
Differential Modulation of the Phosphoproteome by the MAP Kinases Isoforms p38α and p38β


Abstract
1. Introduction
2. Results
2.1. Large Phosphoproteomes and Proteomes Were Obtained from MEFs Expressing Intrinsically Active Variants p38α or p38β or Treated with Anisomycin
2.2. Differential Modulation of the Phosphoproteomes by the Intrinsically Active p38α and p38β
2.3. Differential Effects of p38α and p38β on the Stress-Induced Dynamics of the Phosphoproteome
2.4. Specific Effects of the p38s Activation on Known MAPK Substrates
2.5. p38α and p38β Differently Affected HSP27 Phosphorylation
2.6. Differential Effects of the p38s on the Cytoskeleton
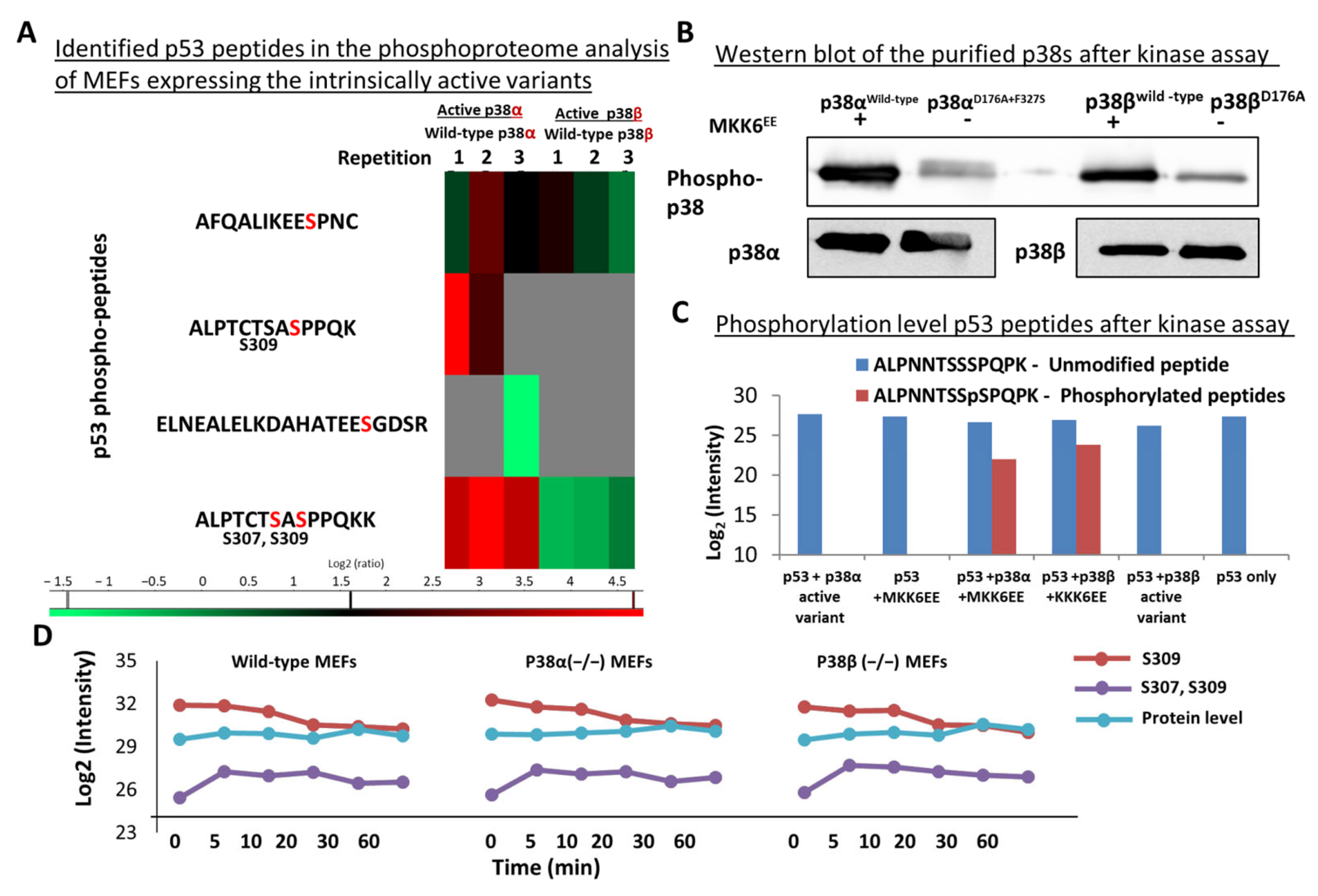
2.7. Differential Phosphorylation of Cancer-Related Proteins, including p53, by p38α and p38β
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Infection and Transfection of Cultured Cells with Constitutively Active p38 Mutants
4.3. Anisomycin Treatment
4.4. SILAC Labeling
4.5. Proteins Extraction and Phosphatase Inhibition
4.6. In-Solution Protein Digestion
4.7. Fractionation of Peptides by Strong Cation Exchange Chromatography
4.8. Phosphopeptides Enrichment on Titanium Beads (MOAC)
4.9. Peptides Analysis by LC-MS/MS
4.10. LC-MS/MS Data Analysis
4.11. Western Blot Analysis
4.12. Protein Expression in Bacteria
4.13. In Vitro Kinase Assays
4.14. Confocal Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef]
- Kudaravalli, S.; den Hollander, P.; Mani, S.A. Role of P38 MAP Kinase in Cancer Stem Cells and Metastasis. Oncogene 2022, 41, 3177–3185. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP Kinase Signalling Cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Roche, O.; Fernández-Aroca, D.M.; Arconada-Luque, E.; García-Flores, N.; Mellor, L.F.; Ruiz-Hidalgo, M.J.; Sánchez-Prieto, R. P38β and Cancer: The Beginning of the Road. Int. J. Mol. Sci. 2020, 21, 7524. [Google Scholar] [CrossRef]
- Benhar, M.; Dalyot, I.; Engelberg, D.; Levitzki, A. Enhanced ROS Production in Oncogenically Transformed Cells Potentiates C-Jun N-Terminal Kinase and P38 Mitogen-Activated Protein Kinase Activation and Sensitization to Genotoxic Stress. Mol. Cell. Biol. 2001, 21, 6913–6926. [Google Scholar] [CrossRef]
- Benhar, M.; Engelberg, D.; Levitzki, A. ROS, Stress-Activated Kinases and Stress Signaling in Cancer. EMBO Rep. 2002, 3, 420–425. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and Signaling of the P38 MAP Kinase Pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef]
- Cuenda, A.; Sanz-Ezquerro, J.J. P38γ and P38δ: From Spectators to Key Physiological Players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef]
- Han, J.; Wu, J.; Silke, J. An Overview of Mammalian P38 Mitogen-Activated Protein Kinases, Central Regulators of Cell Stress and Receptor Signaling. F1000Research 2020, 9, F1000. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and Versatility of P38 Kinase Signalling in Health and Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The P38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef]
- Beamer, E.; Corrêa, S.A.L. The P38MAPK-MK2 Signaling Axis as a Critical Link Between Inflammation and Synaptic Transmission. Front. Cell Dev. Biol. 2021, 9, 635636. [Google Scholar] [CrossRef]
- Jiménez, A.; Vázquez, D. Anisomycin and Related Antibiotics. In Mechanism of Action of Antieukaryotic and Antiviral Compounds; Springer: Berlin/Heidelberg, Germany, 1979; pp. 1–19. [Google Scholar]
- Hazzalin, C.A.; Le Panse, R.; Cano, E.; Mahadevan, L.C. Anisomycin Selectively Desensitizes Signalling Components Involved in Stress Kinase Activation and Fos and Jun Induction. Mol. Cell. Biol. 1998, 18, 1844–1854. [Google Scholar] [CrossRef]
- Cano, E.; Hazzalin, C.A.; Mahadevan, L.C. Anisomycin-Activated Protein Kinases P45 and P55 but Not Mitogen-Activated Protein Kinases ERK-1 and -2 Are Implicated in the Induction of c-fos and c-jun. Mol. Cell. Biol. 1994, 14, 7352–7362. [Google Scholar] [CrossRef]
- Sobin, B.A.; Tanner, F.W. Anisomycin, a New Anti-Protozoan Antibiotic. J. Am. Chem. Soc. 1954, 76, 4053. [Google Scholar] [CrossRef]
- Korb, A.; Tohidast-Akrad, M.; Cetin, E.; Axmann, R.; Smolen, J.; Schett, G. Differential Tissue Expression and Activation of P38 MAPK Alpha, Beta, Gamma, and Delta Isoforms in Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 2745–2756. [Google Scholar] [CrossRef]
- Qin, J.-Z.; Xin, H.; Qi, X.-M.; Chen, G. Isoform-Specific and Cell/Tissue-Dependent Effects of P38 MAPKs in Regulating Inflammation and Inflammation-Associated Oncogenesis. Front. Biosci.-Landmark 2022, 27, 31. [Google Scholar] [CrossRef]
- Hale, K.K.; Trollinger, D.; Rihanek, M.; Manthey, C.L. Differential Expression and Activation of P38 Mitogen-Activated Protein Kinase Alpha, Beta, Gamma, and Delta in Inflammatory Cell Lineages. J. Immunol. 1999, 162, 4246–4252. [Google Scholar] [CrossRef]
- Tamura, K.; Sudo, T.; Senftleben, U.; Dadak, A.M.; Johnson, R.; Karin, M. Requirement for P38alpha in Erythropoietin Expression: A Role for Stress Kinases in Erythropoiesis. Cell 2000, 102, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Mudgett, J.S.; Ding, J.; Guh-Siesel, L.; Chartrain, N.A.; Yang, L.; Gopal, S.; Shen, M.M. Essential Role for P38alpha Mitogen-Activated Protein Kinase in Placental Angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10454–10459. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Porras, A.; Alonso, G.; Jones, M.; Vintersten, K.; Panelli, S.; Valladares, A.; Perez, L.; Klein, R.; Nebreda, A.R. Essential Role of P38alpha MAP Kinase in Placental but Not Embryonic Cardiovascular Development. Mol. Cell 2000, 6, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Beardmore, V.A.; Hinton, H.J.; Eftychi, C.; Apostolaki, M.; Armaka, M.; Darragh, J.; McIlrath, J.; Carr, J.M.; Armit, L.J.; Clacher, C.; et al. Generation and Characterization of P38β (MAPK11) Gene-Targeted Mice. Mol. Cell. Biol. 2005, 25, 10454–10464. [Google Scholar] [CrossRef]
- Del Barco Barrantes, I.; Coya, J.M.; Maina, F.; Arthur, J.S.C.; Nebreda, A.R.; Barrantes, I.D.B.; Coya, J.M.; Maina, F.; Arthurd, J.S.C.; Nebreda, A.R. Genetic Analysis of Specific and Redundant Roles for P38alpha and P38beta MAPKs during Mouse Development. Proc. Natl. Acad. Sci. USA 2011, 108, 12764–12769. [Google Scholar] [CrossRef]
- Remy, G.; Risco, A.M.; Iñesta-Vaquera, F.A.; González-Terán, B.; Sabio, G.; Davis, R.J.; Cuenda, A. Differential Activation of P38MAPK Isoforms by MKK6 and MKK3. Cell. Signal. 2010, 22, 660–667. [Google Scholar] [CrossRef]
- Darlyuk-Saadon, I.; Bai, C.; Heng, C.K.M.; Gilad, N.; Yu, W.-P.; Lim, P.Y.; Cazenave-Gassiot, A.; Zhang, Y.; Wong, W.S.F.; Engelberg, D. Active P38α Causes Macrovesicular Fatty Liver in Mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2018069118. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. P38 MAP-Kinases Pathway Regulation, Function and Role in Human Diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and Functions of P38 MAPK Signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Igea, A.; Nebreda, A.R. The Stress Kinase P38α as a Target for Cancer Therapy. Cancer Res. 2015, 75, 3997–4002. [Google Scholar] [CrossRef]
- Zou, X.; Blank, M. Targeting P38 MAP Kinase Signaling in Cancer through Post-Translational Modifications. Cancer Lett. 2017, 384, 19–26. [Google Scholar] [CrossRef]
- Sanz-Ezquerro, J.J.; Cuenda, A. P38 Signalling Pathway. Int. J. Mol. Sci. 2021, 22, 1003. [Google Scholar] [CrossRef]
- Patterson, H.; Nibbs, R.; Mcinnes, I.; Siebert, S. Protein Kinase Inhibitors in the Treatment of Inflammatory and Autoimmune Diseases. Clin. Exp. Immunol. 2014, 176, 1–10. [Google Scholar] [CrossRef]
- Gupta, J.; Nebreda, A.R. Roles of P38α Mitogen-Activated Protein Kinase in Mouse Models of Inflammatory Diseases and Cancer. FEBS J. 2015, 282, 1841–1857. [Google Scholar] [CrossRef]
- Olson, J.M.; Hallahan, A.R. P38 MAP Kinase: A Convergence Point in Cancer Therapy. Trends Mol. Med. 2004, 10, 125–129. [Google Scholar] [CrossRef]
- Ge, B. MAPKK-Independent Activation of P38alpha Mediated by TAB1-Dependent Autophosphorylation of P38alpha. Science 2002, 295, 1291–1294. [Google Scholar] [CrossRef]
- Bell, M.; Capone, R.; Pashtan, I.; Levitzki, A.; Engelberg, D. Isolation of Hyperactive Mutants of the MAPK P38/Hog1 That Are Independent of MAPK Kinase Activation. J. Biol. Chem. 2001, 276, 25351–25358. [Google Scholar] [CrossRef]
- Levin-Salomon, V.; Kogan, K.; Ahn, N.G.; Livnah, O.; Engelberg, D. Isolation of Intrinsically Active (MEK-Independent) Variants of the ERK Family of Mitogen-Activated Protein (MAP) Kinases. J. Biol. Chem. 2008, 283, 34500–34510. [Google Scholar] [CrossRef]
- Diskin, R.; Askari, N.; Capone, R.; Engelberg, D.; Livnah, O. Active Mutants of the Human P38alpha Mitogen-Activated Protein Kinase. J. Biol. Chem. 2004, 279, 47040–47049. [Google Scholar] [CrossRef]
- Avitzour, M.; Diskin, R.; Raboy, B.; Askari, N.; Engelberg, D.; Livnah, O. Intrinsically Active Variants of All Human P38 Isoforms. FEBS J 2007, 274, 963–975. [Google Scholar] [CrossRef]
- Beenstock, J.; Ben-Yehuda, S.; Melamed, D.; Admon, A.; Livnah, O.; Ahn, N.G.; Engelberg, D. The P38β Mitogen-Activated Protein Kinase Possesses an Intrinsic Autophosphorylation Activity, Generated by a Short Region Composed of the α-G Helix and MAPK Insert. J. Biol. Chem. 2014, 289, 23546–23556. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Mann, M. A Practical Recipe for Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC). Nat. Protoc. 2006, 1, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Bell, M.; Engelberg, D. Phosphorylation of Tyr-176 of the Yeast MAPK Hog1/P38 Is Not Vital for Hog1 Biological Activity. J. Biol. Chem. 2003, 278, 14603–14606. [Google Scholar] [CrossRef]
- Askari, N.; Diskin, R.; Avitzour, M.; Yaakov, G.; Livnah, O.; Engelberg, D. MAP-Quest: Could We Produce Constitutively Active Variants of MAP Kinases? Mol. Cell. Endocrinol. 2006, 252, 231–240. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein–Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Trempolec, N.; Dave-Coll, N.; Nebreda, A.R. SnapShot: P38 MAPK Substrates. Cell 2013, 152, 924.e1. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. Yeast Biochemical Pathways. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Freshney, N.W.; Rawlinson, L.; Guesdon, F.; Jones, E.; Cowley, S.; Hsuan, J.; Saklatvala, J. Interleukin-1 Activates a Novel Protein Kinase Cascade That Results in the Phosphorylation of Hsp27. Cell 1994, 78, 1039–1049. [Google Scholar] [CrossRef]
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A Novel Kinase Cascade Triggered by Stress and Heat Shock That Stimulates MAPKAP Kinase-2 and Phosphorylation of the Small Heat Shock Proteins. Cell 1994, 78, 1027–1037. [Google Scholar] [CrossRef]
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat Shock Protein 27 (HSP27): Biomarker of Disease and Therapeutic Target. Fibrogenesis Tissue Repair 2012, 5, 7. [Google Scholar] [CrossRef]
- Dan, Y.; Radic, N.; Gay, M.; Fernández-Torras, A.; Arauz, G.; Vilaseca, M.; Aloy, P.; Canovas, B.; Nebreda, A.R. Characterization of P38α Signaling Networks in Cancer Cells Using Quantitative Proteomics and Phosphoproteomics. Mol. Cell. Proteom. 2023, 22, 100527. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A Database of Phosphorylation Sites--Update 2011. Nucleic Acids Res. 2011, 39, D261-7. [Google Scholar] [CrossRef]
- Magrane, M. UniProt Consortium UniProt Knowledgebase: A Hub of Integrated Protein Data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 Years of PhosphoSitePlus®: Integrating Post-Translationally Modified Sites, Disease Variants and Isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of Actin Filament Dynamics by P38 Map Kinase-Mediated Phosphorylation of Heat Shock Protein 27. J. Cell Sci. 1997, 110 Pt 3, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Rada, C.C.; Mejia-Pena, H.; Grimsey, N.J.; Cordova, I.C.; Olson, J.; Wozniak, J.M.; Gonzalez, D.J.; Nizet, V.; Trejo, J.A. Heat Shock Protein 27 Activity Is Linked to Endothelial Barrier Recovery after Proinflammatory GPCR-Induced Disruption. Sci. Signal. 2021, 14, eabc1044. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Dominguez, R. Regulation of Actin Cytoskeleton Dynamics in Cells. Mol. Cells 2010, 29, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Askari, N.; Diskin, R.; Avitzour, M.; Capone, R.; Livnah, O.; Engelberg, D. Hyperactive Variants of P38alpha Induce, Whereas Hyperactive Variants of P38gamma Suppress, Activating Protein 1-Mediated Transcription. J. Biol. Chem. 2007, 282, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Stramucci, L.; Pranteda, A.; Bossi, G. Insights of Crosstalk between P53 Protein and the MKK3/MKK6/P38 MAPK Signaling Pathway in Cancer. Cancers 2018, 10, 131. [Google Scholar] [CrossRef]
- Nordgaard, C.; Tollenaere, M.A.X.; Val, A.M.D.; Bekker-Jensen, D.B.; Blasius, M.; Olsen, J.V.; Bekker-Jensen, S. Regulation of the Golgi Apparatus by P38 and JNK Kinases during Cellular Stress Responses. Int. J. Mol. Sci. 2021, 22, 9595. [Google Scholar] [CrossRef]
- Borisova, M.E.; Voigt, A.; Tollenaere, M.A.X.; Sahu, S.K.; Juretschke, T.; Kreim, N.; Mailand, N.; Choudhary, C.; Bekker-Jensen, S.; Akutsu, M.; et al. P38-MK2 Signaling Axis Regulates RNA Metabolism after UV-Light-Induced DNA Damage. Nat. Commun. 2018, 9, 1017. [Google Scholar] [CrossRef]
- Molinar-Inglis, O.; Wozniak, J.M.; Grimsey, N.J.; Orduña-Castillo, L.B.; Cheng, N.; Lin, Y.; Gonzalez Ramirez, M.L.; Birch, C.A.; Lapek, J.D.; Gonzalez, D.J.; et al. Phosphoproteomic Analysis of Thrombin- and P38 MAPK-Regulated Signaling Networks in Endothelial Cells. J. Biol. Chem. 2022, 298, 101801. [Google Scholar] [CrossRef]
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.-Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M.; et al. An Atlas of Substrate Specificities for the Human Serine/Threonine Kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Needham, E.J. Targets Mapped for Almost All Human Kinase Enzymes. Nature 2023, 613, 637–638. [Google Scholar] [CrossRef]
- Knight, J.D.R.; Tian, R.; Lee, R.E.C.; Wang, F.; Beauvais, A.; Zou, H.; Megeney, L.A.; Gingras, A.-C.; Pawson, T.; Figeys, D.; et al. A Novel Whole-Cell Lysate Kinase Assay Identifies Substrates of the P38 MAPK in Differentiating Myoblasts. Skelet. Muscle 2012, 2, 5. [Google Scholar] [CrossRef]
- Prikas, E.; Poljak, A.; Ittner, A. Mapping P38α Mitogen-Activated Protein Kinase Signaling by Proximity-Dependent Labeling. Protein Sci. 2020, 29, 1196–1210. [Google Scholar] [CrossRef]
- Shiryaev, A.; Dumitriu, G.; Moens, U. Distinct Roles of MK2 and MK5 in CAMP/PKA- and Stress/P38MAPK-Induced Heat Shock Protein 27 Phosphorylation. J. Mol. Signal. 2011, 6, 4. [Google Scholar] [CrossRef]
- Wettstein, G.; Bellaye, P.S.; Micheau, O.; Bonniaud, P. Small Heat Shock Proteins and the Cytoskeleton: An Essential Interplay for Cell Integrity? Int. J. Biochem. Cell Biol. 2012, 44, 1680–1686. [Google Scholar] [CrossRef]
- Li, T.; Ying, L.; Wang, H.; Li, N.; Fu, W.; Guo, Z.; Xu, L. Microcystin-Lr Induces Ceramide to Regulate PP2A and Destabilize Cytoskeleton in HEK293 Cells. Toxicol. Sci. 2012, 128, 147–157. [Google Scholar] [CrossRef]
- Clarke, J.P.; Mearow, K.M. Cell Stress Promotes the Association of Phosphorylated HspB1 with F-Actin. PLoS ONE 2013, 8, e68978. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling Mechanisms of P53-Mediated Tumour Suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Luciani, M.G.; Hutchins, J.R.; Zheleva, D.; Hupp, T.R. The C-Terminal Regulatory Domain of P53 Contains a Functional Docking Site for Cyclin A. J. Mol. Biol. 2000, 300, 503–518. [Google Scholar] [CrossRef]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.-M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by Aurora Kinase a Induces Mdm2-Mediated Destabilization and Inhibition of P53. Nat. Genet. 2004, 36, 55–62. [Google Scholar] [CrossRef]
- Heng, C.K.M.; Gilad, N.; Darlyuk-Saadon, I.; Wong, W.S.F.; Engelberg, D. Targeting the P38α Pathway in Chronic Inflammatory Diseases: Could Activation, Not Inhibition, Be the Appropriate Therapeutic Strategy? Pharmacol. Ther. 2022, 235, 108153. [Google Scholar] [CrossRef]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.-J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of P38 MAP Kinase Activation in Vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Rappsilber, J.; Mann, M. Modular Stop and Go Extraction Tips with Stacked Disks for Parallel and Multidimensional Peptide Fractionation in Proteomics. J. Proteome Res. 2006, 5, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Rappsilber, J.; Andersen, J.S.; Mann, M. Microcolumns with Self-Assembled Particle Frits for Proteomics. J. Chromatogr. A 2002, 979, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Phosphorylation Sites | Regulated Sites | |
|---|---|---|
| Intrinsically active p38α experiment | 12,085 | |
| Upregulated 2139 | ||
| Downregulated 2464 | ||
| Intrinsically active p38β experiment | 11,661 | |
| Upregulated 739 | ||
| Downregulated 1056 | ||
| Anisomycin treatment | 21,507 | |
| Upregulated 663 | ||
| Downregulated 436 | ||
| Total | 23,101 |
| Gene Names | Phosphosite Positions | Intrinsically Active Variants | Anisomycin Treatment |
|---|---|---|---|
| Atf2 | 51, 53, 72, 149, 94, 118, 310, 212, 191, 150, 118, 98, 209, 44 | + | + |
| Foxo3 | 7, 12, 279, 252, 424, 283, 299, 298, 285 | + | + |
| Jun | 62, 63 | + | + |
| Mef2a | 98, 108, 479, 464, 400 | + | + |
| Mef2c | 222 | + | + |
| Mitf | 237 | + | + |
| Trp53; Tp53 | 307, 309, 375, 350, 353 | + | + |
| Stat1 | 727 | + | |
| Cdt1 | 165, 164, 114, 403, 414 | + | |
| H2afx | 121, 124 | + | |
| Rb1 | 31, 243, 364, 599, 605, 367, 601, 617, 800, 814, 819, 816 | + | + |
| Rbm17 | 155, 169, 222, 224 | + | + |
| Gsk3a; Gsk3b | 215, 216 | + | + |
| Mapkapk2 | 208, 211 | + | |
| Rps6ka4 | 745, 347, 681, 682, 687, 771, 542, 773, 678 | + | + |
| Casp3 | 12, 26 | + | + |
| Casp8 | 188, 213 | + | + |
| Cdc25b | 186, 325, 346 | + | + |
| Ccnd3 | 68 | + | + |
| Gys1 | 589, 593, 608, 588 | + | + |
| Spag9 | 569, 566, 53, 39, 150, 21, 201, 151, 19, 153, 61, 62, 80, 568, 185, 107, 97, 113, 97, 113 | + | + |
| Cdkn1c | 6 | + | |
| Pip4k2b | 326, 322 | + | |
| Psmd1 | 315, 311, 273, 270 | + | + |
| Tab1 | 7 | + | + |
| Egfr | 695, 697, 993 | + | |
| Adam17 | 794 | + | |
| Zfyve20 | 218, 216, 21, 229, 214, 225, 213, 635, 642 | + | + |
| Hspb1 | 86, 13, 15 | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melamed Kadosh, D.; Beenstock, J.; Engelberg, D.; Admon, A. Differential Modulation of the Phosphoproteome by the MAP Kinases Isoforms p38α and p38β. Int. J. Mol. Sci. 2023, 24, 12442. https://doi.org/10.3390/ijms241512442
Melamed Kadosh D, Beenstock J, Engelberg D, Admon A. Differential Modulation of the Phosphoproteome by the MAP Kinases Isoforms p38α and p38β. International Journal of Molecular Sciences. 2023; 24(15):12442. https://doi.org/10.3390/ijms241512442
Chicago/Turabian StyleMelamed Kadosh, Dganit, Jonah Beenstock, David Engelberg, and Arie Admon. 2023. "Differential Modulation of the Phosphoproteome by the MAP Kinases Isoforms p38α and p38β" International Journal of Molecular Sciences 24, no. 15: 12442. https://doi.org/10.3390/ijms241512442
APA StyleMelamed Kadosh, D., Beenstock, J., Engelberg, D., & Admon, A. (2023). Differential Modulation of the Phosphoproteome by the MAP Kinases Isoforms p38α and p38β. International Journal of Molecular Sciences, 24(15), 12442. https://doi.org/10.3390/ijms241512442