

Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. Case Presentations
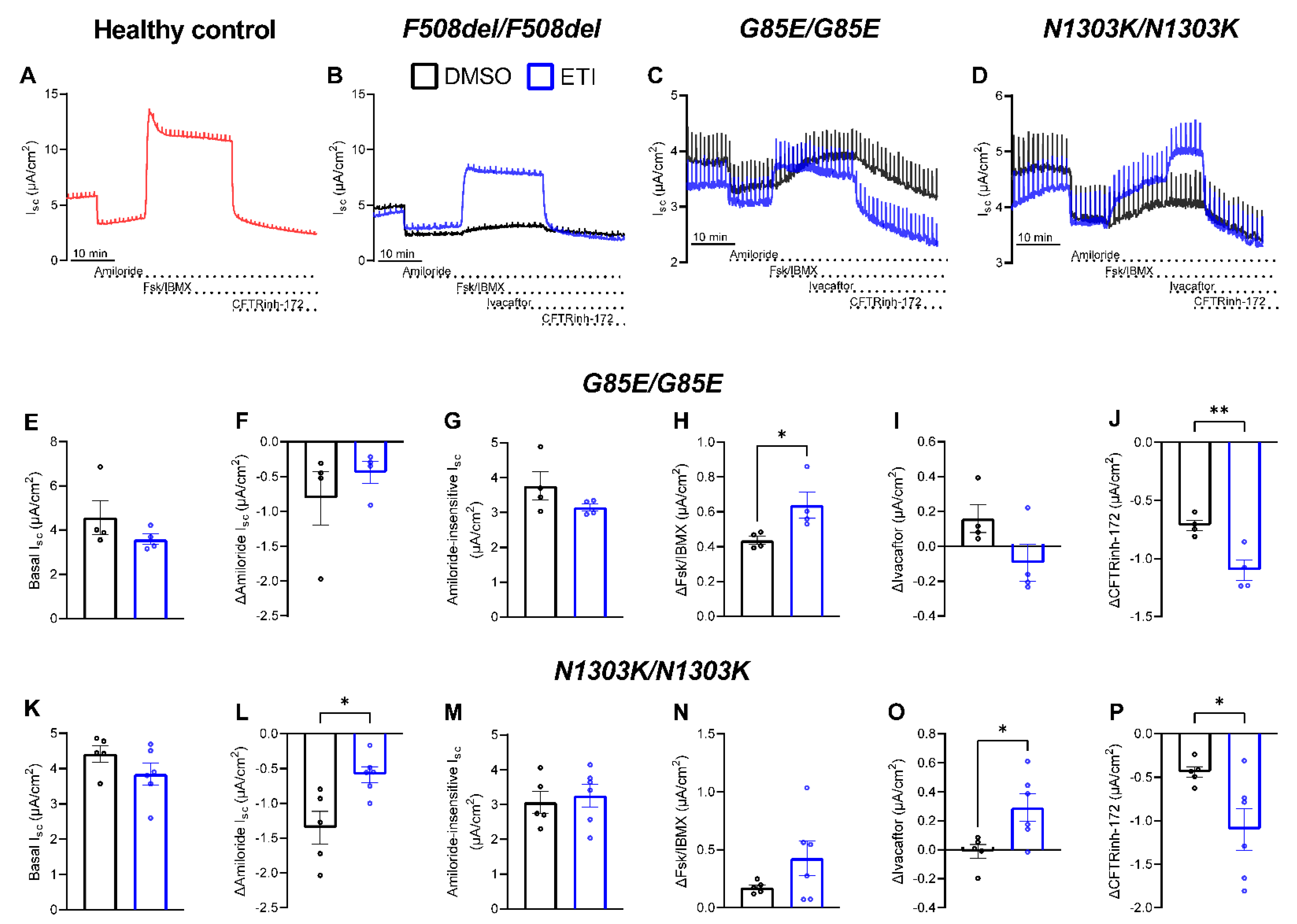
2.2. Preclinical Testing of ETI Therapy In Vitro
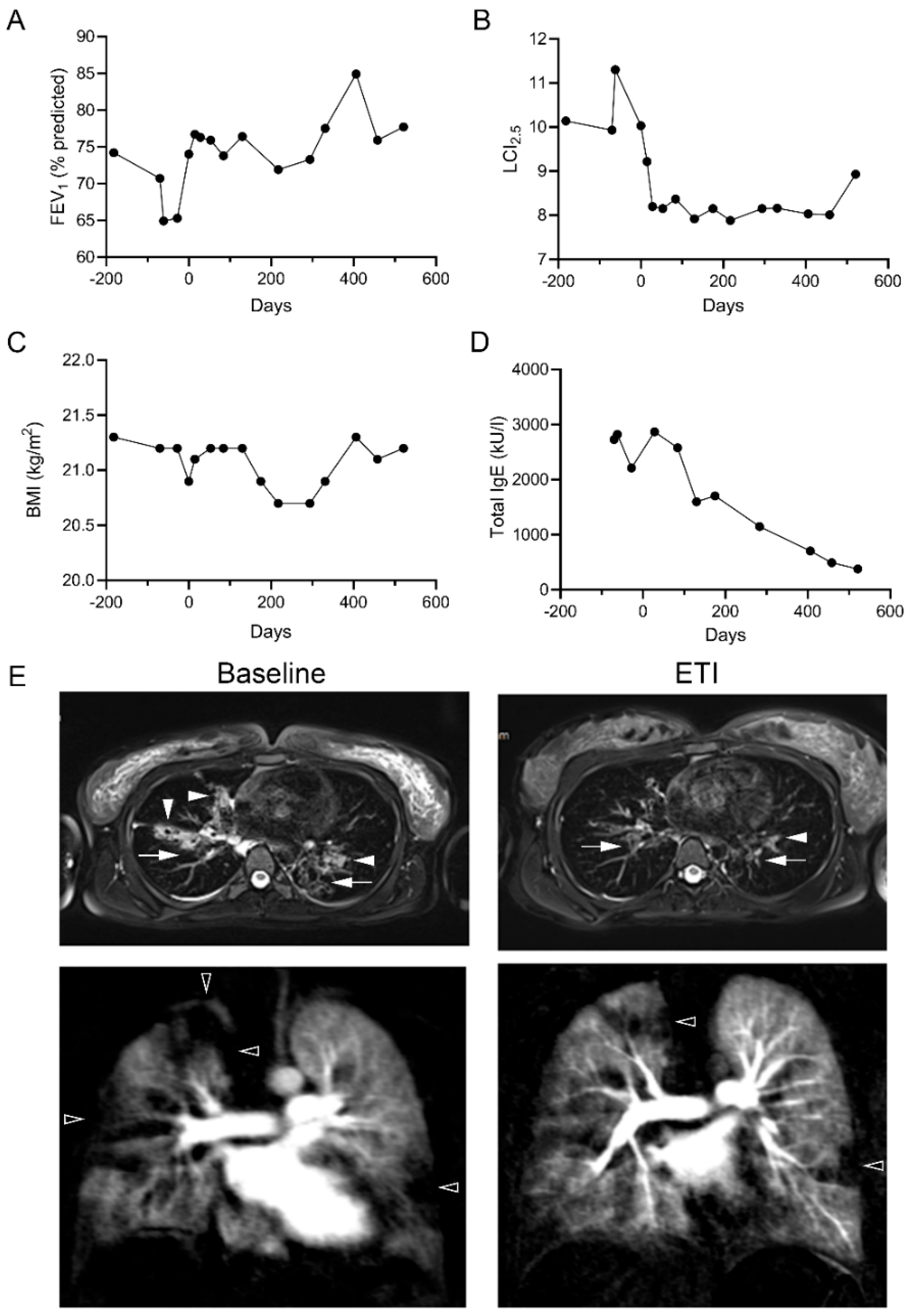
2.3. Improvements in CFTR Function In Vivo
2.4. Clinical Improvements following Treatment with ETI
3. Discussion
4. Materials and Methods
4.1. Study Design and Participants
4.2. Nasal Epithelial Cell Cultures
4.3. Ussing Chamber Experiments in pHNECs
4.4. Lung Function and Multiple-Breath Washout (MBW) Measurements
4.5. Morpho-Functional Chest Magnetic Resonance Imaging (MRI)
4.6. Sweat Chloride
4.7. NPD Measurements
4.8. Intestinal Current Measurements
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.J.; Mall, M.A.; Alvarez, A.; Colombo, C.; de Winter-de Groot, K.M.; Fajac, I.; McBennett, K.A.; McKone, E.F.; Ramsey, B.W.; Sutharsan, S.; et al. Triple Therapy for Cystic Fibrosis Phe508del-Gating and -Residual Function Genotypes. N. Engl. J. Med. 2021, 385, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Mall, M.A.; Brugha, R.; Gartner, S.; Legg, J.; Moeller, A.; Mondejar-Lopez, P.; Prais, D.; Pressler, T.; Ratjen, F.; Reix, P.; et al. Efficacy and Safety of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 Through 11 Years of Age with Cystic Fibrosis Heterozygous for F508del and a Minimal Function Mutation: A Phase 3b, Randomized, Placebo-controlled Study. Am. J. Respir. Crit. Care Med. 2022, 206, 1361–1369. [Google Scholar] [CrossRef]
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Sutharsan, S.; McKone, E.F.; Downey, D.G.; Duckers, J.; MacGregor, G.; Tullis, E.; Van Braeckel, E.; Wainwright, C.E.; Watson, D.; Ahluwalia, N.; et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: A 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir. Med. 2022, 10, 267–277. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Taylor-Cousar, J.L.; Davies, J.; Gibson, R.L.; Mall, M.A.; McKone, E.F.; McNally, P.; Ramsey, B.W.; Rayment, J.H.; Rowe, S.M.; et al. A Phase 3 Open-Label Study of ELX/TEZ/IVA in Children 6 Through 11 Years of Age With CF and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2021, 203, 1522–1532. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Renz, D.M.; Stahl, M.; Pallenberg, S.T.; Sommerburg, O.; Naehrlich, L.; Berges, J.; Dohna, M.; Ringshausen, F.C.; Doellinger, F.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on Lung Clearance Index and Magnetic Resonance Imaging in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2022, 206, 311–320. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Vitzthum, C.; Pallenberg, S.T.; Naehrlich, L.; Stahl, M.; Rohrbach, A.; Drescher, M.; Minso, R.; Ringshausen, F.C.; Rueckes-Nilges, C.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on CFTR Function in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2022, 205, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castanos, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Sermet-Gaudelus, I.; Durieu, I.; Kanaan, R.; Macey, J.; Grenet, D.; Porzio, M.; Coolen-Allou, N.; Chiron, R.; Marguet, C.; et al. The French Compassionate Program of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis with advanced lung disease and no F508del CFTR variant. Eur. Respir. J. 2023, 61, 2202437. [Google Scholar] [CrossRef] [PubMed]
- Awatade, N.T.; Uliyakina, I.; Farinha, C.M.; Clarke, L.A.; Mendes, K.; Solé, A.; Pastor, J.; Ramos, M.M.; Amaral, M.D. Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis. EBioMedicine 2015, 2, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Ensinck, M.; De Keersmaecker, L.; Heylen, L.; Ramalho, A.S.; Gijsbers, R.; Farre, R.; De Boeck, K.; Christ, F.; Debyser, Z.; Carlon, M.S. Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells 2020, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, S.; Gees, M.; Hwang, T.C. Physiological and pharmacological characterization of the N1303K mutant CFTR. J. Cyst. Fibros. 2018, 17, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+ tezacaftor+ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur. Respir. J. 2020, 57, 2002774. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Huang, Y.; Paul, G.; Lee, J.; Yarlagadda, S.; McCoy, K.; Naren, A.P. Elexacaftor/Tezacaftor/Ivacaftor Improved Clinical Outcomes in a Patient with N1303K-CFTR Based on In Vitro Experimental Evidence. Am. J. Respir. Crit. Care Med. 2021, 204, 1231–1235. [Google Scholar] [CrossRef]
- Stekolchik, E.; Saul, D.; Chidekel, A. Clinical efficacy of elexacaftor-tezacaftor-ivacaftor in an adolescent with homozygous G85E cystic fibrosis. Respir. Med. Case Rep. 2022, 40, 101775. [Google Scholar] [CrossRef]
- Sadras, I.; Kerem, E.; Livnat, G.; Sarouk, I.; Breuer, O.; Reiter, J.; Gileles-Hillel, A.; Inbar, O.; Cohen, M.; Gamliel, A.; et al. Clinical and functional efficacy of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis carrying the N1303K mutation. J. Cyst. Fibros. 2023, in press. [Google Scholar] [CrossRef]
- Balazs, A.; Millar-Buchner, P.; Mulleder, M.; Farztdinov, V.; Szyrwiel, L.; Addante, A.; Kuppe, A.; Rubil, T.; Drescher, M.; Seidel, K.; et al. Age-Related Differences in Structure and Function of Nasal Epithelial Cultures From Healthy Children and Elderly People. Front. Immunol. 2022, 13, 822437. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Dopfer, C.; Naehrlich, L.; Gyulumyan, L.; Scheuermann, H.; Hirtz, S.; Wege, S.; Mairbaurl, H.; Dorda, M.; Hyde, R.; et al. Effects of Lumacaftor-Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Hug, M.J.; Sommerburg, O.; Hirtz, S.; Hentschel, J.; Heinzmann, A.; Dopfer, C.; Schulz, A.; Mainz, J.G.; Tummler, B.; et al. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am. J. Respir. Crit. Care Med. 2015, 192, 1252–1255. [Google Scholar] [CrossRef]
- Ensinck, M.M.; De Keersmaecker, L.; Ramalho, A.S.; Cuyx, S.; Van Biervliet, S.; Dupont, L.; Christ, F.; Debyser, Z.; Vermeulen, F.; Carlon, M.S. Novel CFTR modulator combinations maximise rescue of G85E and N1303K in rectal organoids. ERJ Open Res. 2022, 8, 00716–2021. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Trikafta Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217660s000lbl.pdf (accessed on 16 July 2023).
- European Medical Agency. Assessment Report Kaftrio. Available online: https://www.ema.europa.eu/en/documents/assessment-report/kaftrio-epar-public-assessment-report_en.pdf (accessed on 16 July 2023).
- Stutts, M.J.; Canessa, C.M.; Olsen, J.C.; Hamrick, M.; Cohn, J.A.; Rossier, B.C.; Boucher, R.C. CFTR as a cAMP-dependent regulator of sodium channels. Science 1995, 269, 847–850. [Google Scholar] [CrossRef]
- Mall, M.; Hipper, A.; Greger, R.; Kunzelmann, K. Wild type but not Delta F508 CFTR inhibits Na+ conductance when coexpressed in Xenopus oocytes. FEBS Lett. 1996, 381, 47–52. [Google Scholar] [CrossRef]
- Mall, M.; Bleich, M.; Schurlein, M.; Kuhr, J.; Seydewitz, H.H.; Brandis, M.; Greger, R.; Kunzelmann, K. Cholinergic ion secretion in human colon requires coactivation by cAMP. Am. J. Physiol. 1998, 275, G1274–G1281. [Google Scholar] [CrossRef]
- Mall, M.A. ENaC inhibition in cystic fibrosis: Potential role in the new era of CFTR modulator therapies. Eur. Respir. J. 2020, 56, 2000946. [Google Scholar] [CrossRef]
- Mall, M.; Bleich, M.; Greger, R.; Schreiber, R.; Kunzelmann, K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Investig. 1998, 102, 15–21. [Google Scholar] [CrossRef]
- Mall, M.; Wissner, A.; Gonska, T.; Calenborn, D.; Kuehr, J.; Brandis, M.; Kunzelmann, K. Inhibition of amiloride-sensitive epithelial Na(+) absorption by extracellular nucleotides in human normal and cystic fibrosis airways. Am. J. Respir. Cell Mol. Biol. 2000, 23, 755–761. [Google Scholar] [CrossRef] [PubMed]
- O’Brodovich, H.; Yang, P.; Gandhi, S.; Otulakowski, G. Amiloride-insensitive Na+ and fluid absorption in the mammalian distal lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L401–L408. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of Organoid Swelling and In Vivo Biomarkers of CFTR Function to Determine Effects of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592. [Google Scholar] [CrossRef]
- Taylor-Robinson, D.; Whitehead, M.; Diderichsen, F.; Olesen, H.V.; Pressler, T.; Smyth, R.L.; Diggle, P. Understanding the natural progression in %FEV1 decline in patients with cystic fibrosis: A longitudinal study. Thorax 2012, 67, 860–866. [Google Scholar] [CrossRef]
- Frauchiger, B.S.; Ramsey, K.A.; Usemann, J.; Kieninger, E.; Casaulta, C.; Sirtes, D.; Yammine, S.; Spycher, B.; Moeller, A.; Latzin, P. Variability of clinically measured lung clearance index in children with cystic fibrosis. Pediatr. Pulmonol. 2023, 58, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Elson, E.C.; Capel, P.; Haynes, J.; Duehlmeyer, S.; Fischer, M.; Escobar, H. CFTR Modulator Therapy in an Individual With Cystic Fibrosis Caused by a N1303K CFTR Variant and Infected With Mycobacterium abscessus. J. Pediatr. Pharmacol. Ther. 2022, 27, 396–399. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Mandrup Bertozzi, S.; et al. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef]
- Gentzsch, M.; Boyles, S.E.; Cheluvaraju, C.; Chaudhry, I.G.; Quinney, N.L.; Cho, C.; Dang, H.; Liu, X.; Schlegel, R.; Randell, S.H. Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2017, 56, 568–574. [Google Scholar] [CrossRef]
- Salomon, J.J.; Albrecht, T.; Graeber, S.Y.; Scheuermann, H.; Butz, S.; Schatterny, J.; Mairbaurl, H.; Baumann, I.; Mall, M.A. Chronic rhinosinusitis with nasal polyps is associated with impaired TMEM16A-mediated epithelial chloride secretion. J. Allergy Clin. Immunol. 2021, 147, 2191–2201.e2192. [Google Scholar] [CrossRef]
- Beydon, N.; Davis, S.D.; Lombardi, E.; Allen, J.L.; Arets, H.G.; Aurora, P.; Bisgaard, H.; Davis, G.M.; Ducharme, F.M.; Eigen, H.; et al. An official American Thoracic Society/European Respiratory Society statement: Pulmonary function testing in preschool children. Am. J. Respir. Crit. Care Med. 2007, 175, 1304–1345. [Google Scholar] [CrossRef] [PubMed]
- Quanjer, P.H.; Stanojevic, S.; Cole, T.J.; Baur, X.; Hall, G.L.; Culver, B.H.; Enright, P.L.; Hankinson, J.L.; Ip, M.S.; Zheng, J.; et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: The global lung function 2012 equations. Eur. Respir. J. 2012, 40, 1324–1343. [Google Scholar] [CrossRef]
- Stahl, M.; Joachim, C.; Kirsch, I.; Uselmann, T.; Yu, Y.; Alfeis, N.; Berger, C.; Minso, R.; Rudolf, I.; Stolpe, C.; et al. Multicentre feasibility of multiple-breath washout in preschool children with cystic fibrosis and other lung diseases. ERJ Open Res. 2020, 6, 00408–2020. [Google Scholar] [CrossRef]
- Stahl, M.; Wielputz, M.O.; Graeber, S.Y.; Joachim, C.; Sommerburg, O.; Kauczor, H.U.; Puderbach, M.; Eichinger, M.; Mall, M.A. Comparison of Lung Clearance Index and Magnetic Resonance Imaging for Assessment of Lung Disease in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Wyler, F.; Oestreich, M.A.; Frauchiger, B.S.; Ramsey, K.A.; Latzin, P. Correction of sensor crosstalk error in Exhalyzer D multiple-breath washout device significantly impacts outcomes in children with cystic fibrosis. J. Appl. Physiol. 2021, 131, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Wielputz, M.O.; Puderbach, M.; Kopp-Schneider, A.; Stahl, M.; Fritzsching, E.; Sommerburg, O.; Ley, S.; Sumkauskaite, M.; Biederer, J.; Kauczor, H.U.; et al. Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2014, 189, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Eichinger, M.; Optazaite, D.E.; Kopp-Schneider, A.; Hintze, C.; Biederer, J.; Niemann, A.; Mall, M.A.; Wielputz, M.O.; Kauczor, H.U.; Puderbach, M. Morphologic and functional scoring of cystic fibrosis lung disease using MRI. Eur. J. Radiol. 2012, 81, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Naehrlich, L.; Stuhrmann-Spangenberg, M.; Barben, J.; Bargon, J.; Blankenstein, O.; Bremer, W.; Brunsmann, F.; Buchholz, T.; Ellemunter, H.; Fusch, C.; et al. S2-Konsensus-Leitlinie “Diagnose der Mukoviszidose” (AWMF 026-023). Available online: http://www.awmf.org/leitlinien/detail/ll/026-023.html (accessed on 21 June 2023).
- LeGrys, V.A. Sweat Testing: Sample Collection and Quantitative Chloride Analysis, Approved Guideline, 4th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2019. [Google Scholar]
- Rowe, S.M.; Clancy, J.P.; Wilschanski, M. Nasal potential difference measurements to assess CFTR ion channel activity. Methods Mol. Biol. 2011, 741, 69–86. [Google Scholar] [CrossRef]
- Sermet-Gaudelus, I.; Girodon, E.; Sands, D.; Stremmler, N.; Vavrova, V.; Deneuville, E.; Reix, P.; Bui, S.; Huet, F.; Lebourgeois, M.; et al. Clinical phenotype and genotype of children with borderline sweat test and abnormal nasal epithelial chloride transport. Am. J. Respir. Crit. Care Med. 2010, 182, 929–936. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Vitzthum, C.; Mall, M.A. Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis. J. Pers. Med. 2021, 11, 384. [Google Scholar] [CrossRef]
- Mall, M.; Wissner, A.; Seydewitz, H.H.; Kuehr, J.; Brandis, M.; Greger, R.; Kunzelmann, K. Defective cholinergic Cl(-) secretion and detection of K(+) secretion in rectal biopsies from cystic fibrosis patients. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G617–G624. [Google Scholar] [CrossRef] [PubMed]
- Hirtz, S.; Gonska, T.; Seydewitz, H.H.; Thomas, J.; Greiner, P.; Kuehr, J.; Brandis, M.; Eichler, I.; Rocha, H.; Lopes, A.I.; et al. CFTR Cl- channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 2004, 127, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Roth, E.K.; Hirtz, S.; Duerr, J.; Wenning, D.; Eichler, I.; Seydewitz, H.H.; Amaral, M.D.; Mall, M.A. The K+ channel opener 1-EBIO potentiates residual function of mutant CFTR in rectal biopsies from cystic fibrosis patients. PLoS ONE 2011, 6, e24445. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 1 August 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristic | G85E/G85E | N1303K/N1303K |
|---|---|---|
| Age (years) | 15.0 | 19.8 |
| Sex | Female | Male |
| Pancreatic insufficiency | Yes | Yes |
| Sweat chloride (mmol/L) | 102.0 | 99.0 |
| FEV1 (% predicted) | 74.0 | 54.0 |
| LCI2.5 | 10.0 | 14.5 |
| BMI (kg/m2) | 20.9 | 19.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graeber, S.Y.; Balázs, A.; Ziegahn, N.; Rubil, T.; Vitzthum, C.; Piehler, L.; Drescher, M.; Seidel, K.; Rohrbach, A.; Röhmel, J.; et al. Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis. Int. J. Mol. Sci. 2023, 24, 12365. https://doi.org/10.3390/ijms241512365
Graeber SY, Balázs A, Ziegahn N, Rubil T, Vitzthum C, Piehler L, Drescher M, Seidel K, Rohrbach A, Röhmel J, et al. Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis. International Journal of Molecular Sciences. 2023; 24(15):12365. https://doi.org/10.3390/ijms241512365
Chicago/Turabian StyleGraeber, Simon Y., Anita Balázs, Niklas Ziegahn, Tihomir Rubil, Constanze Vitzthum, Linus Piehler, Marika Drescher, Kathrin Seidel, Alexander Rohrbach, Jobst Röhmel, and et al. 2023. "Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis" International Journal of Molecular Sciences 24, no. 15: 12365. https://doi.org/10.3390/ijms241512365
APA StyleGraeber, S. Y., Balázs, A., Ziegahn, N., Rubil, T., Vitzthum, C., Piehler, L., Drescher, M., Seidel, K., Rohrbach, A., Röhmel, J., Thee, S., Duerr, J., Mall, M. A., & Stahl, M. (2023). Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis. International Journal of Molecular Sciences, 24(15), 12365. https://doi.org/10.3390/ijms241512365