124403. [Google Scholar] [CrossRef] [PubMed]
- Giusti, B.; Lucarini, L.; Pietroni, V.; Lucioli, S.; Bandinelli, B.; Sabatelli, P.; Squarzoni, S.; Petrini, S.; Gartioux, C.; Talim, B.; et al. Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann. Neurol. 2005, 58, 400–410. [Google Scholar] [CrossRef]
- Zeevi, D.A.; Chung, K.W.; Levi, C.; Scher, S.Y.; Ekstein, J. Recommendation of premarital genetic screening in the Syrian Jewish community based on mutation carrier frequencies within Syrian Jewish cohorts. Mol. Gen. 2021, 9, e1756. [Google Scholar] [CrossRef]
- Deconinck, N.; Richard, P.; Allamand, V.; Behin, A.; Laforet, P.; Ferreiro, A.; de Becdelievre, A.; Ledeuil, C.; Gartioux, C.; Nelson, I.; et al. Bethlem myopathy: Long-term follow-up identifies COL6 mutations predicting severe clinical evolution. J. Neurol. Neurosurg. Psychiatry 2014, 86, 1337–1346. [Google Scholar] [CrossRef]
- Inoue, M.; Saito, Y.; Yonekawa, T.; Ogawa, M.; Iida, A.; Nishino, I.; Noguchi, S. Causative variant profile of collagen VI-related dystrophy in Japan. Orphanet J. Rare Dis. 2021, 16, 284. [Google Scholar] [CrossRef]
- Yonekawa, T.; Nishino, I. Ullrich congenital muscular dystrophy: Clinicopathological features, natural history and pathomechanism(s). J. Neurol. Neurosurg. Psychiatry 2015, 86, 280–287. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Zhao, D.H.; Yang, H.P.; Liu, A.J.; Chang, X.Z.; Hong, D.J.; Bonnemann, C.; Yuan, Y.; Wu, X.R.; Xiong, H. Novel collagen VI mutations identified in Chinese patients with Ullrich congenital muscular dystrophy. World J. Pediatr. WJP 2014, 10, 126–132. [Google Scholar] [CrossRef]
- Jobsis, G.J.; Boers, J.M.; Barth, P.G.; de Visser, M. Bethlem myopathy: A slowly progressive congenital muscular dystrophy with contractures. Brain J. Neurol. 1999, 122 Pt 4, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.U.; Speer, M.C.; Chu, M.L.; Tandan, R. Respiratory muscle involvement in Bethlem myopathy. Neurology 1999, 52, 174–176. [Google Scholar] [CrossRef] [PubMed]
- van der Kooi, A.J.; de Voogt, W.G.; Bertini, E.; Merlini, L.; Talim, F.B.; Ben Yaou, R.; Urtziberea, A.; de Visser, M. Cardiac and pulmonary investigations in Bethlem myopathy. Arch. Neurol. 2006, 63, 1617–1621. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Na, H.J.; Ding, Y.; Wang, Z.; Lee, S.J.; Choi, M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J. Biol. Chem. 2012, 287, 11677–11688. [Google Scholar] [CrossRef]
- Castroflorio, E.; Perez Berna, A.J.; Lopez-Marquez, A.; Badosa, C.; Loza-Alvarez, P.; Roldan, M.; Jimenez-Mallebrera, C. The Capillary Morphogenesis Gene 2 Triggers the Intracellular Hallmarks of Collagen VI-Related Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 7651. [Google Scholar] [CrossRef]
- Burgi, J.; Kunz, B.; Abrami, L.; Deuquet, J.; Piersigilli, A.; Scholl-Burgi, S.; Lausch, E.; Unger, S.; Superti-Furga, A.; Bonaldo, P.; et al. CMG2/ANTXR2 regulates extracellular collagen VI which accumulates in hyaline fibromatosis syndrome. Nat. Commun. 2017, 8, 15861. [Google Scholar] [CrossRef]
- Sabatelli, P.; Sardone, F.; Traina, F.; Merlini, L.; Santi, S.; Wagener, R.; Faldini, C. TGF-beta1 differentially modulates the collagen VI alpha5 and alpha6 chains in human tendon cultures. J. Biol. Regul. Homeost. Agents 2016, 30, 107–113. [Google Scholar]
- Furukawa, T.; Toyokura, Y. Congenital, hypotonic-sclerotic muscular dystrophy. J. Med. Genet. 1977, 14, 426–429. [Google Scholar] [CrossRef]
- Stoeber, E. Uber atonisch-sklerotische Muskeldystrophie (Typ Ullrich). Z. Kinderheilkd. 1939, 60, 279–284. [Google Scholar] [CrossRef]
- Pepe, G.; de Visser, M.; Bertini, E.; Bushby, K.; Vanegas, O.C.; Chu, M.L.; Lattanzi, G.; Merlini, L.; Muntoni, F.; Urtizberea, A. Bethlem myopathy (BETHLEM) 86th ENMC international workshop, 10–11 November 2000, Naarden, The Netherlands. Neuromuscul. Disord. NMD 2002, 12, 296–305. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Abresch, R.T.; Carter, G.T.; Fowler, W.M., Jr.; Johnson, E.R.; Kilmer, D.D.; Sigford, B.J. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 1995, 74, S70–S92. [Google Scholar] [CrossRef] [PubMed]
- Gajdosik, R.; Lusin, G. Hamstring muscle tightness. Reliability of an active-knee-extension test. Phys. Ther. 1983, 63, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Beenakker, E.A.; van der Hoeven, J.H.; Fock, J.M.; Maurits, N.M. Reference values of maximum isometric muscle force obtained in 270 children aged 4-16 years by hand-held dynamometry. Neuromuscul. Disord. NMD 2001, 11, 441–446. [Google Scholar] [CrossRef]
- van der Ploeg, R.J.; Fidler, V.; Oosterhuis, H.J. Hand-held myometry: Reference values. J. Neurol. Neurosurg. Psychiatry 1991, 54, 244–247. [Google Scholar] [CrossRef]
- Merlini, L.; Mazzone, E.S.; Solari, A.; Morandi, L. Reliability of hand-held dynamometry in spinal muscular atrophy. Muscle Nerve 2002, 26, 64–70. [Google Scholar] [CrossRef]
- Sabatelli, P.; Merlini, L.; Di Martino, A.; Cenni, V.; Faldini, C. Early Morphological Changes of the Rectus Femoris Muscle and Deep Fascia in Ullrich Congenital Muscular Dystrophy. Int. J. Environ. Res. Public Health 2022, 19, 1252. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contractures | Muscle Strength | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Phenotype | Onset | Age at First Visit, y | Age at Last Visit, y | Age at FU, y | CK, Times Normal | MO | NF | EF | FF | KF | AF | First Steps, m | Lost Walk, y | NIV, Age, y | %FVC | %KE | %EF | %HG | Mutation |
| 1 | BM | B | 2 | 16 | 20 | 2 | 0 | 2 | 0 | 0 | 0 | 2 | 42 | - | - | 91 | 41 | 15 | 28 | H COL6A2 c.1970-9G>A [47] |
| 2 | BM | B | 21 | 35 | - | ≤1 | 0 | 2 | 1 | 2 | 1 | 3 | 18 | - | 24 | 38 | 24 | 13 | 19 | h COL6A1 c.868G>C |
| 3 | BM | As | 10 | 18 | 26 | 24 | 0 | 0 | 0 | 1 | 1 | 0 | 12 | - | - | 77 | 85 | 65 | 59 | h COL6A3 c.4928T>G [48] |
| 4 | BM | B | 15 | 28 | 32 | 28 | 0 | 1 | 1 | 1 | 1 | 1 | 14 | - | - | 91 | 85 | 58 | 58 | h COL6A3 c.4928T>G [48] |
| 5 | BM | As | 45 | 56 | - | 4 | 0 | 0 | 1 | 1 | 1 | 1 | 12 | - | - | - | - | - | 28 | h COL6A3 c.4928T>G [48] |
| 6 | BM | B | 51 | 64 | 65 | ≤1 | 0 | 0 | 1 | 2 | 1 | 1 | 12 | 51 | - | 69 | 21 | 23 | 87 | h COL6A2 c.875G>C |
| 7 | BM | Ch | 53 | 54 | 57 | ≤1 | 0 | 1 | 0 | 2 | 0 | 2 | 12 | - | - | - | 36 | - | 41 | h COL6A1 c.1022G>A |
| 8 | BM | Ad | 47 | 51 | 55 | ≤1 | 0 | 1 | 3 | 3 | 1 | 2 | 12 | 45 | - | 41 | 16 | 11 | 9 | H COL6A2 c.2240T>A |
| 9 | BM | Ch | 17 | 29 | 38 | 18 | 1 | 2 | 2 | 2 | 2 | 1 | 12 | - | - | 79 | 61 | 42 | 43 | h COL6A2 c.2098G>A [19] |
| 10 | BM | Ch | 17 | 30 | - | 3.5 | 0 | 2 | 1 | 1 | 2 | 1 | 15 | - | - | 53 | 31 | 16 | 18 | ch COL6A2 c.2947_2952del/chr21 del [49] |
| 11 | BM | Ch | 20 | 58 | - | 1.5 | 1 | 2 | 1 | 1 | 2 | 2 | 12 | 56 | 49 | 44 | 24 | - | 50 | h COL6A2 c.802G>A [48] |
| 12 | BM | B | 11 | 35 | - | 2 | 0 | 1 | 1 | 2 | 2 | 1 | 36 | - | - | 53 | 34 | 14 | 36 | h COL6A3 c.6158G>T [48] |
| 13 | BM | Ch | 41 | 41 | - | UK | 0 | 1 | 2 | 2 | 2 | 3 | 12 | 40 | - | 58 | 28 | 26 | 17 | h COL6A2 c.1770+1G>A |
| 14 | BM | Ch | 33 | 33 | - | ≤1 | 0 | 0 | 2 | 1 | 2 | 2 | 12 | - | - | 80 | 35 | 24 | 46 | h COL6A1 c.1056+1G>A [50] |
| 15 | BM | B | 10 | 35 | - | 4 | 0 | 1 | 0 | 1 | 2 | 2 | 13 | - | - | 89 | 61 | 26 | 33 | H COL6A3 c.1393C>T [9,48] |
| 16 | BM | Ta | 29 | 37 | - | 13 | 0 | 1 | 1 | 2 | 1 | 2 | 12 | - | - | 81 | 14 | 48 | 46 | h COL6A2 c.1970-3C>A [48] |
| 17 | BM | Ad | 41 | 41 | - | 2.2 | 0 | 0 | 1 | 1 | 1 | 0 | 12 | - | - | - | - | - | - | h COL6A3 c.6166G>T [51] |
| 18 | BM | Ch | 11 | 13 | 25 | 2.5 | 0 | 0 | 1 | 1 | 1 | 1 | 16 | - | - | 85 | - | - | - | h COL6A3 c.6166G>T [51] |
| 19 | BM | B | 19 | 25 | - | ≤1 | 0 | 0 | 1 | 1 | 1 | 2 | 30 | - | - | 65 | 49 | 22 | 44 | h COL6A2 c.883G>A [48] |
| 20 | BM | B | 31 | 42 | - | 2 | 0 | 2 | 2 | 1 | 2 | 1 | 12 | - | - | 67 | 24 | 17 | 44 | h COL6A3 c.6230G>A [48] |
| 21 | BM | Ta | 17 | 17 | 21 | UK | 0 | 0 | 0 | 1 | 0 | 0 | 12 | - | - | - | - | - | 79 | c COL6A2 c.679G>A |
| 22 | BM | B | 34 | 40 | - | 3 | 0 | 1 | 2 | 2 | 2 | 2 | 12 | 31 | - | 63 | 14 | 12 | 12 | h COL6A2 c.847G>A |
| 23 | BM | Ad | 40 | 40 | - | 4 | 0 | 2 | 0 | 1 | 0 | 1 | 12 | - | - | - | - | - | - | h COL6A2 c.1861G>A |
| 24 | BM | Ch | 35 | 67 | - | 2 | 0 | 0 | 1 | 1 | 3 | 1 | 12 | - | - | 93 | 27 | 27 | 56 | h COL6A1 c.428+1G>A [52] |
| 25 | BM | Ad | 59 | 59 | - | 2 | 0 | 0 | 2 | 3 | 1 | 0 | 12 | - | - | - | - | - | - | h COL6A1 c.428+1G>A [52] |
| 26 | BM | Ch | 5 | 33 | 42 | 9 | 0 | 0 | 2 | 2 | 3 | 2 | 12 | - | - | - | 36 | 16 | 26 | h COL6A1 c.428+1G>A [52] |
| 27 | BM | Ch | 9 | 32 | 44 | 11 | 0 | 0 | 1 | 2 | 3 | 2 | 12 | - | - | - | 54 | 38 | - | h COL6A1 c.428+1G>A [52] |
| 28 | BM | As | 12 | 12 | 38 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | - | - | - | - | - | - | h COL6A1 c.428+1G>A [52] |
| 29 | BM | As | 17 | 28 | 30 | 5 | 0 | 1 | 1 | 1 | 3 | 0 | 12 | - | - | 100 | 61 | 35 | 76 | h COL6A1 c.428+1G>A [52] |
| 30 | BM | Ad | 14 | 47 | 49 | 15 | 0 | 0 | 1 | 0 | 1 | 1 | 12 | - | - | 74 | 66 | 44 | 60 | h COL6A1 c.428+1G>A [52] |
| 31 | BM | Ad | 65 | 65 | - | UK | 0 | 1 | 2 | 2 | 1 | 1 | 12 | - | - | - | - | - | - | h COL6A1 c.428+1G>A [52] |
| 32 | BM | Ch | 38 | 61 | 67 | 5 | 0 | 0 | 1 | 2 | 1 | 1 | 12 | - | - | 83 | 31 | 24 | 58 | h COL6A1 c.428+1G>A [52] |
| 33 | BM | B | 2.5 | 2.5 | 19 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 16 | - | - | - | - | - | - | h COL6A3 c.6158G>T |
| 34 | MM | Ch | 11 | 22 | 36 | 1.5 | 2 | 3 | 3 | 3 | 3 | 3 | 15 | - | - | 41 | - | - | - | H COL6A2 c.2455C>T [13] |
| 35 | MM | Ch | 16 | 55 | 56 | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 14 | 48 | 33 | - | 26 | 13 | 22 | H COL6A2 c.2455C>T [13] |
| 36 | MM | B | 6 | 12 | 32 | 1.5 | 3 | 3 | 3 | 3 | 3 | 3 | 36 | 11 | 13 * | 30 | 40 | 19 | 14 | H COL6A2 c.2455C>T [13] |
| 37 | MM | Ch | 32 | 32 | 37 | 1.5 | 3 | 3 | 3 | 3 | 3 | 3 | 12 | - | - | 55 | - | - | - | ch COL6A2 c.2455C>T/c.2489G>A [53] |
| 38 | MM | B | 18 | 49 | 60 | 2.5 | 3 | 2 | 3 | 3 | 3 | 3 | 14 | 48 | - | 45 | 33 | 29 | 37 | ch COL6A2 c.1096 C>T/c.2611 G>A [53] |
| 39 | INTM | B | 2.5 | 11 | - | 2.5 | 0 | 1 | 2 | 1 | 2 | 2 | 20 | - | - | 40 | 12 | 6 | 9 | h COL6A1 c.896G>A [54] |
| 40 | INTM | B | 13 | 21 | - | ≤1 | 2 | 2 | 2 | 1 | 1 | 2 | 17 | 19 | - | 47 | 20 | 17 | 28 | h COL6A1 c.896G>A [54] |
| 41 | INTM | B | 25 | 25 | 28 | 2 | 1 | 3 | 3 | 3 | 3 | 3 | 18 | 17 | 18 | 21 | - | - | - | h COL6A3 c.6210+1G>A |
| 42 | INTM | B | 21 | 21 | 26 | UK | 1 | 3 | 2 | 3 | 3 | 3 | 18 | 14 | 15 | 9 | - | - | - | h COL6A2 c.875G>A |
| 43 | INTM | B | 8 | 17 | - | 2.5 | 0 | 1 | 1 | 1 | 2 | 3 | 15 | 17 | - | 59 | 15 | 15 | 19 | ch COL6A2 c.1096 C>T/c.927+5G>A [47] |
| 44 | INTM | B | 45 | 45 | 51 | UK | 0 | 3 | 3 | 2 | 2 | 2 | 18 | 18 | 30 | 29 | 5 | 8 | 18 | h COL6A1 c.877G>A |
| 45 | INTM | B | 11 | 11 | 15 | 4 | 0 | 2 | 2 | 1 | 2 | 1 | 23 | - | - | 48 | 21 | 19 | 8 | h COL6A1 c.877G>A |
| 46 | UCMD | B | 5 | 5 | 9 | 3 | 0 | 0 | 0 | 1 | 1 | 0 | 14 | 8 * | - | - | 12 | 11 | 14 | h COL6A1 c.877G>A |
| 47 | UCMD | B | 5 | 7 | 24 | 1.5 | 0 | 1 | 0 | 1 | 0 | 1 | 24 | 10 * | 12 * | - | 23 | 11 | 12 | h COL6A1 c.850G>A |
| 48 | UCMD | B | 12 | 20 | 29 | 3 | 0 | 3 | 2 | 1 | 1 | 3 | 20 | 6 | 11 | 13 | 18 | 6.5 | 12 | h COL6A1 c.798_804+8del [47] |
| 49 | UCMD | B | 5.5 | 9 | 22 | 4 | 0 | 1 | 1 | 1 | 2 | 2 | 18 | 6 | 10* | - | 11 | 14 | 18 | H COL6A2 c.2626 C>A |
| 50 | UCMD | B | 3 | 10 | 12 | 1.5 | 0 | 2 | 2 | 2 | 2 | 2 | 24 | 6.5 | 10.5 * | 35 | 17 | 7 | - | h COL6A1 c.930+189C>T [55] |
| 51 | UCMD | B | 4 | 13 | 14 | 2 | 0 | 1 | 2 | 2 | 2 | 2 | 24 | 12.5 | 14 * | 40 | 22 | 8 | 17 | h COL6A3 c.6210+1G>A [48] |
| 52 | UCMD | B | 8 | 12 | 21 | UK | 0 | 2 | 2 | 2 | 2 | 2 | 20 | 8 | 11 | 32 | 18 | 8 | 6 | ch COL6A2 c.1459-2A>G/c.1771-1G>A [48] |
| 53 | UCMD | B | 7 | 9 | 10 | ≤1 | 0 | 3 | 2 | 2 | 3 | 2 | 18 | 7 | 10 * | 30 | 13 | 12 | 8 | ch COL6A2 c.1459-2A>G/c.1771-1G>A [48] |
| 54 | UCMD | B | 5 | 10 | 27 | ≤1 | 0 | 1 | 2 | 2 | 3 | 2 | NW | - | 13 * | 13 | 13 | 13 | 17 | h COL6A1 c.850G>A [27] |
| 55 | UCMD | B | 5 | 8 | 11 | ≤1 | 1 | 3 | 2 | 2 | 3 | 3 | NW | - | 4 | 29 | 15 | 5 | 11 | H COL6A2 c. 2572C>T [48] |
| 56 | UCMD | B | 6.5 | 6.5 | 9 | ≤1 | 0 | 1 | 0 | 1 | 1 | 2 | 15 | 9 * | - | 64 | 14 | 11 | - | ch COL6A2 c.2098G>A/c.2381C>A |
| 57 | UCMD | B | 3 | 11 | 15 | 2.5 | 0 | 2 | 2 | 1 | 3 | 3 | 14 | 9 | 12 * | 40 | 20 | 4 | 12 | h COL6A1 c.930+189C>T [55] |
| 58 | UCMD | B | 4 | 11 | 14 | 1.5 | 3 | 3 | 2 | 2 | 3 | 3 | NW | - | 13 * | - | 12 | 17 | 8 | h COL6A1 c.819_833del [48] |
| 59 | UCMD | B | 8.5 | 8.5 | 10 | 1.5 | 1 | 3 | 2 | 2 | 3 | 2 | 36 | 6 | 8.5 | 45 | - | - | - | h COL6A3 c.6871G>C |
| 60 | UCMD | B | 1.5 | 10 | 11 | ≤1 | 1 | 2 | 2 | 2 | 3 | 3 | NW | - | 10 | 18 | - | - | - | H COL6A1 c.1465del [48,56] |
| 61 | UCMD | B | 3 | 3 | 6 | ≤1 | 0 | 0 | 1 | 1 | 1 | 1 | NW | - | - | - | - | - | - | h COL6A1 c.904-2A>G |
| 62 | UCMD | B | 8 | 8 | 20 | UK | 0 | 0 | 2 | 2 | 2 | 0 | NW | - | 14 * | - | 22 | 13 | 12 | h COL6A2 c.1336dup |
| 63 | UCMD | B | 3.5 | 8.5 | 12.5 | 3 | 3 | 3 | 3 | 2 | 3 | 3 | 18 | 4.5 | 11 * | 22 | - | - | - | h COL6A1 c.930+189C>T; [55] |
| 64 | UCMD | B | 11 | 29 | 36 | UK | 1 | 2 | 3 | 2 | 3 | 3 | 16 | 6 | 11 | - | - | - | 12 | h COL6A2 c.875G>T |
| 65 | UCMD | B | 19 | 19 | 23 | 1.5 | 1 | 2 | 3 | 3 | 3 | 3 | 30 | 12 | 10 | - | - | - | - | H COL6A2 c.348dup |
| 66 | UCMD | B | 10 | 10 | 12 | UK | 1 | 2 | 2 | 1 | 2 | 1 | 14 | 12 * | 11.5 * | 57 | 23 | 9 | - | h COL6A1 c.850G>A |
| 67 | UCMD | B | 3 | 4 | 8 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | 15 | - | - | - | 12 | - | - | h COL6A1 c.850G>A |
| 68 | UCMD | B | 1.5 | 3 | 4 | UK | 0 | 0 | 0 | 1 | 1 | 1 | 30 | - | - | - | - | - | - | H COL6A2 c.1402C>T |
| 69 | UCMD | B | 2 | 2 | 7 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 15 | - | - | - | - | - | - | h COL6A2 c.911G>T |
| Patient | Phenotype | Mutation h, ch, H | I Biopsy Pattern: N, mR, MR, A (Age, Years) | II Biopsy Pattern: N, mR, MR, A (Age, Years) | III Biopsy Pattern: N, mR, MR, A (Age, Years) |
|---|---|---|---|---|---|
| 4 | BM | h | N (16) | N (21) | - |
| 5 | BM | h | N (46) | - | - |
| 7 | BM | h | N (54) | - | - |
| 9 | BM | h | N (16) | N (29) | N (30) |
| 11 | BM | h | N (27) | N (48) | N (49) |
| 12 | BM | h | mR (11) | N (29) | - |
| 16 | BM | h | N (29) | N (36) | N (37) |
| 18 | BM | h | N (11) | - | - |
| 19 | BM | h | N (23) | N (24) | - |
| 20 | BM | h | N (29) | N (41) | N (42) |
| 22 | BM | h | N (40) | - | - |
| 26 | BM | h | N (15) | - | - |
| 27 | BM | h | N (10) | - | - |
| 32 | BM | h | N (58) | - | - |
| 33 | BM | h | MR (3) | - | - |
| 1 | BM | H | MR (1) | - | - |
| 8 | BM | H | mR (47) | - | - |
| 10 | BM | ch | mR (19) | - | - |
| 15 | BM | H | mR (10) | mR (24) | - |
| 34 | MM | H | mR (17) | - | - |
| 35 | MM | H | mR (31) | - | - |
| 36 | MM | H | mR (6) | - | - |
| 38 | MM | ch | mR (36) | - | - |
| 40 | INTM | h | MR (3) | N (14) | - |
| 43 | INTM | ch | MR (9) | - | - |
| 48 | UCMD | h | mR (20) | mR (21) | - |
| 50 | UCMD | h | MR (3) | - | - |
| 51 | UCMD | h | MR (5) | mR (14) | - |
| 52 | UCMD | ch | A (1) | MR (10) | - |
| 54 | UCMD | h | mR (8) | mR (11) | - |
| 55 | UCMD | H | MR (3) | mR (11) | - |
| 57 | UCMD | h | MR (3) | - | - |
| 58 | UCMD | h | MR (4) | - | - |
| 60 | UCMD | H | A (2) | - | - |
| 62 | UCMD | ch | MR (3) | - | - |
| 63 | UCMD | h | MR (6) | - | - |
| 66 | UCMD | h | MR (12) | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merlini, L.; Sabatelli, P.; Gualandi, F.; Redivo, E.; Di Martino, A.; Faldini, C. New Clinical and Immunofluorescence Data of Collagen VI-Related Myopathy: A Single Center Cohort of 69 Patients. Int. J. Mol. Sci. 2023, 24, 12474. https://doi.org/10.3390/ijms241512474
Merlini L, Sabatelli P, Gualandi F, Redivo E, Di Martino A, Faldini C. New Clinical and Immunofluorescence Data of Collagen VI-Related Myopathy: A Single Center Cohort of 69 Patients. International Journal of Molecular Sciences. 2023; 24(15):12474. https://doi.org/10.3390/ijms241512474
Chicago/Turabian StyleMerlini, Luciano, Patrizia Sabatelli, Francesca Gualandi, Edoardo Redivo, Alberto Di Martino, and Cesare Faldini. 2023. "New Clinical and Immunofluorescence Data of Collagen VI-Related Myopathy: A Single Center Cohort of 69 Patients" International Journal of Molecular Sciences 24, no. 15: 12474. https://doi.org/10.3390/ijms241512474
APA StyleMerlini, L., Sabatelli, P., Gualandi, F., Redivo, E., Di Martino, A., & Faldini, C. (2023). New Clinical and Immunofluorescence Data of Collagen VI-Related Myopathy: A Single Center Cohort of 69 Patients. International Journal of Molecular Sciences, 24(15), 12474. https://doi.org/10.3390/ijms241512474