
Cholesterol-Depletion-Induced Membrane Repair Carries a Raft Conformer of P-Glycoprotein to the Cell Surface, Indicating Enhanced Cholesterol Trafficking in MDR Cells, Which Makes Them Resistant to Cholesterol Modifications
 ,
,  ,
,  , ,
, ,  ,
,
Abstract
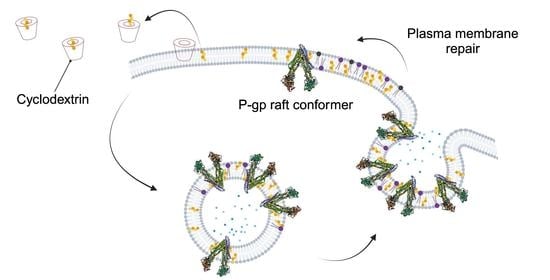
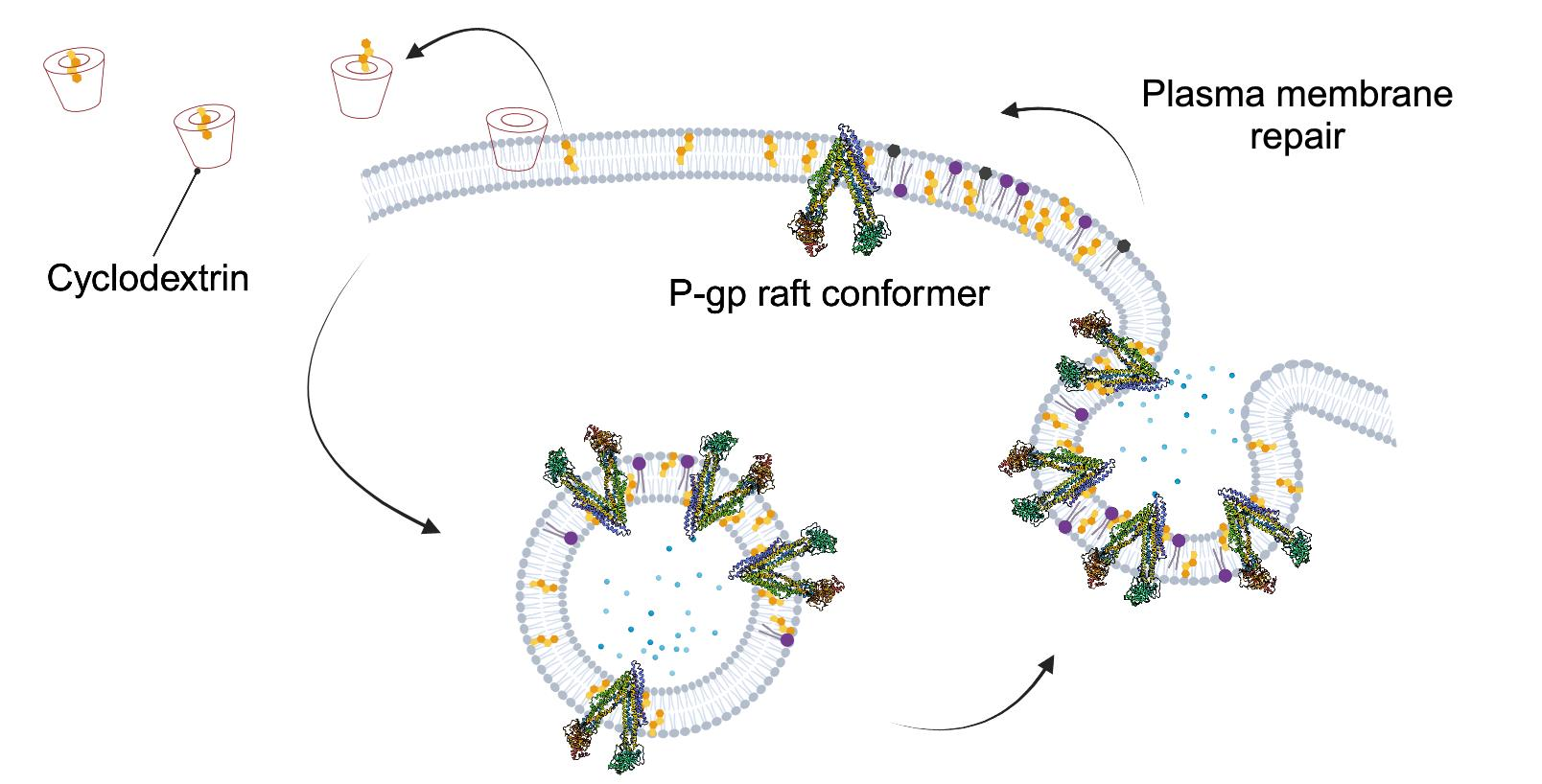{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
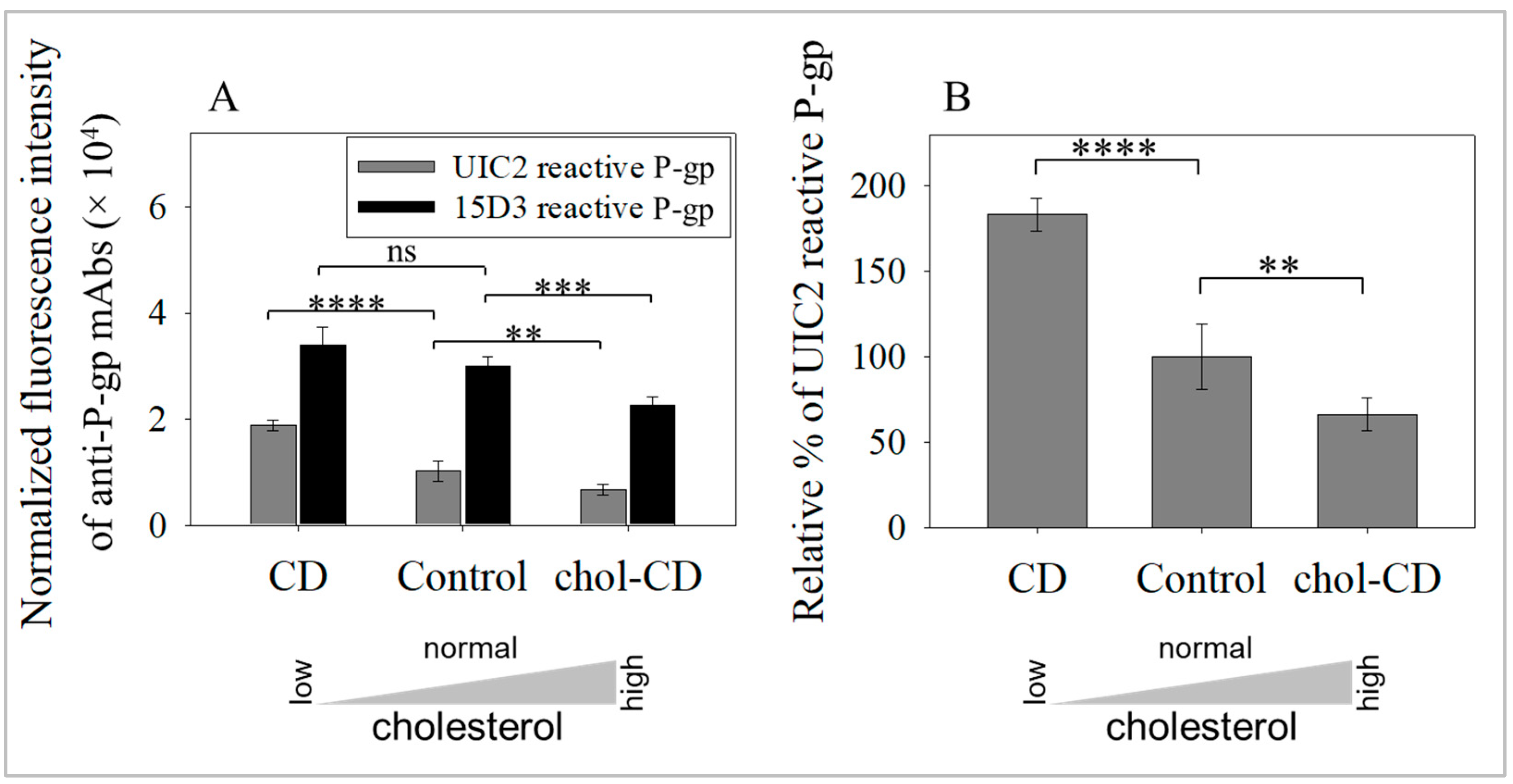
2.1. Cholesterol Extraction Increased the Amount of Raft-Associated, UIC2-Binding Conformer of P-gp in Cell Surface Membranes
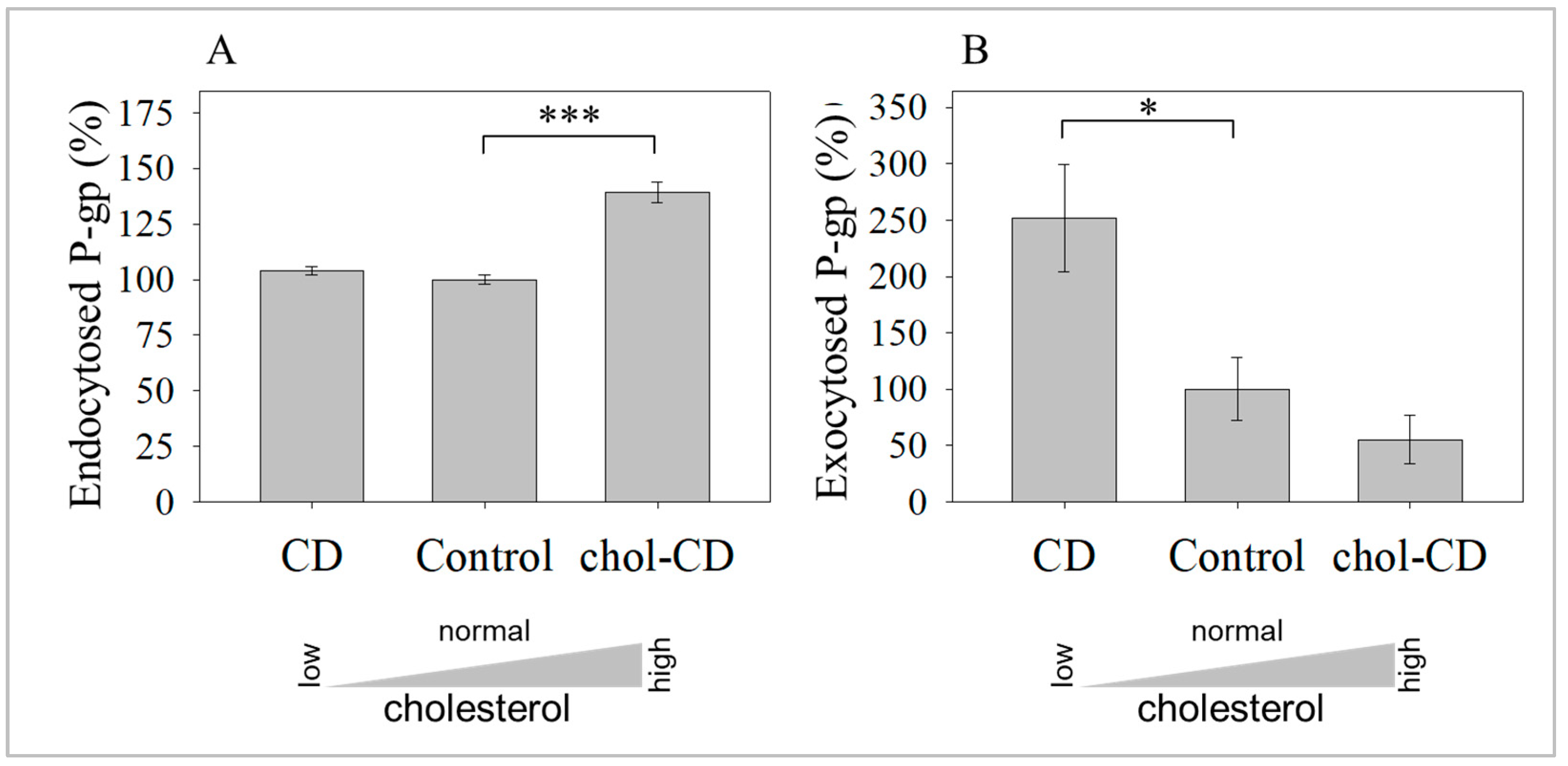
2.2. Cholesterol Depletion-Induced Rise in the UIC2-Binding P-gp Conformer Is the Result of Increased Exocytosis and Not Decreased Endocytosis
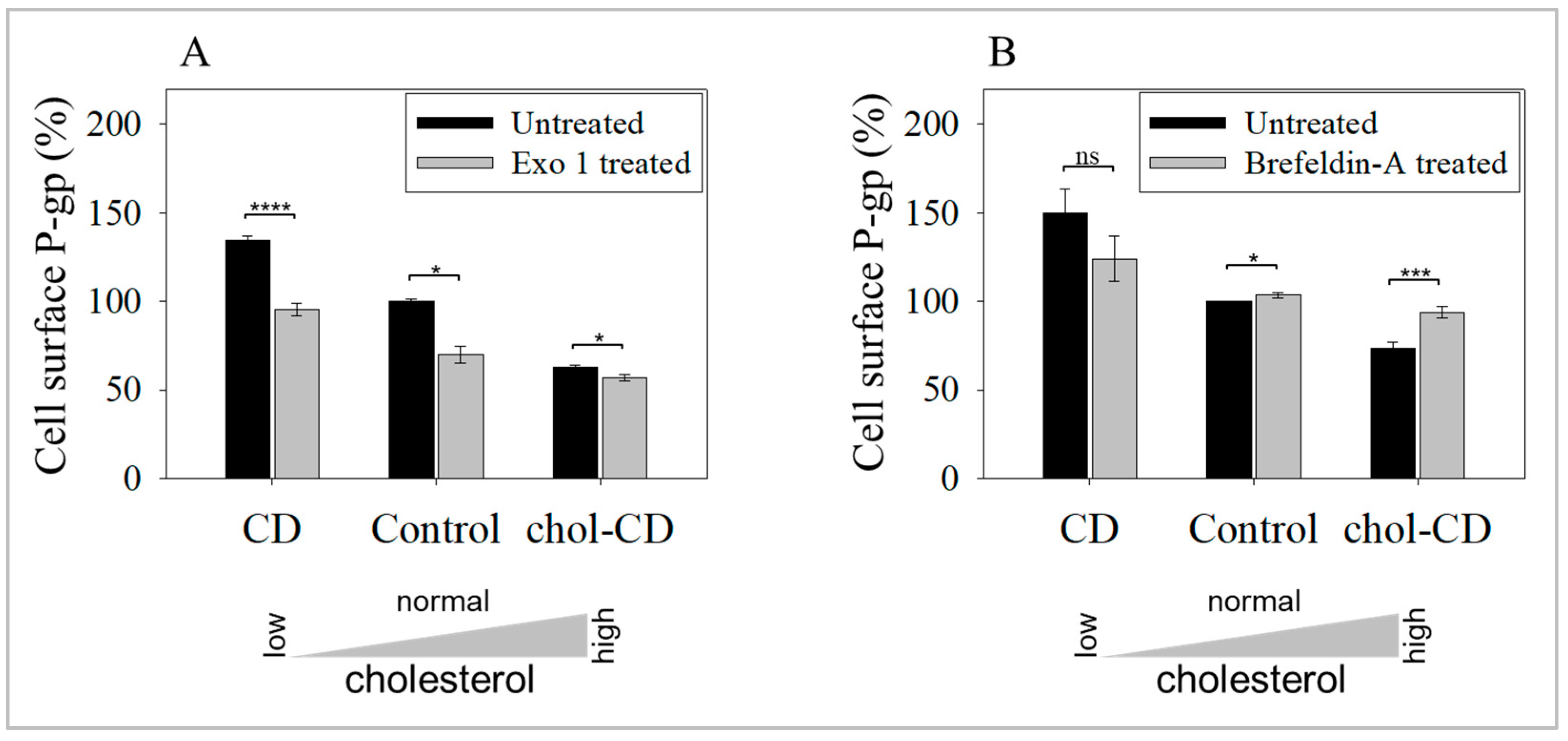
2.3. Inhibition of Exocytosis Prevents the Increase in UIC2-Bound P-gp Caused by Cholesterol Depletion
2.4. UIC2-Binding P-gp Represents the Cholesterol-Dependent Trafficking of Rafts
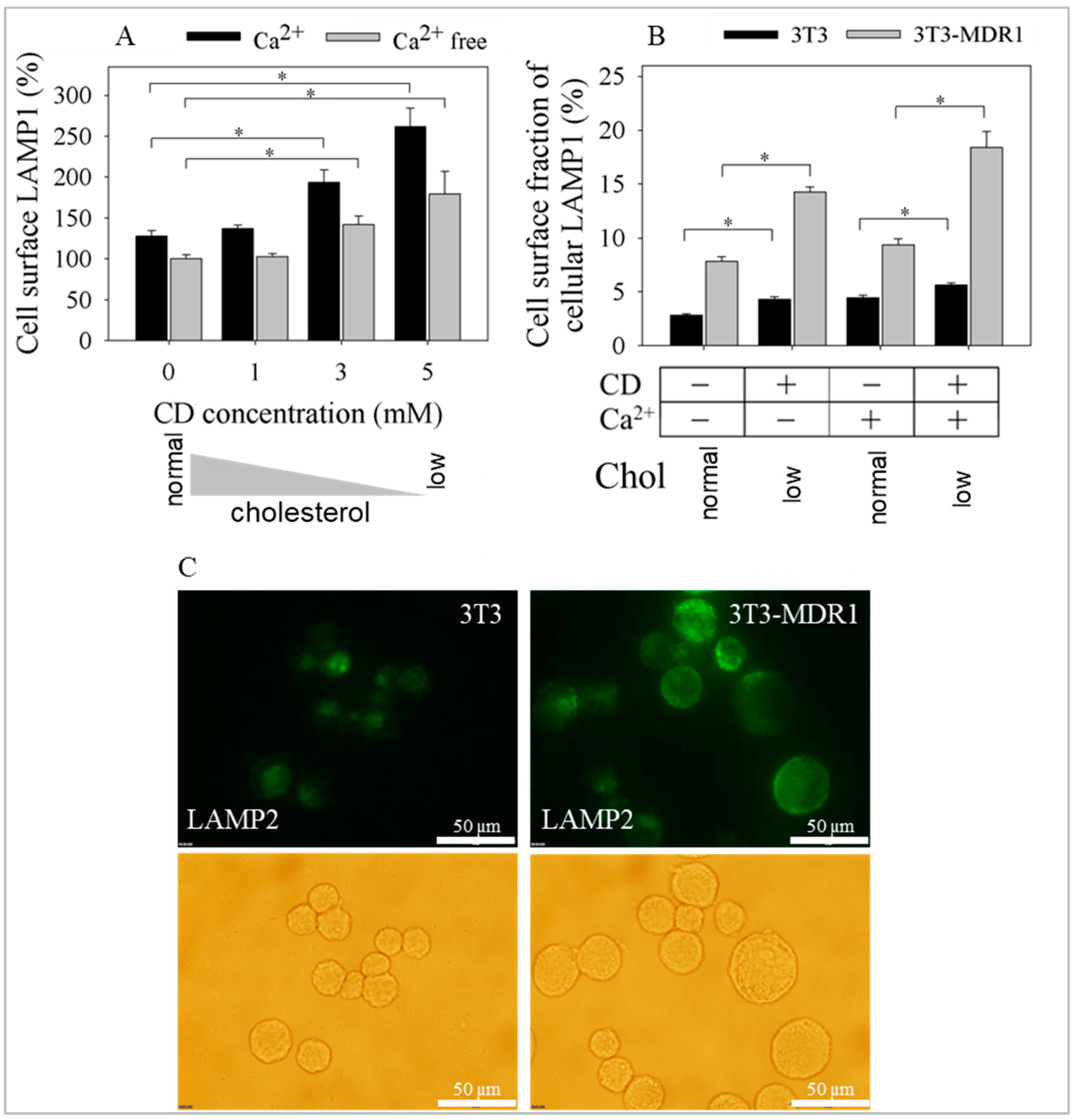
2.5. Exocytosis of LAMP1 and LAMP2 Was Enhanced after Cyclodextrin Treatment in a Calcium and Cholesterol-Dependent Manner
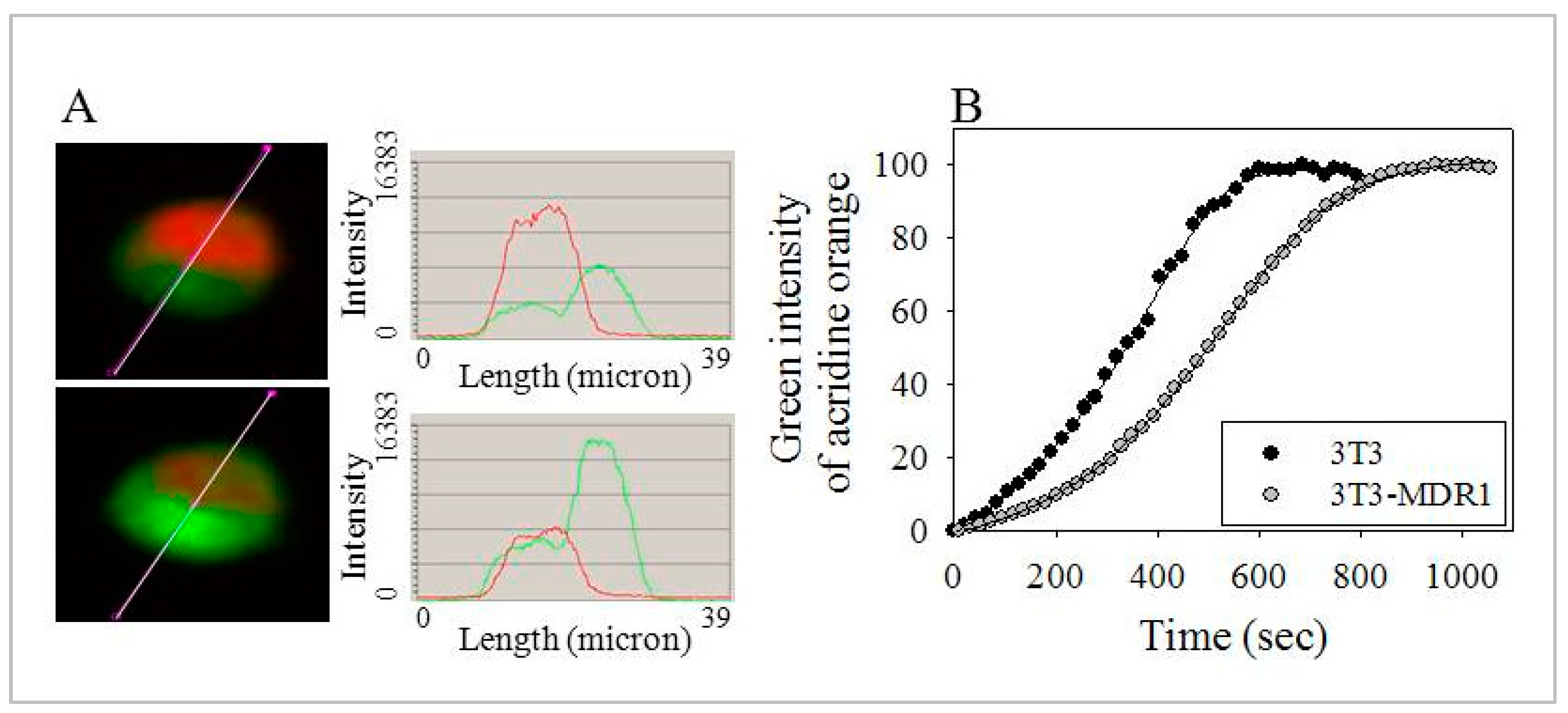
2.6. Enhanced Number and Increased Stability of Lysosomes in 3T3-MDR1 Cells
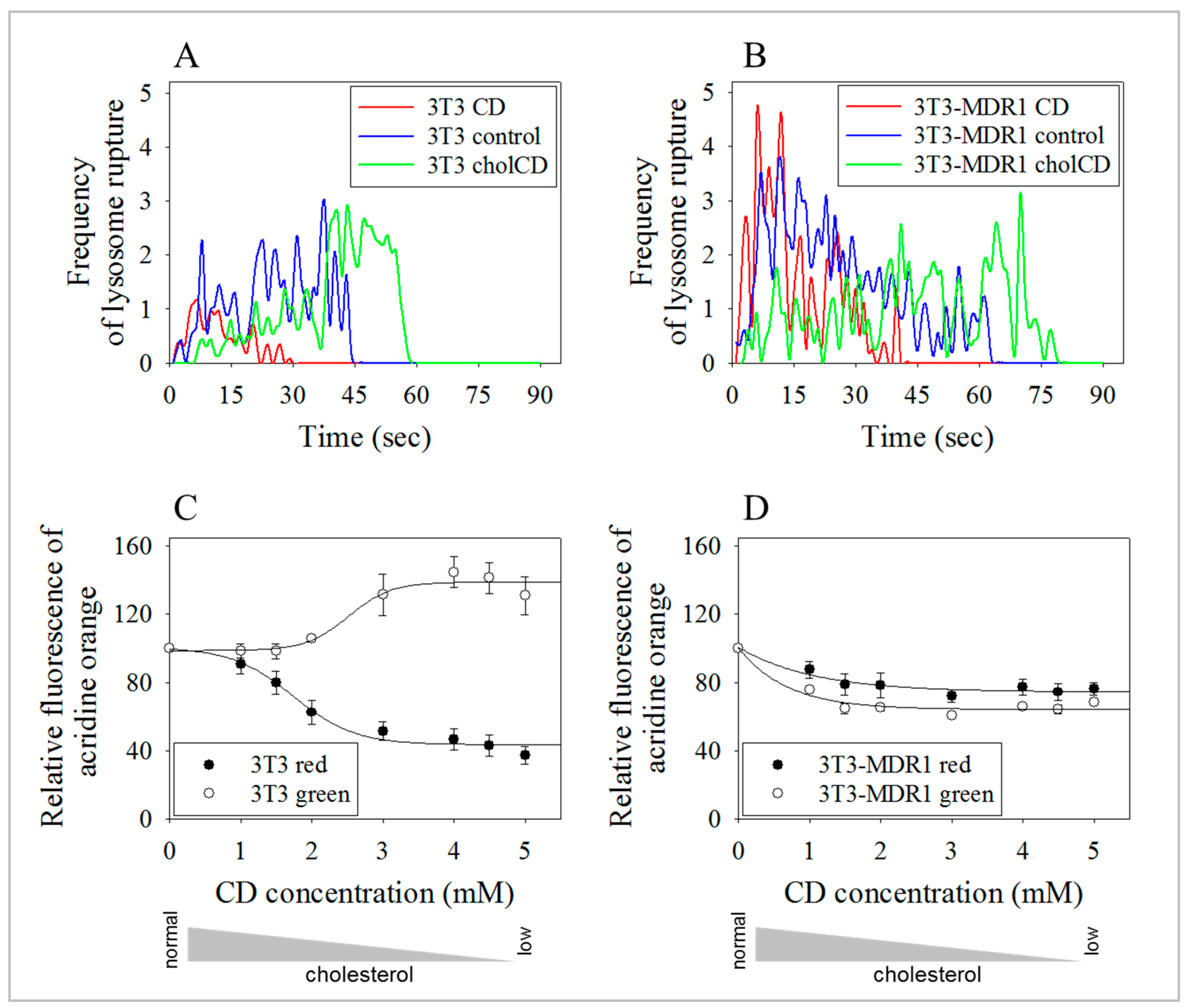
2.7. Lysosomes of 3T3-MDR1 Cells Were More Resistant to Photolysis Than Lysosomes of Parental NIH-3T3 Cells, even after Cholesterol Depletion
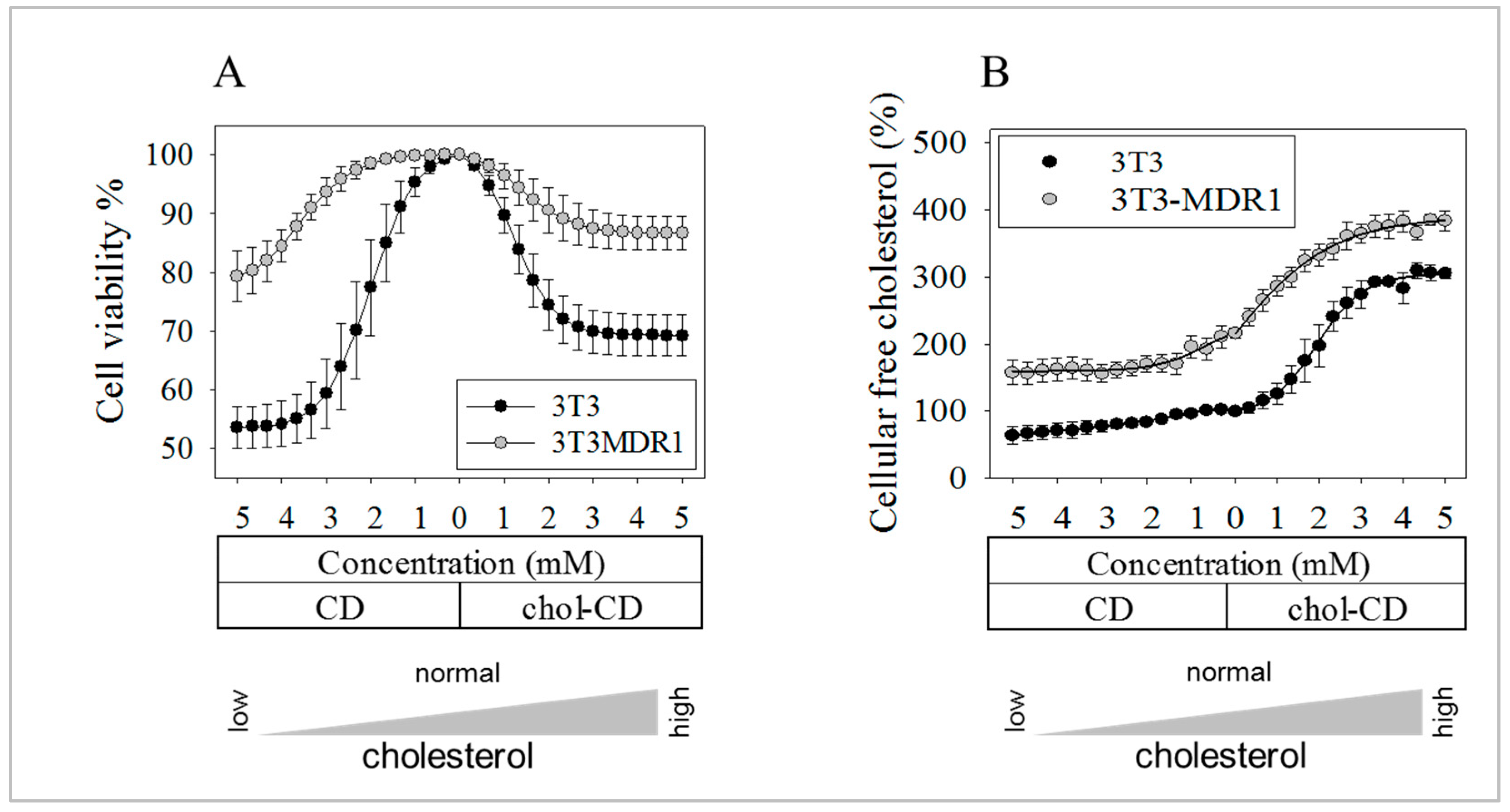
2.8. The More Effective Repair Mechanisms of 3T3-MDR1 Cells Result in Higher Viability after Cholesterol Modulation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals, Antibodies
4.3. Kd and Bmax Values of Antibodies, Cell Membrane Cholesterol Modulations
4.4. Endocytosis Measurements
4.5. Exocytosis of the UIC2 Reactive Fraction
4.6. Inhibition of Exocytosis
4.7. Cell Surface Appearance of LAMP1 and LAMP2 after Cholesterol Extraction
4.8. Measurement of Cellular Cholesterol Levels and Cell Viability of MDR1 Transfected and Non-Transfected Cell Lines
4.9. Determination of Lysosome Content of Cells
4.10. Measurements of Photo-Destruction of Lysosomes
4.11. Analysis of Image Series
4.12. Cholesterol Extraction Effect on Lysosomotropic Dye Accumulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Das, A.; Brown, M.S.; Anderson, D.D.; Goldstein, J.L.; Radhakrishnan, A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 2014, 3, e02882. [Google Scholar] [CrossRef] [PubMed]
- Endapally, S.; Frias, D.; Grzemska, M.; Gay, A.; Tomchick, D.R.; Radhakrishnan, A. Molecular Discrimination between Two Conformations of Sphingomyelin in Plasma Membranes. Cell 2019, 176, 1040–1053.e17. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.; Ralko, A.; Sharma, A. An In Situ Fluorescence Assay for Cholesterol Transporter Activity of the Patched. Methods Mol. Biol. 2022, 2374, 37–47. [Google Scholar]
- Liu, S.L.; Sheng, R.; Jung, J.H.; Wang, L.; Stec, E.; O’Connor, M.J.; Song, S.; Bikkavilli, R.K.; Winn, R.A.; Lee, D.; et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nat. Chem. Biol. 2017, 13, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, F.; Kano, F.; Murata, M.; Kimura, Y.; Kioka, N.; Ueda, K. Changes in the asymmetric distribution of cholesterol in the plasma membrane influence streptolysin O pore formation. Sci. Rep. 2019, 9, 4548. [Google Scholar] [CrossRef]
- Steck, T.L.; Lange, Y. Transverse distribution of plasma membrane bilayer cholesterol: Picking sides. Traffic 2018, 19, 750–760. [Google Scholar] [CrossRef]
- Suzuki, K.G.N.; Kusumi, A. Refinement of Singer-Nicolson fluid-mosaic model by microscopy imaging: Lipid rafts and actin-induced membrane compartmentalization. Biochim. Biophys. Acta Biomembr. 2023, 1865, 184093. [Google Scholar] [CrossRef]
- Sheng, R.; Kim, H.; Lee, H.; Xin, Y.; Chen, Y.; Tian, W.; Cui, Y.; Choi, J.C.; Doh, J.; Han, J.K.; et al. Cholesterol selectively activates canonical Wnt signalling over non-canonical Wnt signalling. Nat. Commun. 2014, 5, 4393. [Google Scholar] [CrossRef]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175, 1352–1364.e14. [Google Scholar] [CrossRef]
- Naito, T.; Ercan, B.; Krshnan, L.; Triebl, A.; Koh, D.H.Z.; Wei, F.Y.; Tomizawa, K.; Torta, F.T.; Wenk, M.R.; Saheki, Y. Movement of accessible plasma membrane cholesterol by the GRAMD1 lipid transfer protein complex. eLife 2019, 8, e51401. [Google Scholar] [CrossRef]
- Boucher, P.; Matz, R.L.; Terrand, J. Atherosclerosis: Gone with the Wnt? Atherosclerosis 2020, 301, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Gautier, E.L.; Ganda, A.; Molusky, M.M.; Wang, W.; Fotakis, P.; Wang, N.; Randolph, G.J.; D’Agati, V.D.; Yvan-Charvet, L.; et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab. 2017, 25, 1294–1304.e6. [Google Scholar] [CrossRef] [PubMed]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009. [Google Scholar] [CrossRef]
- Goda, K.; Bacso, Z.; Szabo, G. Multidrug resistance through the spectacle of P-glycoprotein. Curr. Cancer Drug Targets 2009, 9, 281–297. [Google Scholar] [CrossRef]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol. Ther. 2000, 85, 217–229. [Google Scholar] [CrossRef]
- Guo, H.; Dong, J.; Hu, S.; Cai, X.; Tang, G.; Dou, J.; Tian, M.; He, F.; Nie, Y.; Fan, D. Biased random walk model for the prioritization of drug resistance associated proteins. Sci. Rep. 2015, 5, 10857. [Google Scholar] [CrossRef] [PubMed]
- May, G.L.; Wright, L.C.; Dyne, M.; Mackinnon, W.B.; Fox, R.M.; Mountford, C.E. Plasma membrane lipid composition of vinblastine sensitive and resistant human leukaemic lymphoblasts. Int. J. Cancer. 1988, 42, 728–733. [Google Scholar] [CrossRef]
- Pallares-Trujillo, J.; Domenech, C.; Grau-Oliete, M.R.; Rivera-Fillat, M.P. Role of cell cholesterol in modulating vincristine uptake and resistance. Int. J. Cancer 1993, 55, 667–671. [Google Scholar] [CrossRef]
- Escriba, P.V.; Ferrer-Montiel, A.V.; Ferragut, J.A.; Gonzalez-Ros, J.M. Role of membrane lipids in the interaction of daunomycin with plasma membranes from tumor cells: Implications in drug-resistance phenomena. Biochemistry 1990, 29, 7275–7282. [Google Scholar] [CrossRef]
- Ramu, A.; Glaubiger, D.; Weintraub, H. Differences in lipid composition of doxorubicin-sensitive and -resistant P388 cells. Cancer Treat. Rep. 1984, 68, 637–641. [Google Scholar] [PubMed]
- Lavie, Y.; Cao, H.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar] [CrossRef] [PubMed]
- Lucci, A.; Cho, W.I.; Han, T.Y.; Giuliano, A.E.; Morton, D.L.; Cabot, M.C. Glucosylceramide: A marker for multiple-drug resistant cancers. Anticancer Res. 1998, 18, 475–480. [Google Scholar]
- Morjani, H.; Aouali, N.; Belhoussine, R.; Veldman, R.J.; Levade, T.; Manfait, M. Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets. Int. J. Cancer 2001, 94, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Lavie, Y.; Fiucci, G.; Liscovitch, M. Up-regulation of caveolae and caveolar constituents in multidrug-resistant cancer cells. J. Biol. Chem. 1998, 273, 32380–32383. [Google Scholar] [CrossRef]
- Belanger, M.M.; Gaudreau, M.; Roussel, E.; Couet, J. Role of caveolin-1 in etoposide resistance development in A549 lung cancer cells. Cancer Biol. Ther. 2004, 3, 954–959. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hehlgans, S.; Cordes, N. Caveolin-1: An essential modulator of cancer cell radio-and chemoresistance. Am. J. Cancer Res. 2011, 1, 521–530. [Google Scholar]
- Yang, C.P.; Galbiati, F.; Volonte, D.; Horwitz, S.B.; Lisanti, M.P. Upregulation of caveolin-1 and caveolae organelles in Taxol-resistant A549 cells. FEBS Lett. 1998, 439, 368–372. [Google Scholar] [CrossRef]
- Orlowski, S.; Martin, S.; Escargueil, A. P-glycoprotein and ‘lipid rafts’: Some ambiguous mutual relationships (floating on them, building them or meeting them by chance?). Cell. Mol. Life Sci. CMLS 2006, 63, 1038–1059. [Google Scholar] [CrossRef]
- Fiucci, G.; Czarny, M.; Lavie, Y.; Zhao, D.; Berse, B.; Blusztajn, J.K.; Liscovitch, M. Changes in phospholipase D isoform activity and expression in multidrug-resistant human cancer cells. Int. J. Cancer 2000, 85, 882–888. [Google Scholar] [CrossRef]
- Yi, J.S.; Mun, D.G.; Lee, H.; Park, J.S.; Lee, J.W.; Lee, J.S.; Kim, S.J.; Cho, B.R.; Lee, S.W.; Ko, Y.G. PTRF/cavin-1 is essential for multidrug resistance in cancer cells. J. Proteome Res. 2013, 12, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef]
- George, K.S.; Wu, S. Lipid raft: A floating island of death or survival. Toxicol. Appl. Pharmacol. 2012, 259, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Hummel, I.; Klappe, K.; Kok, J.W. Up-regulation of lactosylceramide synthase in MDR1 overexpressing human liver tumour cells. FEBS Lett. 2005, 579, 3381–3384. [Google Scholar] [CrossRef]
- Lucci, A.; Giuliano, A.E.; Han, T.Y.; Dinur, T.; Liu, Y.Y.; Senchenkov, A.; Cabot, M.C. Ceramide toxicity and metabolism differ in wild-type and multidrug-resistant cancer cells. Int. J. Oncol. 1999, 15, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J. Biol. Chem. 1999, 274, 1140–1146. [Google Scholar] [CrossRef]
- Dimitroulakos, J.; Yeger, H. HMG-CoA reductase mediates the biological effects of retinoic acid on human neuroblastoma cells: Lovastatin specifically targets P-glycoprotein-expressing cells. Nat. Med. 1996, 2, 326–333. [Google Scholar] [CrossRef]
- Raggers, R.J.; Pomorski, T.; Holthuis, J.C.; Kalin, N.; van Meer, G. Lipid traffic: The ABC of transbilayer movement. Traffic 2000, 1, 226–234. [Google Scholar] [CrossRef]
- Aye, I.L.; Singh, A.T.; Keelan, J.A. Transport of lipids by ABC proteins: Interactions and implications for cellular toxicity, viability and function. Chem.-Biol. Interact. 2009, 180, 327–339. [Google Scholar] [CrossRef]
- Romsicki, Y.; Sharom, F.J. Phospholipid flippase activity of the reconstituted P-glycoprotein multidrug transporter. Biochemistry 2001, 40, 6937–6947. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem. J. 2005, 389 Pt 2, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.; Sharom, F.J. Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: Effects on ATPase activity, drug binding and transport. Biochemistry 2008, 47, 13686–13698. [Google Scholar] [CrossRef] [PubMed]
- van Helvoort, A.; Smith, A.J.; Sprong, H.; Fritzsche, I.; Schinkel, A.H.; Borst, P.; van Meer, G. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 1996, 87, 507–517. [Google Scholar] [CrossRef] [PubMed]
- van Helvoort, A.; Giudici, M.L.; Thielemans, M.; van Meer, G. Transport of sphingomyelin to the cell surface is inhibited by brefeldin A and in mitosis, where C6-NBD-sphingomyelin is translocated across the plasma membrane by a multidrug transporter activity. J. Cell Sci. 1997, 110 Pt 1, 75–83. [Google Scholar] [CrossRef]
- Garrigues, A.; Escargueil, A.E.; Orlowski, S. The multidrug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. Proc. Natl. Acad. Sci. USA 2002, 99, 10347–10352. [Google Scholar] [CrossRef]
- Kapoor, K.; Pant, S.; Tajkhorshid, E. Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein. Chem. Sci. 2021, 12, 6293–6306. [Google Scholar] [CrossRef]
- Kimura, Y.; Kioka, N.; Kato, H.; Matsuo, M.; Ueda, K. Modulation of drug-stimulated ATPase activity of human MDR1/P-glycoprotein by cholesterol. Biochem. J. 2007, 401, 597–605. [Google Scholar] [CrossRef]
- Ghetie, M.A.; Marches, R.; Kufert, S.; Vitetta, E.S. An anti-CD19 antibody inhibits the interaction between P-glycoprotein (P-gp) and CD19, causes P-gp to translocate out of lipid rafts, and chemosensitizes a multidrug-resistant (MDR) lymphoma cell line. Blood 2004, 104, 178–183. [Google Scholar] [CrossRef]
- Kamau, S.W.; Kramer, S.D.; Gunthert, M.; Wunderli-Allenspach, H. Effect of the modulation of the membrane lipid composition on the localization and function of P-glycoprotein in MDR1-MDCK cells. Vitr. Cell. Dev. Biol. Anim. 2005, 41, 207–216. [Google Scholar] [CrossRef][Green Version]
- Bacso, Z.; Nagy, H.; Goda, K.; Bene, L.; Fenyvesi, F.; Matko, J.; Szabo, G. Raft and cytoskeleton associations of an ABC transporter: P-glycoprotein. Cytom. Part A J. Int. Soc. Anal. Cytol. 2004, 61, 105–116. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid rafts: Heterogeneity on the high seas. Biochem. J. 2004, 378 Pt 2, 281–292. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Luker, G.D.; Pica, C.M.; Kumar, A.S.; Covey, D.F.; Piwnica-Worms, D. Effects of cholesterol and enantiomeric cholesterol on P-glycoprotein localization and function in low-density membrane domains. Biochemistry 2000, 39, 8692. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yunomae, K.; Arima, H.; Hirayama, F.; Uekama, K. Involvement of cholesterol in the inhibitory effect of dimethyl-beta-cyclodextrin on P-glycoprotein and MRP2 function in Caco-2 cells. FEBS Lett. 2003, 536, 225–231. [Google Scholar] [CrossRef]
- Arima, H.; Yunomae, K.; Morikawa, T.; Hirayama, F.; Uekama, K. Contribution of cholesterol and phospholipids to inhibitory effect of dimethyl-beta-cyclodextrin on efflux function of P-glycoprotein and multidrug resistance-associated protein 2 in vinblastine-resistant Caco-2 cell monolayers. Pharm. Res. 2004, 21, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Reungpatthanaphong, P.; Marbeuf-Gueye, C.; Le Moyec, L.; Salerno, M.; Garnier-Suillerot, A. Decrease of P-glycoprotein activity in K562/ADR cells by MbetaCD and filipin and lack of effect induced by cholesterol oxidase indicate that this transporter is not located in rafts. J. Bioenerg. Biomembr. 2004, 36, 533–543. [Google Scholar] [CrossRef]
- Meyer dos Santos, S.; Weber, C.C.; Franke, C.; Muller, W.E.; Eckert, G.P. Cholesterol: Coupling between membrane microenvironment and ABC transporter activity. Biochem. Biophys. Res. Commun. 2007, 354, 216–221. [Google Scholar]
- Kimura, Y.; Kodan, A.; Matsuo, M.; Ueda, K. Cholesterol fill-in model: Mechanism for substrate recognition by ABC proteins. J. Bioenerg. Biomembr. 2007, 39, 447–452. [Google Scholar] [CrossRef]
- Fenyvesi, F.; Fenyvesi, E.; Szente, L.; Goda, K.; Bacso, Z.; Bacskay, I.; Varadi, J.; Kiss, T.; Molnar, E.; Janaky, T.; et al. P-glycoprotein inhibition by membrane cholesterol modulation. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2008, 34, 236–242. [Google Scholar] [CrossRef]
- Barsony, O.; Szaloki, G.; Turk, D.; Tarapcsak, S.; Gutay-Toth, Z.; Bacso, Z.; Holb, I.J.; Szekvolgyi, L.; Szabo, G.; Csanady, L.; et al. A single active catalytic site is sufficient to promote transport in P-glycoprotein. Sci. Rep. 2016, 6, 24810. [Google Scholar] [CrossRef]
- Vahedi, S.; Lusvarghi, S.; Pluchino, K.; Shafrir, Y.; Durell, S.R.; Gottesman, M.M.; Ambudkar, S.V. Mapping discontinuous epitopes for MRK-16, UIC2 and 4E3 antibodies to extracellular loops 1 and 4 of human P-glycoprotein. Sci. Rep. 2018, 8, 12716. [Google Scholar] [CrossRef] [PubMed]
- Bacso, Z.; Goda, K.; Nagy, H.; Szabó, Z.; Fenyvesi, F.; Birkó, Z.; Janáky, T.; Bíró, S.; Szabó, G. Rafting Till Freedom (Poster #2073). In Proceedings of the ASCB 47th Annual Meeting, Washington, DC, USA, 1–5 December 2007; Available online: http://www.ascb.org/files/Past-AM-Meetings/2007_Abstracts.pdf (accessed on 1 January 2014).
- Gutayné Tóth, Z.; Bársony, O.; Goda, K.; Szabó, G.; Bacso, Z. Conformational dependent trafficking of P-glycoprotein with rafts (Abstracts of the 8th EBSA European Biophysics Congress (European Biophysical Societies Association). August 23-27, 2011. Budapest, Hungary). Eur. Biophys. J. 2011, 40 (Suppl. S1), 35–256. [Google Scholar]
- Gutay-Toth, Z.; Fenyvesi, F.; Barsony, O.; Szente, L.; Goda, K.; Szabo, G.; Bacso, Z. Cholesterol-dependent conformational changes of P-glycoprotein are detected by the 15D3 monoclonal antibody. Biochim. Biophys. Acta 2016, 1861, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef]
- Chen, F.W.; Li, C.; Ioannou, Y.A. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS ONE 2010, 5, e15054. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Kapoor, K.; Ohnuma, S.; Patel, A.; Swaim, W.; Ambudkar, I.S.; Ambudkar, S.V. Revealing the fate of cell surface human P-glycoprotein (ABCB1): The lysosomal degradation pathway. Biochim. Biophys. Acta 2015, 1853 Pt A, 2361–2370. [Google Scholar] [CrossRef]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef]
- Sehested, M.; Skovsgaard, T.; van Deurs, B.; Winther-Nielsen, H. Increased plasma membrane traffic in daunorubicin resistant P388 leukaemic cells. Effect of daunorubicin and verapamil. Br. J. Cancer 1987, 56, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Jardillier, J.C.; Ordentlich, P. Secretion of lysosomal enzymes by drug-sensitive and multiple drug-resistant cells. Cancer Res. 1991, 51, 1996–2001. [Google Scholar]
- Kilsdonk, E.P.; Yancey, P.G.; Stoudt, G.W.; Bangerter, F.W.; Johnson, W.J.; Phillips, M.C.; Rothblat, G.H. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 1995, 270, 17250–17256. [Google Scholar] [CrossRef]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yu, S.; Lasell, T.K.; Jadhav, A.P.; Macia, E.; Chardin, P.; Melancon, P.; Roth, M.; Mitchison, T.; Kirchhausen, T. Exo1: A new chemical inhibitor of the exocytic pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 6469–6474. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Phair, R.D. Lipids and cholesterol as regulators of traffic in the endomembrane system. Annu. Rev. Biophys. 2010, 39, 559–578. [Google Scholar] [CrossRef] [PubMed]
- Mishev, K.; Dejonghe, W.; Russinova, E. Small molecules for dissecting endomembrane trafficking: A cross-systems view. Chem. Biol. 2013, 20, 475–486. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Finazzi, D.; Klausner, R.D. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 1992, 360, 350–352. [Google Scholar] [CrossRef]
- Breslow, D.K.; Weissman, J.S. Membranes in balance: Mechanisms of sphingolipid homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Sohn, J.; Lin, H.; Fritch, M.R.; Tuan, R.S. Influence of cholesterol/caveolin-1/caveolae homeostasis on membrane properties and substrate adhesion characteristics of adult human mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 86. [Google Scholar] [CrossRef]
- Radulovic, M.; Wenzel, E.M.; Gilani, S.; Holland, L.K.; Lystad, A.H.; Phuyal, S.; Olkkonen, V.M.; Brech, A.; Jaattela, M.; Maeda, K.; et al. Cholesterol transfer via endoplasmic reticulum contacts mediates lysosome damage repair. EMBO J. 2022, 41, e112677. [Google Scholar] [CrossRef]
- Toops, K.A.; Lakkaraju, A. Let’s play a game of chutes and ladders: Lysosome fusion with the epithelial plasma membrane. Commun. Integr. Biol. 2013, 6, e24474. [Google Scholar] [CrossRef][Green Version]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Lane, D.J.; Jansson, P.J.; Richardson, D.R. Glucose Modulation Induces Lysosome Formation and Increases Lysosomotropic Drug Sequestration via the P-Glycoprotein Drug Transporter. J. Biol. Chem. 2016, 291, 3796–3820. [Google Scholar] [CrossRef]
- Warren, L.; Malarska, A.; Jardillier, J.C. The structure of P-glycoprotein and the secretion of lysosomal enzymes in multidrug-resistant cells. Cancer Chemother. Pharmacol. 1995, 35, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zaguilan, R.; Raghunand, N.; Lynch, R.M.; Bellamy, W.; Martinez, G.M.; Rojas, B.; Smith, D.; Dalton, W.S.; Gillies, R.J. pH and drug resistance. I. Functional expression of plasmalemmal V-type H+-ATPase in drug-resistant human breast carcinoma cell lines. Biochem. Pharmacol. 1999, 57, 1037–1046. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Zdolsek, J.M.; Olsson, G.M.; Brunk, U.T. Photooxidative damage to lysosomes of cultured macrophages by acridine orange. Photochem. Photobiol. 1990, 51, 67–76. [Google Scholar] [CrossRef]
- Kagedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359 Pt 2, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kagedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis Int. J. Program. Cell Death 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, F.; Mego, J.L.; Dang, J.; Simon, C. Thyroid lysosomes: The stability of the lysosomal membrane. Eur. J. Cell Biol. 1983, 30, 272–278. [Google Scholar]
- Sydor, M.J.; Kendall, R.L.; Holian, A. Cholesterol content regulates silica-induced lysosomal membrane permeability. Front. Toxicol. 2023, 5, 1112822. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, H.; Sandin, L.; Bjornstrom, K.; Saftig, P.; Garner, B.; Ollinger, K.; Kagedal, K. Sensitivity to lysosome-dependent cell death is directly regulated by lysosomal cholesterol content. PLoS ONE 2012, 7, e50262. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A. Non-vesicular sterol transport in cells. Prog. Lipid Res. 2007, 46, 297–314. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. How cells handle cholesterol. Science 2000, 290, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Heino, S.; Lusa, S.; Somerharju, P.; Ehnholm, C.; Olkkonen, V.M.; Ikonen, E. Dissecting the role of the golgi complex and lipid rafts in biosynthetic transport of cholesterol to the cell surface. Proc. Natl. Acad. Sci. USA 2000, 97, 8375–8380. [Google Scholar] [CrossRef]
- Keller, P.; Simons, K. Post-Golgi biosynthetic trafficking. J. Cell Sci. 1997, 110 Pt 24, 3001–3009. [Google Scholar] [CrossRef]
- Jaipuria, G.; Leonov, A.; Giller, K.; Vasa, S.K.; Jaremko, L.; Jaremko, M.; Linser, R.; Becker, S.; Zweckstetter, M. Cholesterol-mediated allosteric regulation of the mitochondrial translocator protein structure. Nat. Commun. 2017, 8, 14893. [Google Scholar] [CrossRef]
- Slimane, T.A.; Trugnan, G.; Van, I.S.C.; Hoekstra, D. Raft-mediated trafficking of apical resident proteins occurs in both direct and transcytotic pathways in polarized hepatic cells: Role of distinct lipid microdomains. Mol. Biol. Cell 2003, 14, 611–624. [Google Scholar] [CrossRef]
- Sharma, D.K.; Brown, J.C.; Choudhury, A.; Peterson, T.E.; Holicky, E.; Marks, D.L.; Simari, R.; Parton, R.G.; Pagano, R.E. Selective stimulation of caveolar endocytosis by glycosphingolipids and cholesterol. Mol. Biol. Cell 2004, 15, 3114–3122. [Google Scholar] [CrossRef]
- Hubert, M.; Larsson, E.; Vegesna, N.V.G.; Ahnlund, M.; Johansson, A.I.; Moodie, L.W.; Lundmark, R. Lipid accumulation controls the balance between surface connection and scission of caveolae. eLife 2020, 9, e55038. [Google Scholar] [CrossRef]
- Ying, M.; Grimmer, S.; Iversen, T.G.; Van Deurs, B.; Sandvig, K. Cholesterol loading induces a block in the exit of VSVG from the TGN. Traffic 2003, 4, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Clouser, A.F.; Alam, Y.H.; Atkins, W.M. Cholesterol Asymmetrically Modulates the Conformational Ensemble of the Nucleotide-Binding Domains of P-Glycoprotein in Lipid Nanodiscs. Biochemistry 2021, 60, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. P-glycoprotein ATPase activity requires lipids to activate a switch at the first transmission interface. Biochem. Biophys. Res. Commun. 2016, 472, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; Kapoor, K.; Tajkhorshid, E. Probing cholesterol binding and translocation in P-glycoprotein. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183090. [Google Scholar] [CrossRef] [PubMed]
- Jeannesson, E.; Siest, G.; Bastien, B.; Albertini, L.; Aslanidis, C.; Schmitz, G.; Visvikis-Siest, S. Association of ABCB1 gene polymorphisms with plasma lipid and apolipoprotein concentrations in the STANISLAS cohort. Clin. Chim. Acta Int. J. Clin. Chem. 2009, 403, 198–202. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Dong, X.P.; Wang, X.; Shen, D.; Chen, S.; Liu, M.; Wang, Y.; Mills, E.; Cheng, X.; Delling, M.; Xu, H. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J. Biol. Chem. 2009, 284, 32040–32052. [Google Scholar] [CrossRef]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.G.; et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 2013, 26, 511–524. [Google Scholar] [CrossRef]
- Cao, Q.; Yang, Y.; Zhong, X.Z.; Dong, X.P. The lysosomal Ca2+ release channel TRPML1 regulates lysosome size by activating calmodulin. J. Biol. Chem. 2017, 292, 8424–8435. [Google Scholar] [CrossRef]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sorensen, T.; Rafn, B.; Bottzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jaattela, M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Gray, J.; Priestman, D.A.; Wallom, K.L.; Atkins, J.; Olsen, O.D.; Klein, A.; Drndarski, S.; Petersen, N.H.; Ingemann, L.; et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 2016, 8, 355ra118. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Yin, F.; Qi, S.; Shen, L.; Xu, Y.; Luo, L.; Lan, L.; Yin, Z. Parthenolide reverses doxorubicin resistance in human lung carcinoma A549 cells by attenuating NF-kappaB activation and HSP70 up-regulation. Toxicol. Lett. 2013, 221, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.M.; Seebacher, N.A.; Arzuman, L.; Kovacevic, Z.; Lane, D.J.; Richardson, V.; Merlot, A.M.; Lok, H.; Kalinowski, D.S.; Sahni, S.; et al. Lysosomal membrane stability plays a major role in the cytotoxic activity of the anti-proliferative agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT). Biochim. Biophys. Acta 2016, 1863 Pt A, 1665–1681. [Google Scholar] [CrossRef]
- Hu, P.; Li, H.; Sun, W.; Wang, H.; Yu, X.; Qing, Y.; Wang, Z.; Zhu, M.; Xu, J.; Guo, Q.; et al. Cholesterol-associated lysosomal disorder triggers cell death of hematological malignancy: Dynamic analysis on cytotoxic effects of LW-218. Acta Pharm. Sin. B 2021, 11, 3178–3192. [Google Scholar] [CrossRef]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126 Pt 9, 1905–1912. [Google Scholar] [CrossRef]
- Broker, L.E.; Huisman, C.; Span, S.W.; Rodriguez, J.A.; Kruyt, F.A.; Giaccone, G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004, 64, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Groth-Pedersen, L.; Ostenfeld, M.S.; Hoyer-Hansen, M.; Nylandsted, J.; Jaattela, M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 2007, 67, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Groth-Pedersen, L.; Aits, S.; Corcelle-Termeau, E.; Petersen, N.H.; Nylandsted, J.; Jaattela, M. Identification of cytoskeleton-associated proteins essential for lysosomal stability and survival of human cancer cells. PLoS ONE 2012, 7, e45381. [Google Scholar] [CrossRef] [PubMed]
- Ba, Q.; Raghavan, G.; Kiselyov, K.; Yang, G. Whole-Cell Scale Dynamic Organization of Lysosomes Revealed by Spatial Statistical Analysis. Cell Rep. 2018, 23, 3591–3606. [Google Scholar] [CrossRef] [PubMed]
- Lange, Y.; Ye, J.; Steck, T.L. Circulation of cholesterol between lysosomes and the plasma membrane. J. Biol. Chem. 1998, 273, 18915–18922. [Google Scholar] [CrossRef] [PubMed]
- Tessner, T.G.; Stenson, W.F. Overexpression of MDR1 in an intestinal cell line results in increased cholesterol uptake from micelles. Biochem. Biophys. Res. Commun. 2000, 267, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Luker, G.D.; Nilsson, K.R.; Covey, D.F.; Piwnica-Worms, D. Multidrug resistance (MDR1) P-glycoprotein enhances esterification of plasma membrane cholesterol. J. Biol. Chem. 1999, 274, 6979–6991. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, G.; Corsetto, P.A.; Campia, I.; Montorfano, G.; Kopecka, J.; Castella, B.; Gazzano, E.; Ghigo, D.; Rizzo, A.M.; Riganti, C. Omega 3 fatty acids chemosensitize multidrug resistant colon cancer cells by down-regulating cholesterol synthesis and altering detergent resistant membranes composition. Mol. Cancer 2013, 12, 137. [Google Scholar] [CrossRef]
- Lange, Y.; Steck, T.L. Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog. Lipid Res. 2008, 47, 319–332. [Google Scholar] [CrossRef]
- Bruggemann, E.P.; Currier, S.J.; Gottesman, M.M.; Pastan, I. Characterization of the azidopine and vinblastine binding site of P-glycoprotein. J. Biol. Chem. 1992, 267, 21020–21026. [Google Scholar] [CrossRef]
- Kioka, N.; Tsubota, J.; Kakehi, Y.; Komano, T.; Gottesman, M.M.; Pastan, I.; Ueda, K. P-glycoprotein gene (MDR1) cDNA from human adrenal: Normal P-glycoprotein carries Gly185 with an altered pattern of multidrug resistance. Biochem. Biophys. Res. Commun. 1989, 162, 224–231. [Google Scholar] [CrossRef]
- Mechetner, E.B.; Roninson, I.B. Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc. Natl. Acad. Sci. USA 1992, 89, 5824–5828. [Google Scholar] [CrossRef]
- Shi, T.; Wrin, J.; Reeder, J.; Liu, D.; Ring, D.B. High-affinity monoclonal antibodies against P-glycoprotein. Clin. Immunol. Immunopathol. 1995, 76 Pt 1, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.G.; Lefebvre, R.; Bodnar, D.; Legrand, C.; Szentesi, P.; Vincze, J.; Poulard, K.; Bertrand-Michel, J.; Csernoch, L.; Buj-Bello, A.; et al. Phosphoinositide substrates of myotubularin affect voltage-activated Ca(2)(+) release in skeletal muscle. Pflug. Arch. Eur. J. Physiol. 2014, 466, 973–985. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutay-Tóth, Z.; Gellen, G.; Doan, M.; Eliason, J.F.; Vincze, J.; Szente, L.; Fenyvesi, F.; Goda, K.; Vecsernyés, M.; Szabó, G.; et al. Cholesterol-Depletion-Induced Membrane Repair Carries a Raft Conformer of P-Glycoprotein to the Cell Surface, Indicating Enhanced Cholesterol Trafficking in MDR Cells, Which Makes Them Resistant to Cholesterol Modifications. Int. J. Mol. Sci. 2023, 24, 12335. https://doi.org/10.3390/ijms241512335
Gutay-Tóth Z, Gellen G, Doan M, Eliason JF, Vincze J, Szente L, Fenyvesi F, Goda K, Vecsernyés M, Szabó G, et al. Cholesterol-Depletion-Induced Membrane Repair Carries a Raft Conformer of P-Glycoprotein to the Cell Surface, Indicating Enhanced Cholesterol Trafficking in MDR Cells, Which Makes Them Resistant to Cholesterol Modifications. International Journal of Molecular Sciences. 2023; 24(15):12335. https://doi.org/10.3390/ijms241512335
Chicago/Turabian StyleGutay-Tóth, Zsuzsanna, Gabriella Gellen, Minh Doan, James F. Eliason, János Vincze, Lajos Szente, Ferenc Fenyvesi, Katalin Goda, Miklós Vecsernyés, Gábor Szabó, and et al. 2023. "Cholesterol-Depletion-Induced Membrane Repair Carries a Raft Conformer of P-Glycoprotein to the Cell Surface, Indicating Enhanced Cholesterol Trafficking in MDR Cells, Which Makes Them Resistant to Cholesterol Modifications" International Journal of Molecular Sciences 24, no. 15: 12335. https://doi.org/10.3390/ijms241512335
APA StyleGutay-Tóth, Z., Gellen, G., Doan, M., Eliason, J. F., Vincze, J., Szente, L., Fenyvesi, F., Goda, K., Vecsernyés, M., Szabó, G., & Bacso, Z. (2023). Cholesterol-Depletion-Induced Membrane Repair Carries a Raft Conformer of P-Glycoprotein to the Cell Surface, Indicating Enhanced Cholesterol Trafficking in MDR Cells, Which Makes Them Resistant to Cholesterol Modifications. International Journal of Molecular Sciences, 24(15), 12335. https://doi.org/10.3390/ijms241512335