
Deciphering the Biology of Circulating Tumor Cells through Single-Cell RNA Sequencing: Implications for Precision Medicine in Cancer
 , ,
, ,
Abstract
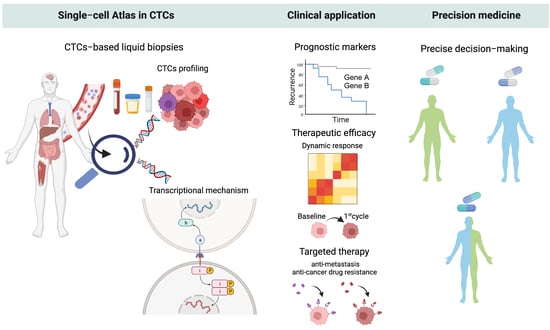
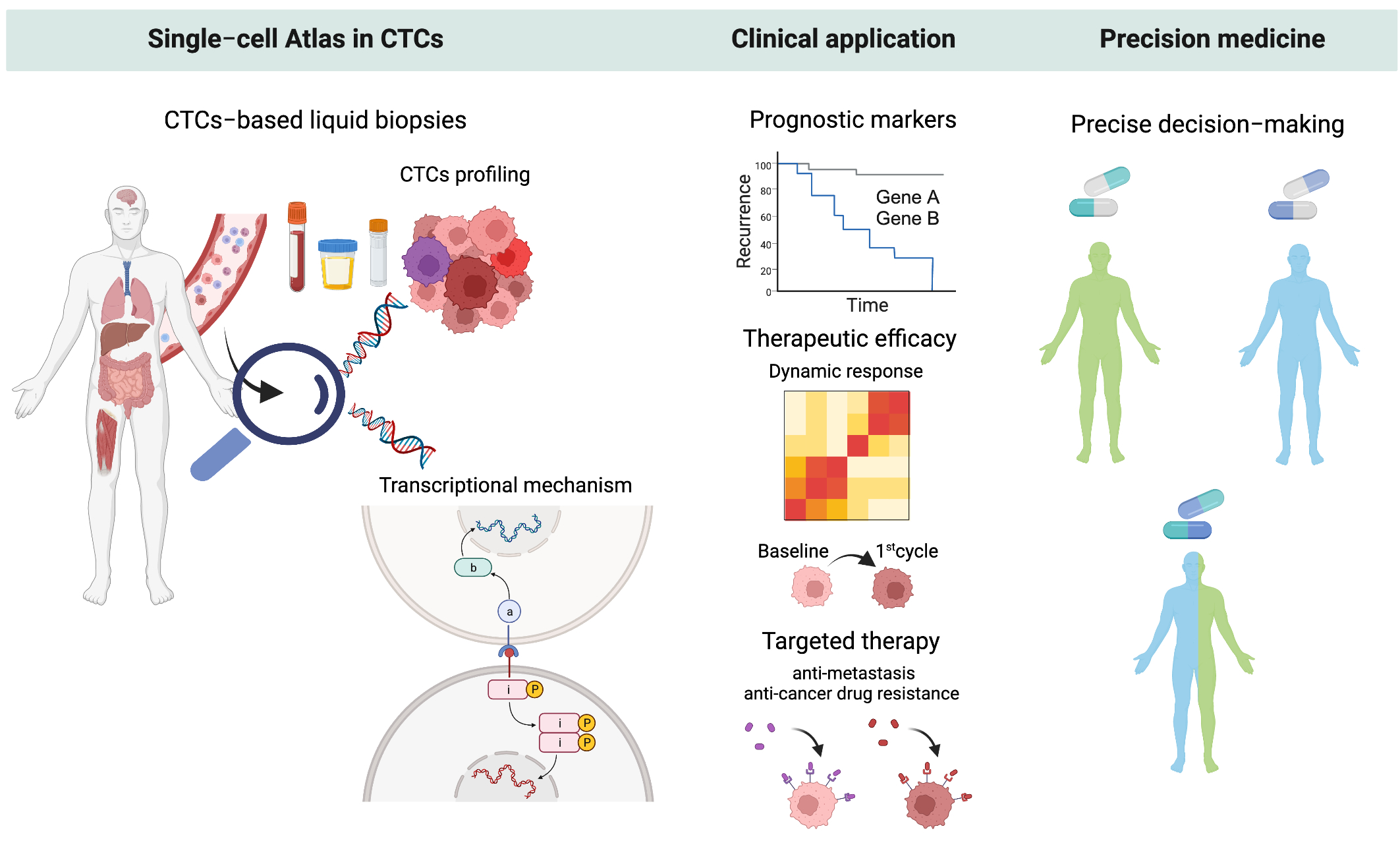
1. Introduction
2. Workflow of scRNA-Seq of CTCs
2.1. Single-Cell Isolation Techniques
2.1.1. Fluorescent-Activated Cell Sorting (FACS) Isolation

{kind=link}
{kind=link}
{kind=link}
| Enrichment Based Methods | Publication | Blood | Samples (Number and Characteristics) | Enrichment Technology | Enrichment Features | Capture Efficiency | Reference |
|---|---|---|---|---|---|---|---|
| Breast cancer | |||||||
| EpCAM | Talasaz et al., 2019 | A total of 9 mL | A total of 17 metastatic cancers | MagSweeper | anti-EpCAM immunomagnetic separation | In total: 12 ± 23 (means ± SD) CTCs per 9 mL. | [27] |
| Riebensahm et al., 2019 | A total of 7.5 mL | A total of 44 brain metastases in breast cancers | CellSearch | anti-EpCAM immunomagnetic separation | In total: 1 to 1800 with median number of 4 CTCs per 7.5 mL blood. | [28] | |
| Nagrath et al., 2007 | A total of 0.9–5.1 mL | A total of 10 | CTC-Chip | anti-EpCAM coated microposts based microfluidic | In total: 5 to 176 with 79 ± 52 (mean ± SD) CTCs per mL. | [29] | |
| Non-EpCAM | Drucker et al., 2020 | A total of 15 mL | A total of 28 metastatic breast cancer | RosetteSep™ | Immunodensity with negative depletion of WBCs | In total: 0.55 CTCs/mL (mean). | [30] |
| A total of 8 metastatic breast cancer | ScreenCell® Cyto filters | Size-based methods | mean count 4.2 CTCs per mL. | ||||
| Colorectal cancer (CRC) | |||||||
| EpCAM | Nagrath et al., 2007 | A total of 0.9–5.1 mL | A total of 10 | CTC-Chip | anti-EpCAM coated microposts based microfluidic | In total: 42 to 375 with 121 ± 127 (mean ± SD) CTCs per mL. | [29] |
| Tsai et al., 2016 | A total of 2 mL | non-metastatic (n = 95), and m-CRC (n = 15) patients. | CellMax (CMx®) | anti-EpCAM coated chip |
| [31] | |
| Dizdar et al., 2019 | A total of 7.5 mL | A total of 80 CRC with M0 and M1 | CellSearch | anti-EpCAM immunomagnetic separation | A total of 135 CTCs from n = 80. | [32] | |
| Wu et al., 2017 | A total of 7.5 mL | A total of 44 CRC | CellSearch | anti-EpCAM immunomagnetic separation |
| [33] | |
| Non-EpCAM | Hendricks et al., 2020 | A total of 8 mL | A total of 31 diagnosed with colon carcinomas and rectal carcinomas | ScreenCell® Cyto filters | Size-based methods | In total: 0.2 to 14.3 with mean count 3.25 CTC per mL. | [34] |
| Vasantharajan et al., 2022 | A total of 8 mL | A total of 15 CRC with various stages of cancer (AJCC stages I-IVB) | MetaCell | size-based separation | In total: 0 to 12 with mean count of 2 CTCs per 7.5 mL. | [35] | |
| Hepatocellular carcinoma (HCC) | |||||||
| EpCAM | Morris et al., 2014 | A total of 7.5 mL | A total of 50 HCC | CellSearch | anti-EpCAM immunomagnetic separation | In total: 0 to 8 CTCs per 7.5 mL. | [36] |
| Zhang et al., 2016 | A total of 2 mL | A total of 36 HCC | CTC-chip | anti-EpCAM coated microposts based microfluidic | In total: 14 ± 10 (mean ± SD) CTCs per 2 mL. | [37] | |
| Non-EpCAM | Morris et al., 2014 | A total of 7.5 mL | A total of 50 HCC | ISET | size-based separation | In total: 0 to 47 CTCs per 7.5 mL. | [36] |
| Zhao et al., 2023 | A total of 7.5 mL | A total of 127 HCC patients With low recurrence (LR) and high recurrence (HR) | Leucosep® with CD45 depletion | Density and immunomagnetic separation with negative depletion of WBCs |
| [38] | |
| Lung cancer | |||||||
| EpCAM | Nagrath et al., 2007 | A total of 0.9–5.1 mL | A total of 55 NSCLC | CTC-Chip | anti-EpCAM coated microposts based microfluidic | In total: 5 to 1281 with 155 ± 236 (mean ± SD) CTCs per mL. | [29] |
| Ke et al., 2015 | A total of 7 NSCLC | NanoVelcro | anti-EpCAM)-coated nanostructured substrates in microfluidic chip | In total: 2 to 7 with 7 ± 4.74 (mean ± SD) CTCs per mL. | [39] | ||
| Non-EpCAM | Hosokawa et al., 2013 | A total of 7.5 mL | NSCLC | miniaturized microcavity array (MCA) system | size-based separation | In total: 0 to 291 with median of 13 CTCs per 7.5 mL. | [40] |
| Sonn et al., 2017 | A total of 5 mL | A total of 66 stage I–III patients and 16 stage IV | ISET | size-based separation |
| [41] | |
2.1.2. Micromanipulation
2.1.3. Laser Capture Microdissection (LCM)
2.1.4. Microfluidic Technology
2.2. Data Analysis in scRNA-Seq
2.2.1. Cellular Subpopulation Identification
2.2.2. Differential Gene Expression Analysis
2.2.3. Pseudotime Analysis
3. Translational Relevance of scRNA-Seq of CTCs
3.1. Decoding Tumor Heterogeneity in CTCs and Their Gene Expression Signatures Implicating Clinical Outcome
3.1.1. Prognostic Molecular Markers
3.1.2. Treatment Response and Disease Progression
3.1.3. CTC Subtyping Analysis in Other Biological Fluids
3.2. Revealing the Altered Molecular Pathways Underlying Cancer Progression for Druggable CTCs
3.2.1. Metastatic Mechanisms
3.2.2. Drug Resistance Mechanisms
4. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: From circulating tumor cells to metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Zhang, X.F.; Peng, J.; Li, X.F.; Wang, A.L.; Bie, Y.Q.; Shi, L.H.; Lin, M.B.; Zhang, X.F. Survival mechanisms and influence factors of circulating tumor cells. BioMed Res. Int. 2018, 2018, 6304701. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.E.; Allan, A.L. Chapter Four—Circulating Tumor Cells and Implications of the Epithelial-to-Mesenchymal Transition. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 83, pp. 121–181. [Google Scholar]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hong, W.; Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef]
- Cognart, H.A.; Viovy, J.-L.; Villard, C. Fluid shear stress coupled with narrow constrictions induce cell type-dependent morphological and molecular changes in SK-BR-3 and MDA-MB-231 cells. Sci. Rep. 2020, 10, 6386. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Pereira-Veiga, T.; Schneegans, S.; Pantel, K.; Wikman, H. Circulating tumor cell-blood cell crosstalk: Biology and clinical relevance. Cell Rep. 2022, 40, 111298. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Crucial roles of circulating tumor cells in the metastatic cascade and tumor immune escape: Biology and clinical translation. J. Immunother. Cancer 2022, 10, e005615. [Google Scholar] [CrossRef]
- Varotsos Vrynas, A.; Perea Paizal, J.; Bakal, C.; Au, S.H. Arresting metastasis within the microcirculation. Clin. Exp. Metastasis 2021, 38, 337–342. [Google Scholar] [CrossRef]
- Krog, B.L.; Henry, M.D. Biomechanics of the circulating tumor cell microenvironment. Adv. Exp. Med. Biol. 2018, 1092, 209–233. [Google Scholar] [CrossRef]
- Heeke, S.; Mograbi, B.; Alix-Panabières, C.; Hofman, P. Never travel alone: The crosstalk of circulating tumor cells and the blood microenvironment. Cells 2019, 8, 714. [Google Scholar] [CrossRef]
- Eslami, S.Z.; Cortés-Hernández, L.E.; Alix-Panabières, C. The metastatic cascade as the basis for liquid biopsy development. Front. Oncol. 2020, 10, 1055. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liao, K.; Yang, X.; Wu, C.; Wu, W. Using single-cell sequencing technology to detect circulating tumor cells in solid tumors. Mol. Cancer 2021, 20, 104. [Google Scholar] [CrossRef]
- Akpe, V.; Kim, T.H.; Brown, C.L.; Cock, I.E. Circulating tumour cells: A broad perspective. J. R. Soc. Interface 2020, 17, 20200065. [Google Scholar] [CrossRef]
- Ried, K.; Eng, P.; Sali, A. Screening for Circulating Tumour Cells Allows Early Detection of Cancer and Monitoring of Treatment Effectiveness: An Observational Study. Asian Pac. J. Cancer Prev. 2017, 18, 2275–2285. [Google Scholar] [CrossRef]
- Vasseur, A.; Kiavue, N.; Bidard, F.-C.; Pierga, J.-Y.; Cabel, L. Clinical utility of circulating tumor cells: An update. Mol. Oncol. 2021, 15, 1647–1666. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Shao, W.; Li, Z.; Zhao, R.; Ye, Z. Strategies for enrichment of circulating tumor cells. Transl. Cancer Res. 2020, 9, 2012–2025. [Google Scholar] [CrossRef] [PubMed]
- Swennenhuis, J.F.; van Dalum, G.; Zeune, L.L.; Terstappen, L.W. Improving the CellSearch® system. Expert Rev. Mol. Diagn. 2016, 16, 1291–1305. [Google Scholar] [CrossRef]
- Grover, P.K.; Cummins, A.G.; Price, T.J.; Roberts-Thomson, I.C.; Hardingham, J.E. Circulating tumour cells: The evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research. Ann. Oncol. 2014, 25, 1506–1516. [Google Scholar] [CrossRef]
- Nicolazzo, C.; Gradilone, A.; Loreni, F.; Raimondi, C.; Gazzaniga, P. EpCAM(low) circulating tumor cells: Gold in the waste. Dis. Markers 2019, 2019, 1718920. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [PubMed]
- Rushton, A.J.; Nteliopoulos, G.; Shaw, J.A.; Coombes, R.C. A review of circulating tumour cell enrichment technologies. Cancers 2021, 13, 970. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Sa, S.; Wang, L.; Dulmage, K.; Bhagwat, N.; Yee, S.S.; Sen, M.; Pletcher, C.H., Jr.; Moore, J.S.; Saksena, S.; et al. An integrated enrichment system to facilitate isolation and molecular characterization of single cancer cells from whole blood. Cytom. A 2018, 93, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, N.; Dulmage, K.; Pletcher, C.H.; Wang, L.; DeMuth, W.; Sen, M.; Balli, D.; Yee, S.S.; Sa, S.; Tong, F.; et al. An integrated flow cytometry-based platform for isolation and molecular characterization of circulating tumor single cells and clusters. Sci. Rep. 2018, 8, 5035. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single cell isolation and analysis. Front. Cell Dev. Biol. 2016, 4, 116. [Google Scholar] [CrossRef]
- Talasaz, A.H.; Powell, A.A.; Huber, D.E.; Berbee, J.G.; Roh, K.H.; Yu, W.; Xiao, W.; Davis, M.M.; Pease, R.F.; Mindrinos, M.N.; et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc. Natl. Acad. Sci. USA 2009, 106, 3970–3975. [Google Scholar] [CrossRef]
- Riebensahm, C.; Joosse, S.A.; Mohme, M.; Hanssen, A.; Matschke, J.; Goy, Y.; Witzel, I.; Lamszus, K.; Kropidlowski, J.; Petersen, C.; et al. Clonality of circulating tumor cells in breast cancer brain metastasis patients. Breast Cancer Res. 2019, 21, 101. [Google Scholar] [CrossRef]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef]
- Drucker, A.; Teh, E.M.; Kostyleva, R.; Rayson, D.; Douglas, S.; Pinto, D.M. Comparative performance of different methods for circulating tumor cell enrichment in metastatic breast cancer patients. PLoS ONE 2020, 15, e0237308. [Google Scholar] [CrossRef]
- Tsai, W.-S.; Chen, J.-S.; Shao, H.-J.; Wu, J.-C.; Lai, J.-M.; Lu, S.-H.; Hung, T.-F.; Chiu, Y.-C.; You, J.-F.; Hsieh, P.-S.; et al. Circulating Tumor Cell Count Correlates with Colorectal Neoplasm Progression and Is a Prognostic Marker for Distant Metastasis in Non-Metastatic Patients. Sci. Rep. 2016, 6, 24517. [Google Scholar] [CrossRef]
- Dizdar, L.; Fluegen, G.; van Dalum, G.; Honisch, E.; Neves, R.P.; Niederacher, D.; Neubauer, H.; Fehm, T.; Rehders, A.; Krieg, A.; et al. Detection of circulating tumor cells in colorectal cancer patients using the GILUPI CellCollector: Results from a prospective, single-center study. Mol. Oncol. 2019, 13, 1548–1558. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, Z.; Gao, X.H.; Shen, Z.; Jing, Y.; Lu, H.; Li, H.; Yang, X.; Cui, X.; Li, Y.; et al. Clinical significance of detecting circulating tumor cells in colorectal cancer using subtraction enrichment and immunostaining-fluorescence in situ hybridization (SE-iFISH). Oncotarget 2017, 8, 21639–21649. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, A.; Brandt, B.; Geisen, R.; Dall, K.; Röder, C.; Schafmayer, C.; Becker, T.; Hinz, S.; Sebens, S. Isolation and Enumeration of CTC in Colorectal Cancer Patients: Introduction of a Novel Cell Imaging Approach and Comparison to Cellular and Molecular Detection Techniques. Cancers 2020, 12, 2643. [Google Scholar] [CrossRef]
- Vasantharajan, S.S.; Barnett, E.; Gray, E.S.; McCall, J.L.; Rodger, E.J.; Eccles, M.R.; Munro, F.; Pattison, S.; Chatterjee, A. Assessment of a Size-Based Method for Enriching Circulating Tumour Cells in Colorectal Cancer. Cancers 2022, 14, 3446. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.L.; Tugwood, J.D.; Khoja, L.; Lancashire, M.; Sloane, R.; Burt, D.; Shenjere, P.; Zhou, C.; Hodgson, C.; Ohtomo, T.; et al. Circulating biomarkers in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2014, 74, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Zhang, J.; Sun, B.; Zheng, L.; Li, J.; Liu, S.; Sui, G.; Yin, Z. Microfluidic chip for isolation of viable circulating tumor cells of hepatocellular carcinoma for their culture and drug sensitivity assay. Cancer Biol. Ther. 2016, 17, 1177–1187. [Google Scholar] [CrossRef]
- Zhao, L.; Zheng, Z.; Liu, Y.; Liu, F.; Li, X.; Wu, Z. The mesenchymal circulating tumor cells as biomarker for prognosis prediction and supervision in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2023, 149, 6035–6048. [Google Scholar] [CrossRef]
- Ke, Z.; Lin, M.; Chen, J.-F.; Choi, J.-s.; Zhang, Y.; Fong, A.; Liang, A.-J.; Chen, S.-F.; Li, Q.; Fang, W.; et al. Programming Thermoresponsiveness of NanoVelcro Substrates Enables Effective Purification of Circulating Tumor Cells in Lung Cancer Patients. ACS Nano 2015, 9, 62–70. [Google Scholar] [CrossRef]
- Hosokawa, M.; Kenmotsu, H.; Koh, Y.; Yoshino, T.; Yoshikawa, T.; Naito, T.; Takahashi, T.; Murakami, H.; Nakamura, Y.; Tsuya, A.; et al. Size-Based Isolation of Circulating Tumor Cells in Lung Cancer Patients Using a Microcavity Array System. PLoS ONE 2013, 8, e67466. [Google Scholar] [CrossRef]
- Sonn, C.H.; Cho, J.H.; Kim, J.W.; Kang, M.S.; Lee, J.; Kim, J. Detection of circulating tumor cells in patients with non-small cell lung cancer using a size-based platform. Oncol. Lett. 2017, 13, 2717–2722. [Google Scholar] [CrossRef]
- Theil, G.; Fischer, K.; Weber, E.; Medek, R.; Hoda, R.; Lücke, K.; Fornara, P. The use of a new CellCollector to isolate circulating tumor cells from the blood of patients with different stages of prostate cancer and clinical outcomes—A proof-of-concept study. PLoS ONE 2016, 11, e0158354. [Google Scholar] [CrossRef] [PubMed]
- Stilwell, J.L.; Varshavskaya, P.; Werbin, J.L.; Nordberg, J.J.; Ramirez, A.B.; Quarre, S.; Tzucker, J.; Chow, J.; Enright, B.; Kaldjian, E.P. RareCyte® CTC analysis step 3: Using the cytepicker® module for individual cell retrieval and subsequent whole genome amplification of circulating tumor cells for genomic analysis. In Circulating Tumor Cells: Methods and Protocols; Magbanua, M.J.M., Park, J.W., Eds.; Springer: New York, NY, USA, 2017; pp. 181–192. [Google Scholar] [CrossRef]
- Kaldjian, E.P.; Ramirez, A.B.; Sun, Y.; Campton, D.E.; Werbin, J.L.; Varshavskaya, P.; Quarre, S.; George, T.; Madan, A.; Blau, C.A.; et al. The RareCyte® platform for next-generation analysis of circulating tumor cells. Cytometry A 2018, 93, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Saremi, S.; Klotz, R.; Iriondo, O.; Amzaleg, Y.; Chairez, Y.; Tulpule, V.; Lang, J.E.; Kang, I.; Yu, M. PIC&RUN: An integrated assay for the detection and retrieval of single viable circulating tumor cells. Sci. Rep. 2019, 9, 17470. [Google Scholar] [CrossRef] [PubMed]
- Tokar, J.J.; Stahlfeld, C.N.; Sperger, J.M.; Niles, D.J.; Beebe, D.J.; Lang, J.M.; Warrick, J.W. Pairing microwell arrays with an affordable, semiautomated single-cell aspirator for the interrogation of circulating tumor cell heterogeneity. SLAS Technol. 2020, 25, 162–176. [Google Scholar] [CrossRef]
- Chen, S.; El-Heliebi, A.; Tauber, G.; Langsenlehner, T.; Pötscher, M.; Kashofer, K.; Czyż, Z.T.; Polzer, B.; Riethdorf, S.; Kuske, A.; et al. Catch and release: Rare cell analysis from a functionalised medical wire. Sci. Rep. 2017, 7, 43424. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Adalsteinsson, V.A.; Cibulskis, K.; Choudhury, A.D.; Rosenberg, M.; Cruz-Gordillo, P.; Francis, J.M.; Zhang, C.-Z.; Shalek, A.K.; Satija, R.; et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat. Biotechnol. 2014, 32, 479–484. [Google Scholar] [CrossRef]
- He, Y.; Shi, J.; Shi, G.; Xu, X.; Liu, Q.; Liu, C.; Gao, Z.; Bai, J.; Shan, B. Using the new cellcollector to capture circulating tumor cells from blood in different groups of pulmonary disease: A cohort study. Sci Rep. 2017, 7, 9542. [Google Scholar] [CrossRef]
- Campton, D.E.; Ramirez, A.B.; Nordberg, J.J.; Drovetto, N.; Clein, A.C.; Varshavskaya, P.; Friemel, B.H.; Quarre, S.; Breman, A.; Dorschner, M.; et al. High-recovery visual identification and single-cell retrieval of circulating tumor cells for genomic analysis using a dual-technology platform integrated with automated immunofluorescence staining. BMC Cancer 2015, 15, 360. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.F.; Wu, L.; Liu, S.P.; Jiang, M.M.; Hu, B.; Zhou, K.Q.; Guo, W.; Xu, Y.; Zhong, Y.; Zhou, X.R.; et al. Dissecting spatial heterogeneity and the immune-evasion mechanism of CTCs by single-cell RNA-seq in hepatocellular carcinoma. Nat. Commun. 2021, 12, 4091. [Google Scholar] [CrossRef]
- Fend, F.; Raffeld, M. Laser capture microdissection in pathology. J. Clin. Pathol. 2000, 53, 666. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Zhao, L.; Shen, Q.; Yu, J.; Ng, C.; Kong, X.; Wu, D.; Song, M.; Shi, X.; Xu, X.; et al. Polymer nanofiber-embedded microchips for detection, isolation, and molecular analysis of single circulating melanoma cells. Angew. Chem. Int. Ed. Engl. 2013, 52, 3379–3383. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Lu, Y.T.; Ho, H.; Li, B.; Chen, J.F.; Lin, M.; Li, F.; Wu, K.; Wu, H.; Lichterman, J.; et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget 2015, 6, 44781–44793. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lu, Y.-T.; Li, F.; Wu, K.; Hou, S.; Yu, J.; Shen, Q.; Wu, D.; Song, M.; OuYang, W.-H.; et al. High-purity prostate circulating tumor cell isolation by a polymer nanofiber-embedded microchip for whole exome sequencing. Adv. Mater. 2013, 25, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.; Lee, D.; Chungwon Lee, A.; Lee, Y.; Bae, H.J.; Lee, H.-B.; Kim, R.N.; Han, W.; Kwon, S. Whole genome sequencing of single circulating tumor cells isolated by applying a pulsed laser to cell-capturing microstructures. Small 2019, 15, 1902607. [Google Scholar] [CrossRef]
- Zhou, W.-M.; Yan, Y.-Y.; Guo, Q.-R.; Ji, H.; Wang, H.; Xu, T.-T.; Makabel, B.; Pilarsky, C.; He, G.; Yu, X.-Y.; et al. Microfluidics applications for high-throughput single cell sequencing. J. Nanobiotechnol. 2021, 19, 312. [Google Scholar] [CrossRef]
- Pauken, C.M.; Kenney, S.R.; Brayer, K.J.; Guo, Y.; Brown-Glaberman, U.A.; Marchetti, D. Heterogeneity of circulating tumor cell neoplastic subpopulations outlined by single-cell transcriptomics. Cancers 2021, 13, 4885. [Google Scholar] [CrossRef]
- D’Avola, D.; Villacorta-Martin, C.; Martins-Filho, S.N.; Craig, A.; Labgaa, I.; von Felden, J.; Kimaada, A.; Bonaccorso, A.; Tabrizian, P.; Hartmann, B.M.; et al. High-density single cell mRNA sequencing to characterize circulating tumor cells in hepatocellular carcinoma. Sci. Rep. 2018, 8, 11570. [Google Scholar] [CrossRef]
- Yamawaki, T.M.; Lu, D.R.; Ellwanger, D.C.; Bhatt, D.; Manzanillo, P.; Arias, V.; Zhou, H.; Yoon, O.K.; Homann, O.; Wang, S.; et al. Systematic comparison of high-throughput single-cell RNA-seq methods for immune cell profiling. BMC Genom. 2021, 22, 66. [Google Scholar] [CrossRef]
- Hu, X.; Zang, X.; Lv, Y. Detection of circulating tumor cells: Advances and critical concerns. Oncol. Lett. 2021, 21, 422. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Chen, Y.C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.T.; Lee, W.; Hao, Z.; Sahoo, S.; Min Kang, H.; et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat. Commun. 2019, 10, 2163. [Google Scholar] [CrossRef]
- Shi, F.; Jia, F.; Wei, Z.; Ma, Y.; Fang, Z.; Zhang, W.; Hu, Z. A microfluidic chip for efficient circulating tumor cells enrichment, screening, and single-cell RNA sequencing. Proteomics 2021, 21, e2000060. [Google Scholar] [CrossRef] [PubMed]
- Hamza, B.; Ng, S.R.; Prakadan, S.M.; Delgado, F.F.; Chin, C.R.; King, E.M.; Yang, L.F.; Davidson, S.M.; DeGouveia, K.L.; Cermak, N.; et al. Optofluidic real-time cell sorter for longitudinal CTC studies in mouse models of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2232–2236. [Google Scholar] [CrossRef]
- Su, M.; Pan, T.; Chen, Q.-Z.; Zhou, W.-W.; Gong, Y.; Xu, G.; Yan, H.-Y.; Li, S.; Shi, Q.-Z.; Zhang, Y.; et al. Data analysis guidelines for single-cell RNA-seq in biomedical studies and clinical applications. Mil. Med. Res. 2022, 9, 68. [Google Scholar] [CrossRef]
- Tian, T.; Wan, J.; Song, Q.; Wei, Z. Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat. Mach. Intell. 2019, 1, 191–198. [Google Scholar] [CrossRef]
- Xiang, R.; Wang, W.; Yang, L.; Wang, S.; Xu, C.; Chen, X. A comparison for dimensionality reduction methods of single-cell RNA-seq data. Front. Genet. 2021, 12, 646936. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Regev, A.; Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 2016, 34, 1145–1160. [Google Scholar] [CrossRef]
- Tsuyuzaki, K.; Sato, H.; Sato, K.; Nikaido, I. Benchmarking principal component analysis for large-scale single-cell RNA-sequencing. Genome Biol. 2020, 21, 9. [Google Scholar] [CrossRef]
- Zhou, B.; Jin, W. Visualization of single cell RNA-seq data using t-SNE in R. Methods Mol. Biol. 2020, 2117, 159–167. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef]
- Hu, H.; Li, Z.; Li, X.; Yu, M.; Pan, X. ScCAEs: Deep clustering of single-cell RNA-seq via convolutional autoencoder embedding and soft K-means. Brief. Bioinform. 2021, 23, bbab321. [Google Scholar] [CrossRef]
- Ding, J.; Condon, A.; Shah, S.P. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 2018, 9, 2002. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Greene, C.S. Parameter tuning is a key part of dimensionality reduction via deep variational autoencoders for single cell RNA transcriptomics. bioRxiv 2018, 385534. [Google Scholar] [CrossRef]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, K.; Lyu, Y.; Pan, H.; Zhang, J.; Stambolian, D.; Susztak, K.; Reilly, M.P.; Hu, G.; Li, M. Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat. Commun. 2020, 11, 2338. [Google Scholar] [CrossRef]
- Jie Zheng, K.W. Emerging deep learning methods for single-cell RNA-seq data analysis. Quant. Biol. 2019, 7, 247–254. [Google Scholar] [CrossRef]
- Poonia, S.; Goel, A.; Chawla, S.; Bhattacharya, N.; Rai, P.; Lee, Y.F.; Yap, Y.S.; West, J.; Bhagat, A.A.; Tayal, J.; et al. Marker-free characterization of single live circulating tumor cell full-length transcriptomes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Lin, J.; Lin, Q.; Wong, K.C. Review of single-cell RNA-seq data clustering for cell-type identification and characterization. RNA 2023, 29, 517–530. [Google Scholar] [CrossRef]
- Das, S.; Rai, A.; Merchant, M.L.; Cave, M.C.; Rai, S.N. A comprehensive survey of statistical approaches for differential expression analysis in single-cell RNA sequencing studies. Genes 2021, 12, 1947. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Vu, T.N.; Wills, Q.F.; Kalari, K.R.; Niu, N.; Wang, L.; Rantalainen, M.; Pawitan, Y. Beta-Poisson model for single-cell RNA-seq data analyses. Bioinformatics 2016, 32, 2128–2135. [Google Scholar] [CrossRef]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.A.; Trapnell, C. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef]
- Miao, Z.; Deng, K.; Wang, X.; Zhang, X. DEsingle for detecting three types of differential expression in single-cell RNA-seq data. Bioinformatics 2018, 34, 3223–3224. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, B.; Nelson, C.E.; Nabavi, S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinform. 2019, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, A.; Baruzzo, G.; Di Camillo, B. Single-cell RNA-sequencing: Assessment of differential expression analysis methods. Front. Genet. 2017, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Robinson, M.D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 2018, 15, 255–261. [Google Scholar] [CrossRef]
- Jaakkola, M.K.; Seyednasrollah, F.; Mehmood, A.; Elo, L.L. Comparison of methods to detect differentially expressed genes between single-cell populations. Brief. Bioinform. 2016, 18, 735–743. [Google Scholar] [CrossRef]
- Mou, T.; Deng, W.; Gu, F.; Pawitan, Y.; Vu, T.N. Reproducibility of methods to detect differentially expressed genes from single-cell RNA sequencing. Front. Genet. 2020, 10, 1331. [Google Scholar] [CrossRef]
- Jeon, H.; Xie, J.; Jeon, Y.; Jung, K.J.; Gupta, A.; Chang, W.; Chung, D. Statistical Power Analysis for Designing Bulk, Single-Cell, and Spatial Transcriptomics Experiments: Review, Tutorial, and Perspectives. Biomolecules 2023, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Vinod-Paul, K.; Gillen, A.E.; Kopin, E.G.; Gibney, K.; Elias, A.D.; Hayashi, M.; Sartorius, C.A.; Kabos, P. Analysis of circulating breast cancer cell heterogeneity and interactions with peripheral blood mononuclear cells. Mol. Carcinog. 2020, 59, 1129–1139. [Google Scholar] [CrossRef]
- Tritschler, S.; Büttner, M.; Fischer, D.S.; Lange, M.; Bergen, V.; Lickert, H.; Theis, F.J. Concepts and limitations for learning developmental trajectories from single cell genomics. Development 2019, 146, dev170506. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, L.; Cannoodt, R.; Saelens, W.; Deplancke, B.; Saeys, Y. Recent advances in trajectory inference from single-cell omics data. Curr. Opin. Syst. Biol. 2021, 27, 100344. [Google Scholar] [CrossRef]
- Van den Berge, K.; Roux de Bézieux, H.; Street, K.; Saelens, W.; Cannoodt, R.; Saeys, Y.; Dudoit, S.; Clement, L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020, 11, 1201. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Liu, S.X.; Gustafson, H.H.; Jackson, D.L.; Pun, S.H.; Trapnell, C. Trajectory analysis quantifies transcriptional plasticity during macrophage polarization. Sci. Rep. 2020, 10, 12273. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; McFaline-Figueroa, J.L.; Ramani, V.; Saunders, L.; Cao, J.; Packer, J.; Pliner, H.A.; Jackson, D.L.; Daza, R.M.; Christiansen, L.; et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 2020, 367, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Setty, M.; Kiseliovas, V.; Levine, J.; Gayoso, A.; Mazutis, L.; Pe’er, D. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 2019, 37, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Risso, D.; Fletcher, R.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Göttgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019, 20, 59. [Google Scholar] [CrossRef]
- Chen, H.; Albergante, L.; Hsu, J.Y.; Lareau, C.A.; Lo Bosco, G.; Guan, J.; Zhou, S.; Gorban, A.N.; Bauer, D.E.; Aryee, M.J.; et al. Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM. Nat. Commun. 2019, 10, 1903. [Google Scholar] [CrossRef]
- Tran, T.N.; Bader, G.D. Tempora: Cell trajectory inference using time-series single-cell RNA sequencing data. PLoS Comput. Biol. 2020, 16, e1008205. [Google Scholar] [CrossRef]
- Fan, J.; Slowikowski, K.; Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 2020, 52, 1452–1465. [Google Scholar] [CrossRef]
- Schissler, A.G.; Li, Q.; Chen, J.L.; Kenost, C.; Achour, I.; Billheimer, D.D.; Li, H.; Piegorsch, W.W.; Lussier, Y.A. Analysis of aggregated cell-cell statistical distances within pathways unveils therapeutic-resistance mechanisms in circulating tumor cells. Bioinformatics 2016, 32, i80–i89. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Enver, T. Single-cell entropy for accurate estimation of differentiation potency from a cell’s transcriptome. Nat. Commun. 2017, 8, 15599. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Mavrommati, I.; Johnson, F.; Echeverria, G.V.; Natrajan, R. Subclonal heterogeneity and evolution in breast cancer. npj Breast Cancer 2021, 7, 155. [Google Scholar] [CrossRef]
- Lüönd, F.; Tiede, S.; Christofori, G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Br. J. Cancer. 2021, 125, 164–175. [Google Scholar] [CrossRef]
- Guo, X.; Lin, F.; Yi, C.; Song, J.; Sun, D.; Lin, L.; Zhong, Z.; Wu, Z.; Wang, X.; Zhang, Y.; et al. Deep transfer learning enables lesion tracing of circulating tumor cells. Nat. Commun. 2022, 13, 7687. [Google Scholar] [CrossRef] [PubMed]
- Burr, R.; Gilles, C.; Thompson, E.W.; Maheswaran, S. Epithelial-mesenchymal plasticity in circulating tumor cells, the precursors of metastasis. Adv. Exp. Med. Biol. 2020, 1220, 11–34. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, M.; Battaglin, F.; Jayachandran, P.; Wang, J.; Arai, H.; Soni, S.; Zhang, W.; Hirai, M.; Matsusaka, S.; Lenz, H.J. Clinical significance of circulating tumor cell induced epithelial-mesenchymal transition in patients with metastatic colorectal cancer by single-cell RNA-sequencing. Cancers 2021, 13, 4862. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef]
- Kaigorodova, E.V.; Savelieva, O.E.; Tashireva, L.A.; Tarabanovskaya, N.A.; Simolina, E.I.; Denisov, E.V.; Slonimskaya, E.M.; Choynzonov, E.L.; Perelmuter, V.M. Heterogeneity of circulating tumor cells in neoadjuvant chemotherapy of breast cancer. Molecules 2018, 23, 727. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.; Jain, N.; Rawal, R. Evidence for circulating cancer stem-like cells and epithelial-mesenchymal transition phenotype in the pleurospheres derived from lung adenocarcinoma using liquid biopsy. Tumour Biol. 2017, 39, 1010428317695915. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.A.; Stoupis, G.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol. Cancer Ther. 2019, 18, 437–447. [Google Scholar] [CrossRef]
- Chen, V.L.; Huang, Q.; Harouaka, R.; Du, Y.; Lok, A.S.; Parikh, N.D.; Garmire, L.X.; Wicha, M.S. A dual-filtration system for single-cell sequencing of circulating tumor cells and clusters in HCC. Hepatol. Commun. 2022, 6, 1482–1491. [Google Scholar] [CrossRef]
- Hong, S.P.; Chan, T.E.; Lombardo, Y.; Corleone, G.; Rotmensz, N.; Bravaccini, S.; Rocca, A.; Pruneri, G.; McEwen, K.R.; Coombes, R.C.; et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy. Nat. Commun. 2019, 10, 3840. [Google Scholar] [CrossRef]
- Kwan, T.T.; Bardia, A.; Spring, L.M.; Giobbie-Hurder, A.; Kalinich, M.; Dubash, T.; Sundaresan, T.; Hong, X.; LiCausi, J.A.; Ho, U.; et al. A digital RNA signature of circulating tumor cells predicting early therapeutic response in localized and metastatic breast cancer. Cancer Discov. 2018, 8, 1286–1299. [Google Scholar] [CrossRef]
- Li, X.; Sun, H.; Liu, Q.; Liu, Y.; Hou, Y.; Jin, W. Conjoint analysis of circulating tumor cells and solid tumors for exploring potential prognostic markers and constructing a robust novel predictive signature for breast cancer. Cancer Cell Int. 2021, 21, 708. [Google Scholar] [CrossRef]
- Cann, G.M.; Gulzar, Z.G.; Cooper, S.; Li, R.; Luo, S.; Tat, M.; Stuart, S.; Schroth, G.; Srinivas, S.; Ronaghi, M.; et al. mRNA-Seq of single prostate cancer circulating tumor cells reveals recapitulation of gene expression and pathways found in prostate cancer. PLoS ONE 2012, 7, e49144. [Google Scholar] [CrossRef]
- Ateeq, B.; Tomlins, S.A.; Laxman, B.; Asangani, I.A.; Cao, Q.; Cao, X.; Li, Y.; Wang, X.; Feng, F.Y.; Pienta, K.J.; et al. Therapeutic targeting of SPINK1-positive prostate cancer. Sci. Transl. Med. 2011, 3, 72ra17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Latham, D.E.; Delaney, M.A.; Chakravarti, A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene 2005, 24, 2474–2482. [Google Scholar] [CrossRef]
- Mazzu, Y.Z.; Armenia, J.; Nandakumar, S.; Chakraborty, G.; Yoshikawa, Y.; Jehane, L.E.; Lee, G.M.; Atiq, M.; Khan, N.; Schultz, N.; et al. Ribonucleotide reductase small subunit M2 is a master driver of aggressive prostate cancer. Mol. Oncol. 2020, 14, 1881–1897. [Google Scholar] [CrossRef] [PubMed]
- Mazzu, Y.Z.; Armenia, J.; Chakraborty, G.; Yoshikawa, Y.; Coggins, S.A.A.; Nandakumar, S.; Gerke, T.A.; Pomerantz, M.M.; Qiu, X.; Zhao, H.; et al. A novel mechanism driving poor-prognosis prostate cancer: Overexpression of the DNA repair gene, ribonucleotide reductase small subunit M2 (RRM2). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4480–4492. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Horning, A.M.; Lieberman, B.; Kim, M.; Lin, C.K.; Hung, C.N.; Chou, C.W.; Wang, C.M.; Lin, C.L.; Kirma, N.B.; et al. Spatial EGFR dynamics and metastatic phenotypes modulated by upregulated EphB2 and Src pathways in advanced prostate cancer. Cancers 2019, 11, 1910. [Google Scholar] [CrossRef]
- Shalaby, T.; Achini, F.; Grotzer, M.A. Targeting cerebrospinal fluid for discovery of brain cancer biomarkers. J. Cancer Metastatis Treat. 2016, 2, 176–187. [Google Scholar] [CrossRef]
- Ruan, H.; Zhou, Y.; Shen, J.; Zhai, Y.; Xu, Y.; Pi, L.; Huang, R.; Chen, K.; Li, X.; Ma, W.; et al. Circulating tumor cell characterization of lung cancer brain metastases in the cerebrospinal fluid through single-cell transcriptome analysis. Clin. Transl. Med. 2020, 10, e246. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, Z.; Shi, Q. Liquid biopsy based single-cell transcriptome profiling characterizes heterogeneity of disseminated tumor cells from lung adenocarcinoma. Proteomics 2020, 20, e1900224. [Google Scholar] [CrossRef]
- Ting, D.T.; Wittner, B.S.; Ligorio, M.; Vincent Jordan, N.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. Wnt signaling in tumors: The way to evade drugs and immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.; Chen, D.; Lang, J.E. The current status of the clinical utility of liquid biopsies in cancer. Expert Rev. Mol. Diagn. 2019, 19, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wu, H. The significant prognostic value of circulating tumor cells in colorectal cancer: A systematic review and meta-analysis. Curr. Probl. Cancer 2018, 42, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.L.; Yang, Y.F.; Yuan, C.H.; Chen, H.; Wang, F.B. Circulating tumor cells for predicting the prognostic of patients with hepatocellular carcinoma: A meta analysis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 37, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, M.; Han, S.; Jin, C.; Yang, J. The prognostic role of circulating tumor cells in gastric cancer: A meta-analysis. Front. Oncol. 2022, 12, 963091. [Google Scholar] [CrossRef]
- Zhang, L.; Riethdorf, S.; Wu, G.; Wang, T.; Yang, K.; Peng, G.; Liu, J.; Pantel, K. Meta-analysis of the prognostic value of circulating tumor cells in breast cancer. Clin. Cancer Res. 2012, 18, 5701–5710. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, S.; Adams, J.; De Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-mesenchymal transition (EMT) as a therapeutic target. Cells Tissues Organs 2022, 211, 157–182. [Google Scholar] [CrossRef]
- Sun, Y.-F.; Guo, W.; Xu, Y.; Shi, Y.-H.; Gong, Z.-J.; Ji, Y.; Du, M.; Zhang, X.; Hu, B.; Huang, A.; et al. Circulating tumor cells from different vascular sites exhibit spatial heterogeneity in epithelial and mesenchymal composition and distinct clinical significance in hepatocellular carcinoma. Clin. Cancer Res. 2018, 24, 547–559. [Google Scholar] [CrossRef]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L.; et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef]
- Thaiparambil, J.T.; Bender, L.; Ganesh, T.; Kline, E.; Patel, P.; Liu, Y.; Tighiouart, M.; Vertino, P.M.; Harvey, R.D.; Garcia, A.; et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer 2011, 129, 2744–2755. [Google Scholar] [CrossRef]
- Medrek, C.; Landberg, G.; Andersson, T.; Leandersson, K. Wnt-5a-CKI{alpha} signaling promotes {beta}-catenin/E-cadherin complex formation and intercellular adhesion in human breast epithelial cells. J. Biol. Chem. 2009, 284, 10968–10979. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, L.; He, C.S.; Xu, F.; Liu, J.L.; Hu, Z.H.; Zhao, L.P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef]
- Wang, X.Q.; Zhang, W.; Lui, E.L.; Zhu, Y.; Lu, P.; Yu, X.; Sun, J.; Yang, S.; Poon, R.T.; Fan, S.T. Notch1-Snail1-E-cadherin pathway in metastatic hepatocellular carcinoma. Int. J. Cancer 2012, 131, E163–E172. [Google Scholar] [CrossRef]
- Lei, J.; Ma, J.; Ma, Q.; Li, X.; Liu, H.; Xu, Q.; Duan, W.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol. Cancer 2013, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Amintas, S.; Bedel, A.; Moreau-Gaudry, F.; Boutin, J.; Buscail, L.; Merlio, J.P.; Vendrely, V.; Dabernat, S.; Buscail, E. Circulating tumor cell clusters: United we stand divided we fall. Int. J. Mol. Sci. 2020, 21, 2653. [Google Scholar] [CrossRef] [PubMed]
- Undevia, S.D.; Vogelzang, N.J.; Mauer, A.M.; Janisch, L.; Mani, S.; Ratain, M.J. Phase I clinical trial of CEP-2563 dihydrochloride, a receptor tyrosine kinase inhibitor, in patients with refractory solid tumors. Investig. New Drugs 2004, 22, 449–458. [Google Scholar] [CrossRef]
- Marshall, J.L.; Kindler, H.; Deeken, J.; Bhargava, P.; Vogelzang, N.J.; Rizvi, N.; Luhtala, T.; Boylan, S.; Dordal, M.; Robertson, P.; et al. Phase I trial of orally administered CEP-701, a novel neurotrophin receptor-linked tyrosine kinase inhibitor. Investig. New Drugs 2005, 23, 31–37. [Google Scholar] [CrossRef]
- Collins, C.; Carducci, M.; Eisenberger, M.; Isaacs, J.T.; Partin, A.; Pili, R.; Sinibaldi, V.; Walczak, J.; Denmeade, S. Preclinical and clinical studies with the multi-kinase inhibitor cep-701 as treatment for prostate cancer demonstrate the inadequacy of psa response as a primary endpoint. Cancer Biol. Ther. 2007, 6, 1360–1367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yaguchi, S.; Fukui, Y.; Koshimizu, I.; Yoshimi, H.; Matsuno, T.; Gouda, H.; Hirono, S.; Yamazaki, K.; Yamori, T. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J. Natl. Cancer Inst. 2006, 98, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.I.; Eccles, S.; Clarke, P.A.; Hayes, A.; Nutley, B.; Alix, S.; Henley, A.; Di-Stefano, F.; Ahmad, Z.; Guillard, S.; et al. Pharmacologic characterization of a potent inhibitor of class i phosphatidylinositide 3-kinases. Cancer Res. 2007, 67, 5840–5850. [Google Scholar] [CrossRef]
- Dan, S.; Tamaki, N.; Namatame, N.; Yoshizawa, Y.; Okamura, M.; Nishimura, Y.; Yamazaki, K.; Yaguchi, S.-i. Abstract 3909: Potential antitumor effect of a pan-PI3K inhibitor ZSTK474 on human sarcoma cell lines. Cancer Res. 2019, 79, 3909. [Google Scholar] [CrossRef]
- Lockhart, A.C.; Olszanski, A.J.; Allgren, R.L.; Yaguchi, S.; Cohen, S.J.; Hilton, J.F.; Wang-Gillam, A.; Shapiro, G.I. Abstract B271: A first-in-human Phase I study of ZSTK474, an oral pan-PI3K inhibitor, in patients with advanced solid malignancies. Mol. Cancer Ther. 2013, 12, B271. [Google Scholar] [CrossRef]
- Peralta, M.; Osmani, N.; Goetz, J.G. Circulating tumor cells: Towards mechanical phenotyping of metastasis. iScience 2022, 25, 103969. [Google Scholar] [CrossRef]
- Tzanakakis, G.N.; Agarwal, K.C.; Vezeridis, M.P. Prevention of human pancreatic cancer cell-induced hepatic metastasis in nude mice by dipyridamole and its analog RA-233. Cancer 1993, 71, 2466–2471. [Google Scholar] [CrossRef]
- Trikha, M.; Zhou, Z.; Timar, J.; Raso, E.; Kennel, M.; Emmell, E.; Nakada, M.T. Multiple roles for platelet GPIIb/IIIa and alphavbeta3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Res. 2002, 62, 2824–2833. [Google Scholar]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T cell dysfunction and exhaustion in cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Smit, D.J.; Pantel, K.; Jücker, M. Circulating tumor cells as a promising target for individualized drug susceptibility tests in cancer therapy. Biochem. Pharmacol. 2021, 188, 114589. [Google Scholar] [CrossRef] [PubMed]
- Bleijs, M.; van de Wetering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 2019, 38, e101654. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.L.; Francescangeli, F.; Nicolazzo, C.; Signore, M.; Giuliani, A.; Colace, L.; Boe, A.; Magri, V.; Baiocchi, M.; Ciardi, A.; et al. An organoid model of colorectal circulating tumor cells with stem cell features, hybrid EMT state and distinctive therapy response profile. J. Exp. Clin. Cancer Res. 2022, 41, 86. [Google Scholar] [CrossRef] [PubMed]
- Mout, L.; van Dessel, L.F.; Kraan, J.; de Jong, A.C.; Neves, R.P.L.; Erkens-Schulze, S.; Beaufort, C.M.; Sieuwerts, A.M.; van Riet, J.; Woo, T.L.C.; et al. Generating human prostate cancer organoids from leukapheresis enriched circulating tumour cells. Eur. J. Cancer 2021, 150, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Hung, Y.P.; Chiu, N.C.; Lee, R.C.; Li, C.P.; Chao, Y.; Shyr, Y.M.; Wang, S.E.; Chen, S.C.; Lin, S.H.; et al. Correlation between drug sensitivity profiles of circulating tumour cell-derived organoids and clinical treatment response in patients with pancreatic ductal adenocarcinoma. Eur. J. Cancer 2022, 166, 208–218. [Google Scholar] [CrossRef]
- Lin, K.C.; Ting, L.L.; Chang, C.L.; Lu, L.S.; Lee, H.L.; Hsu, F.C.; Chiou, J.F.; Wang, P.Y.; Burnouf, T.; Ho, D.C.; et al. Ex Vivo Expanded Circulating Tumor Cells for Clinical Anti-Cancer Drug Prediction in Patients with Head and Neck Cancer. Cancer 2021, 13, 6076. [Google Scholar] [CrossRef]
- Burnouf, T.; Chang, C.-Y.; Chen, S.-H.; Chen, Y.-J.; Chen, Y.-H.; Ho, W.-L.; Liao, Y.-M.; Lin, P.-C.; Liu, Y.-L.; Miser, J.S.; et al. Treatment response prediction with circulating tumor cell-derived organoids for soft tissue sarcoma. J. Clin. Oncol. 2023, 41, e23521. [Google Scholar] [CrossRef]
- Rodríguez-Lee, M.; Kolatkar, A.; McCormick, M.; Dago, A.D.; Kendall, J.; Carlsson, N.A.; Bethel, K.; Greenspan, E.J.; Hwang, S.E.; Waitman, K.R.; et al. Effect of blood collection tube type and time to processing on the enumeration and high-content characterization of circulating tumor cells using the high-definition single-cell assay. Arch. Pathol. Lab. Med. 2018, 142, 198–207. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orrapin, S.; Thongkumkoon, P.; Udomruk, S.; Moonmuang, S.; Sutthitthasakul, S.; Yongpitakwattana, P.; Pruksakorn, D.; Chaiyawat, P. Deciphering the Biology of Circulating Tumor Cells through Single-Cell RNA Sequencing: Implications for Precision Medicine in Cancer. Int. J. Mol. Sci. 2023, 24, 12337. https://doi.org/10.3390/ijms241512337
Orrapin S, Thongkumkoon P, Udomruk S, Moonmuang S, Sutthitthasakul S, Yongpitakwattana P, Pruksakorn D, Chaiyawat P. Deciphering the Biology of Circulating Tumor Cells through Single-Cell RNA Sequencing: Implications for Precision Medicine in Cancer. International Journal of Molecular Sciences. 2023; 24(15):12337. https://doi.org/10.3390/ijms241512337
Chicago/Turabian StyleOrrapin, Santhasiri, Patcharawadee Thongkumkoon, Sasimol Udomruk, Sutpirat Moonmuang, Songphon Sutthitthasakul, Petlada Yongpitakwattana, Dumnoensun Pruksakorn, and Parunya Chaiyawat. 2023. "Deciphering the Biology of Circulating Tumor Cells through Single-Cell RNA Sequencing: Implications for Precision Medicine in Cancer" International Journal of Molecular Sciences 24, no. 15: 12337. https://doi.org/10.3390/ijms241512337
APA StyleOrrapin, S., Thongkumkoon, P., Udomruk, S., Moonmuang, S., Sutthitthasakul, S., Yongpitakwattana, P., Pruksakorn, D., & Chaiyawat, P. (2023). Deciphering the Biology of Circulating Tumor Cells through Single-Cell RNA Sequencing: Implications for Precision Medicine in Cancer. International Journal of Molecular Sciences, 24(15), 12337. https://doi.org/10.3390/ijms241512337