

Contemporary Antiretroviral Therapy Dysregulates Iron Transport and Augments Mitochondrial Dysfunction in HIV-Infected Human Microglia and Neural-Lineage Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
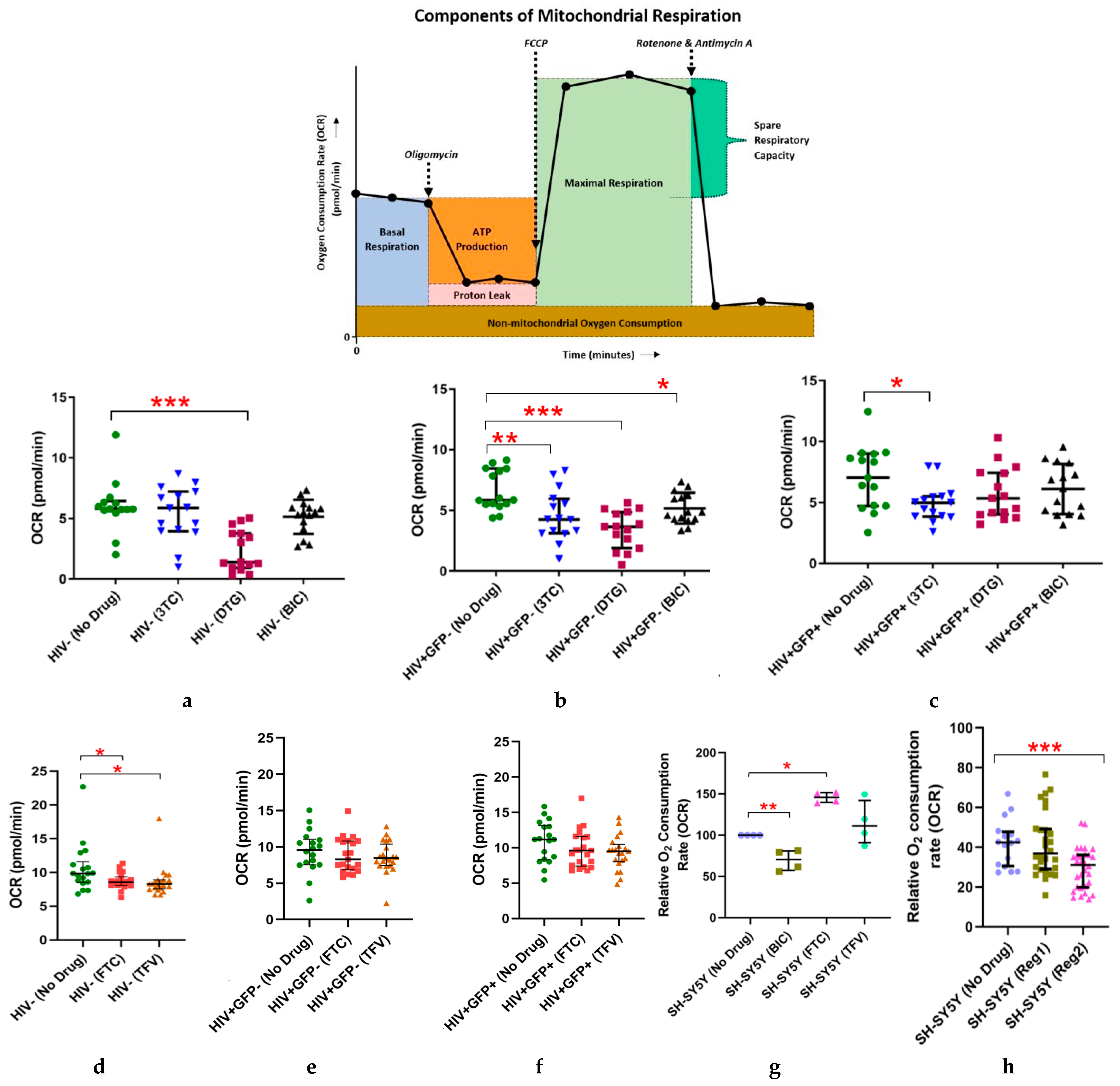
2.1. Effects of Individual ARVs or cART on Basal Oxygen Consumption Rate (OCR) in Uninfected (C20) and C20/HIV+ Human Microglia (Hμglia) and in Neural-Lineage SH-SY5Y Cells
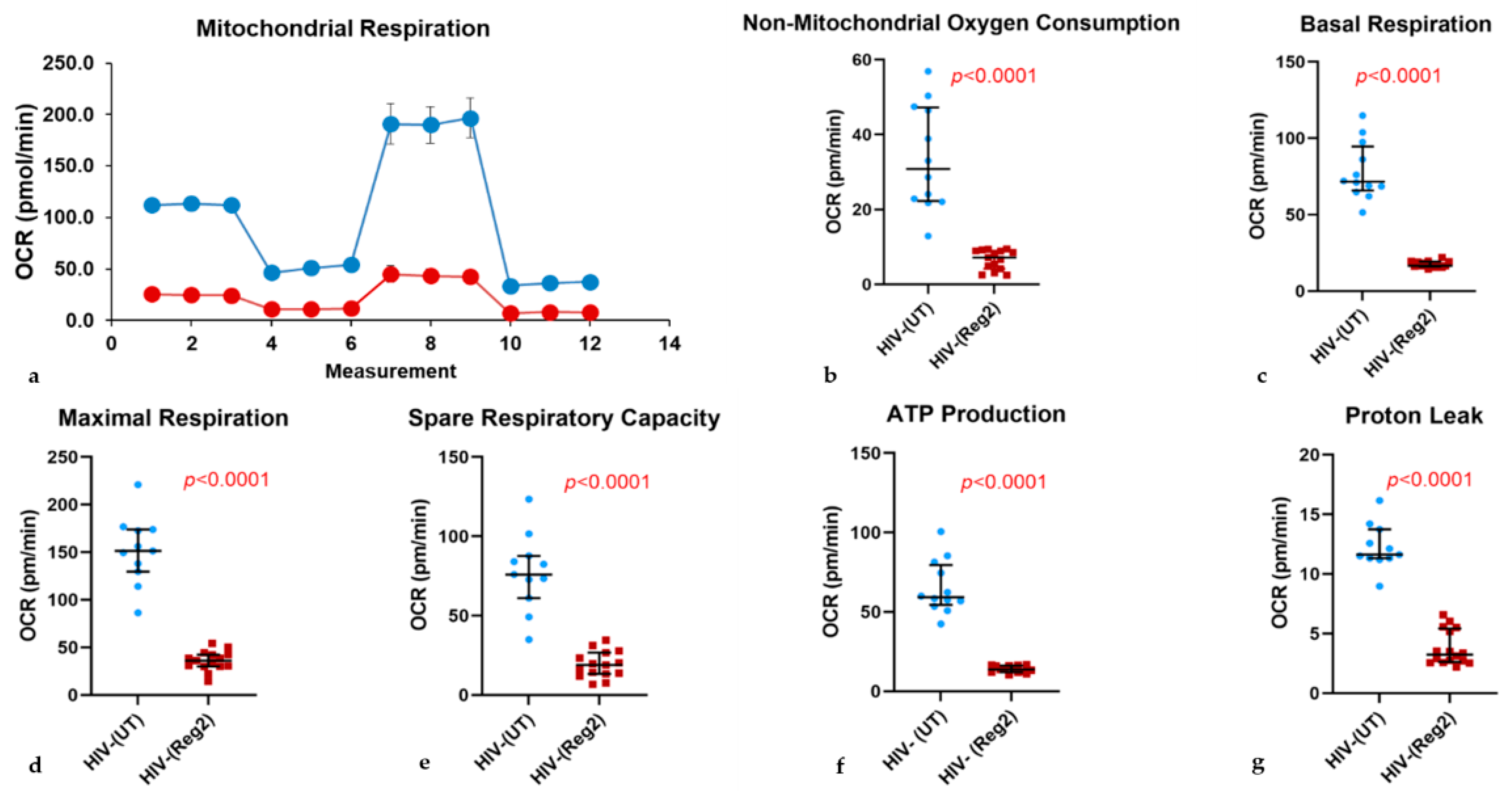
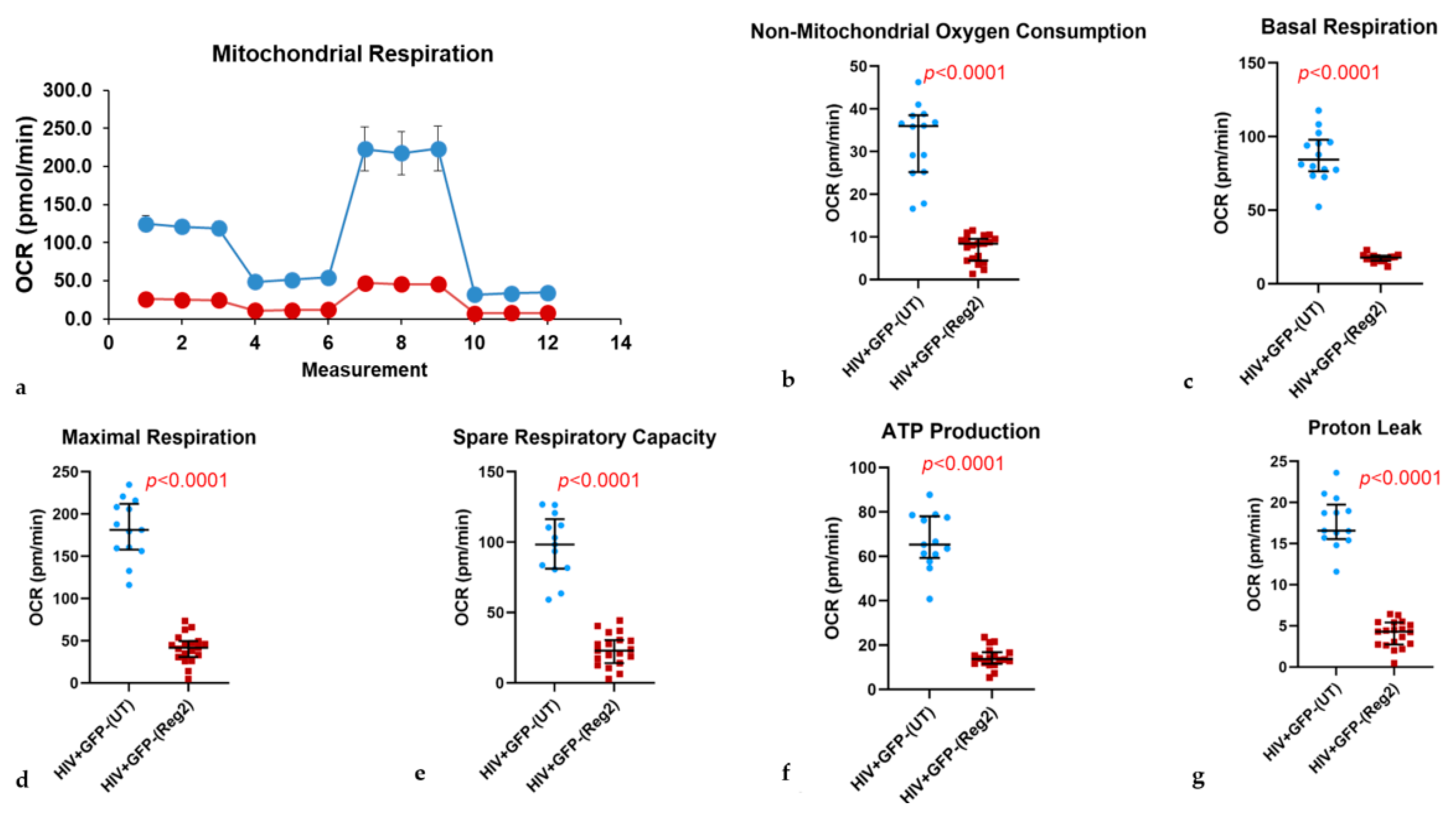
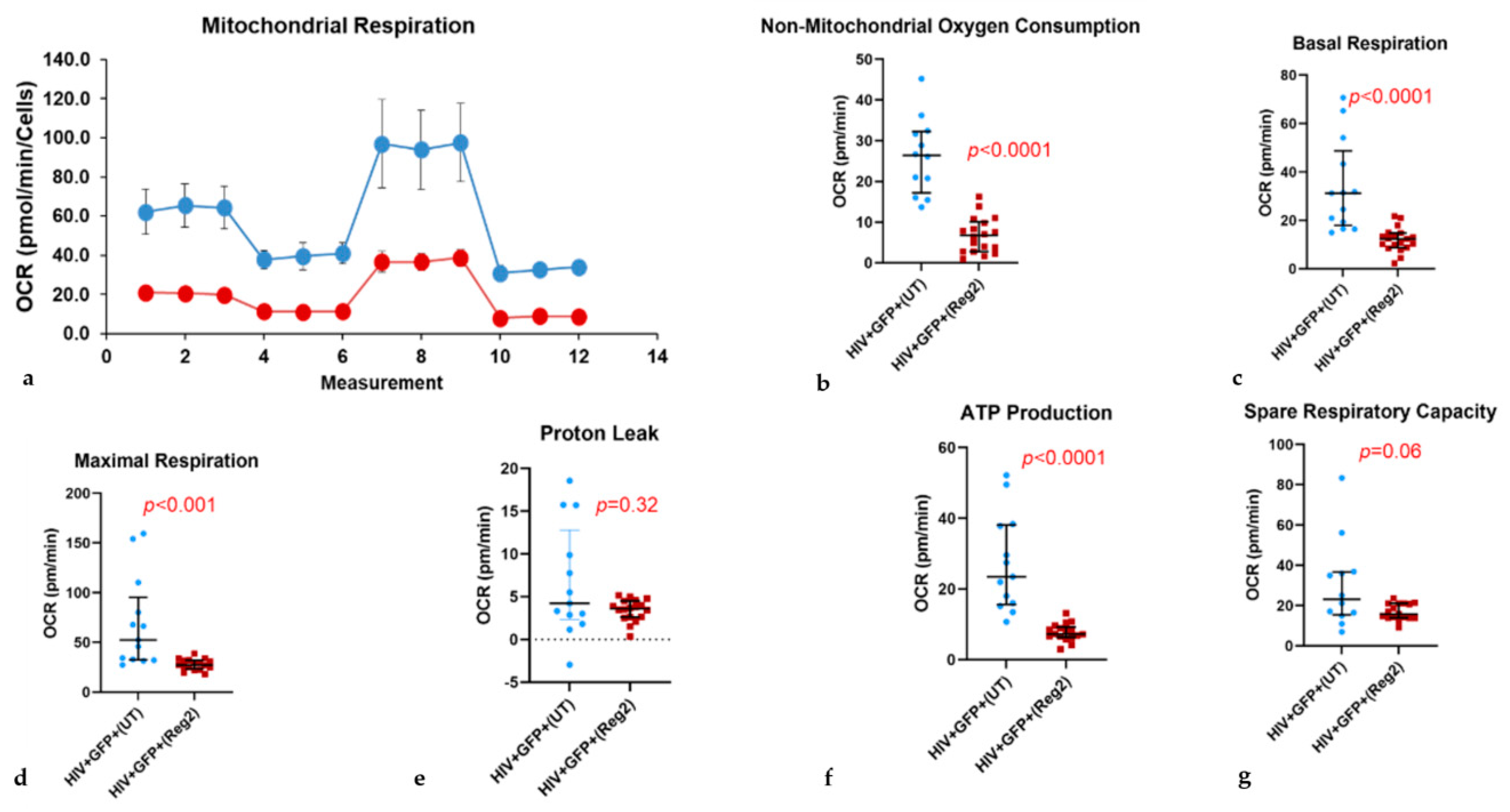
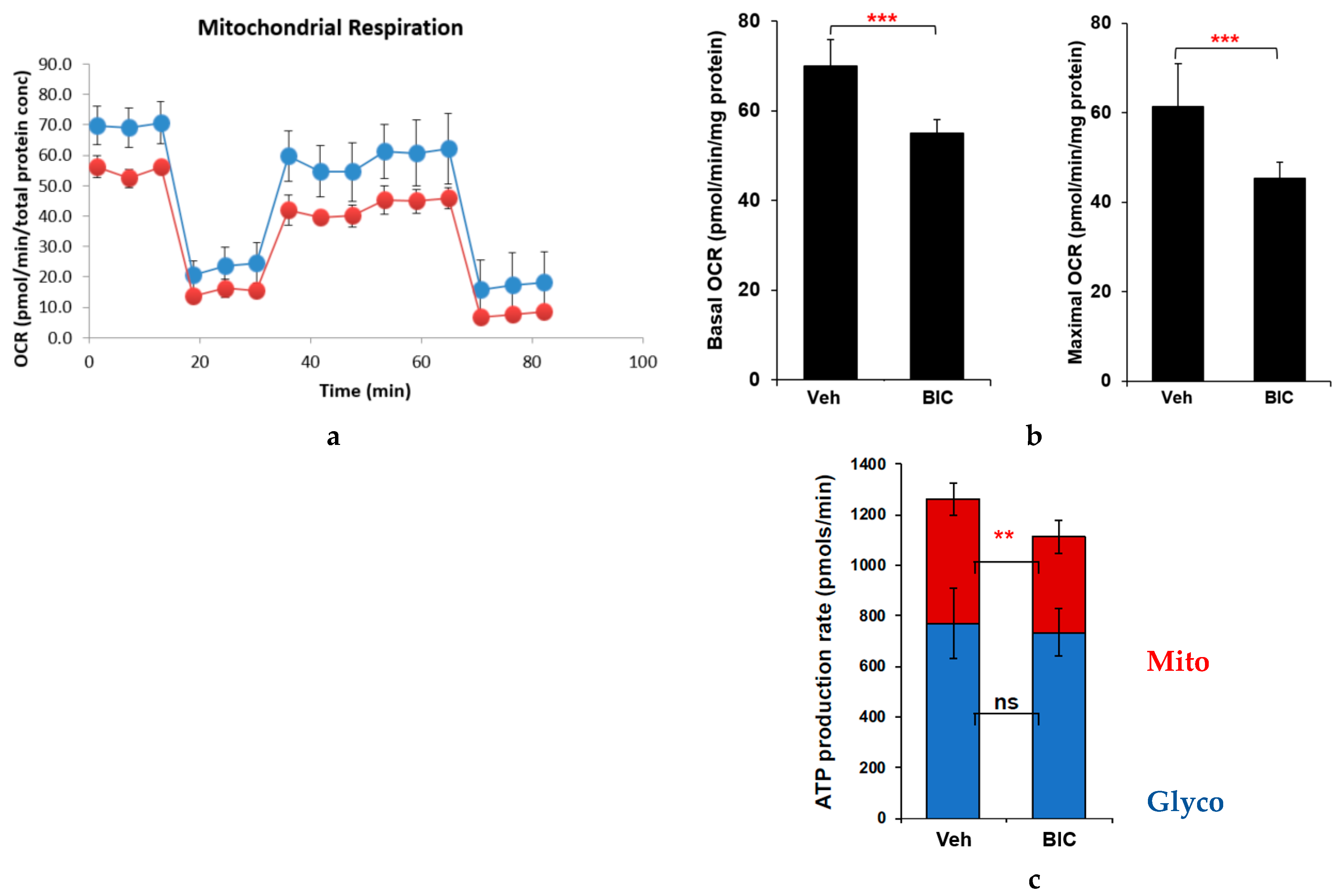
2.2. Effects of Commonly Used ARVs on Mitochondrial Bioenergetics in Uninfected and HIV+ Hµglia and SH-SY5Y Cells
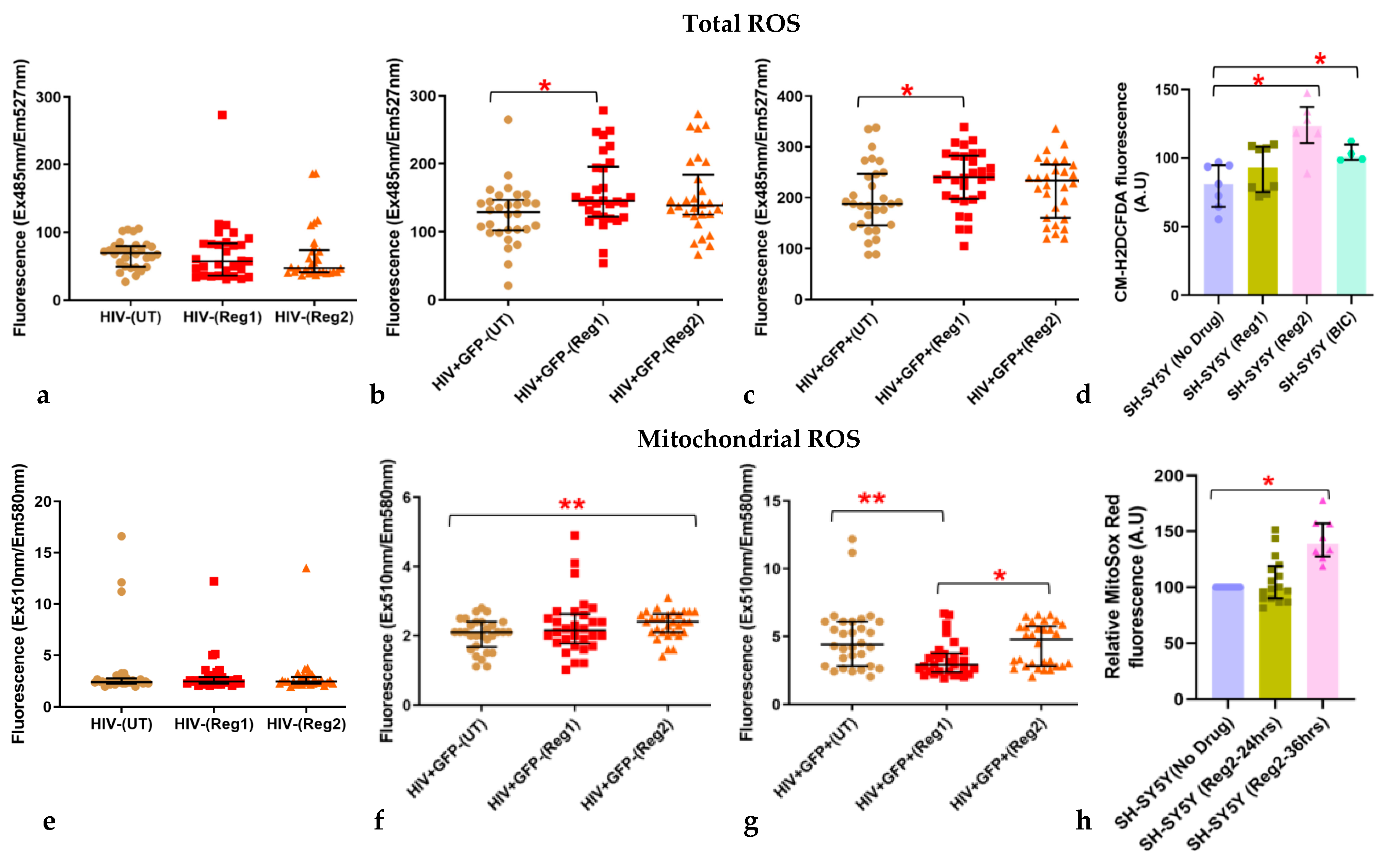
2.3. Effects of Contemporary ARVs on ROS Production in Hµglia and SH-SY5Y Cells
2.4. ARV-Related Changes in Cellular Glycolysis
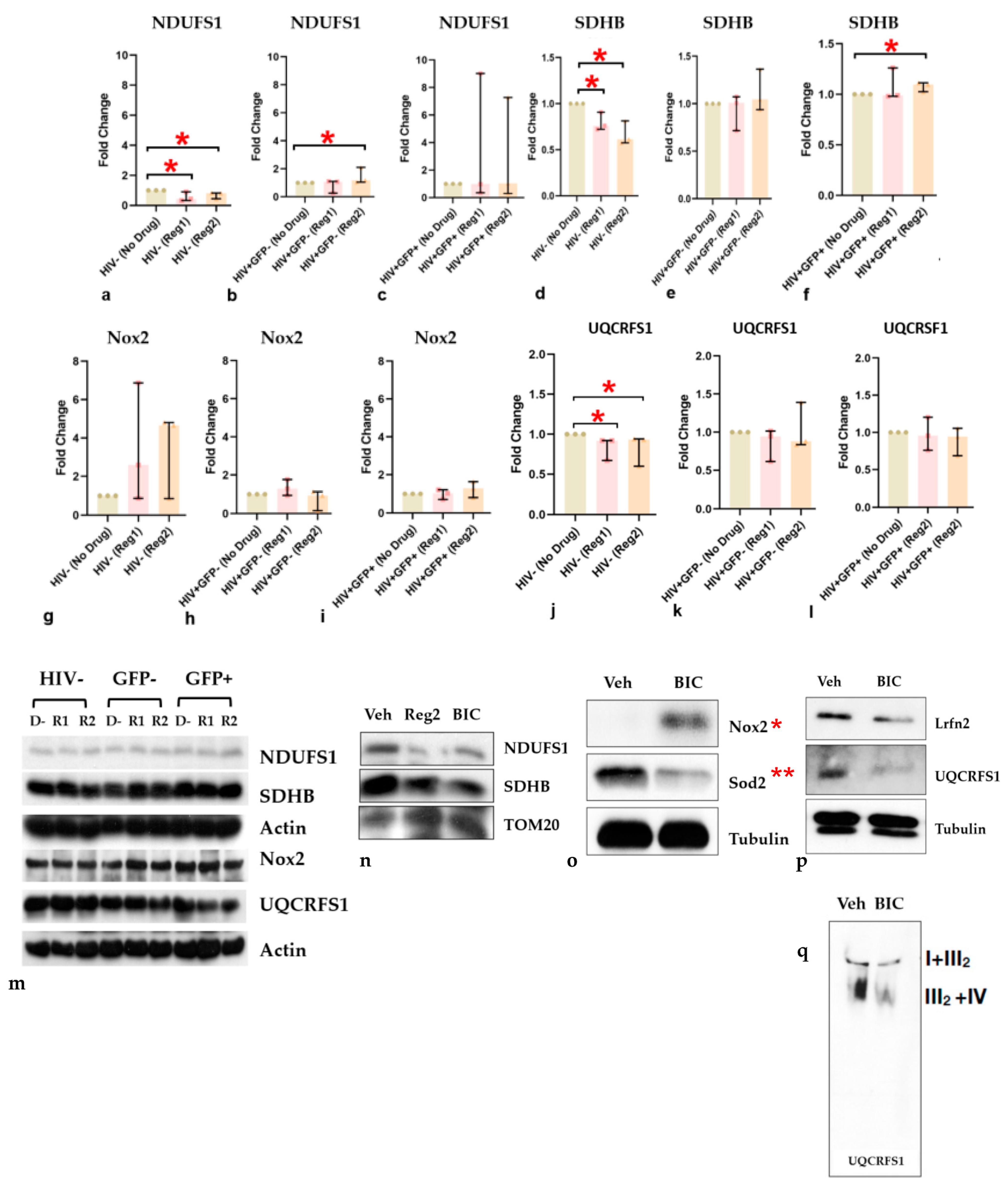
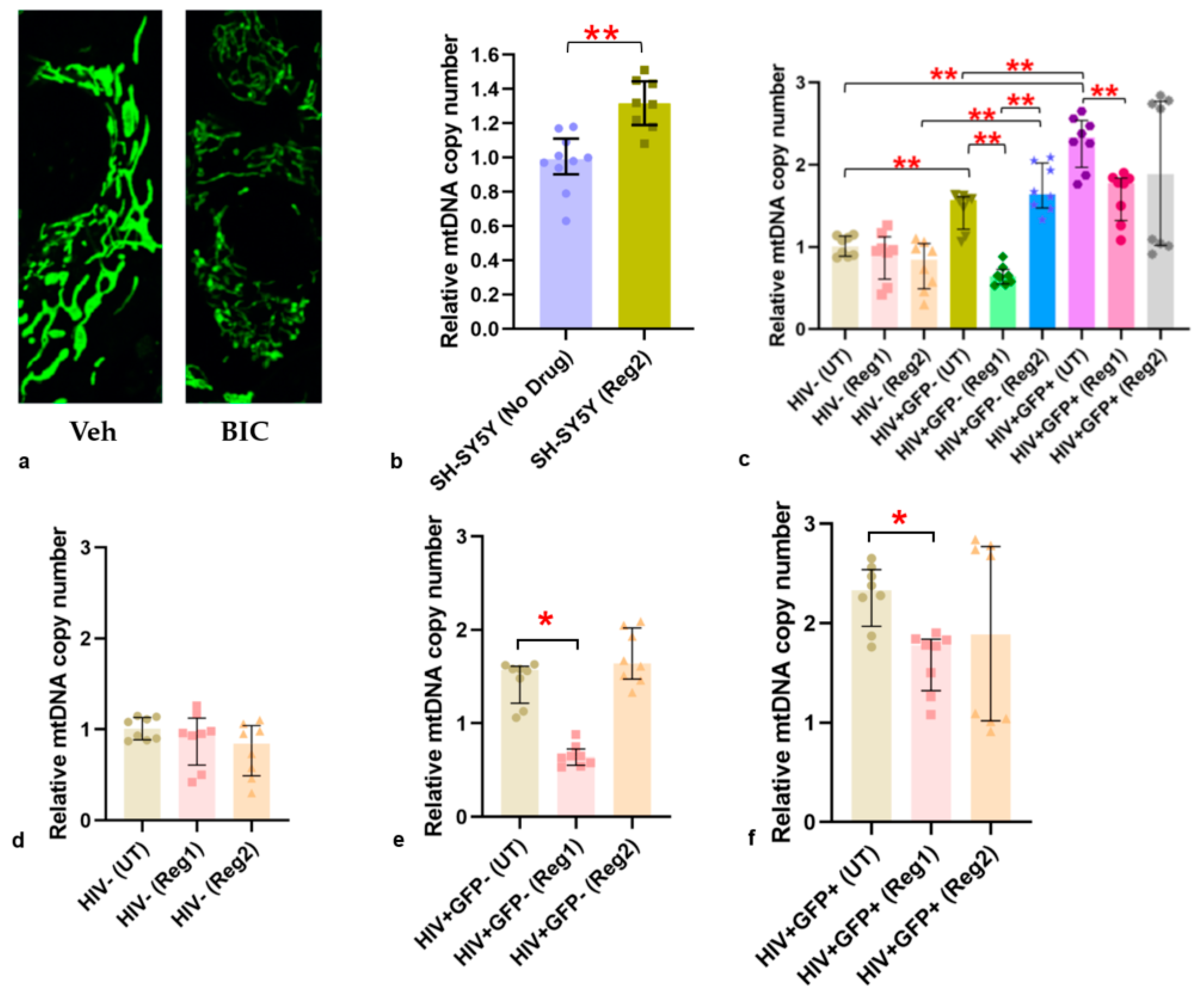
2.5. Impact of ARVs on Mitochondrial Protein Expression, Morphology, and Mitochondrial DNA (mtDNA) Copy Number
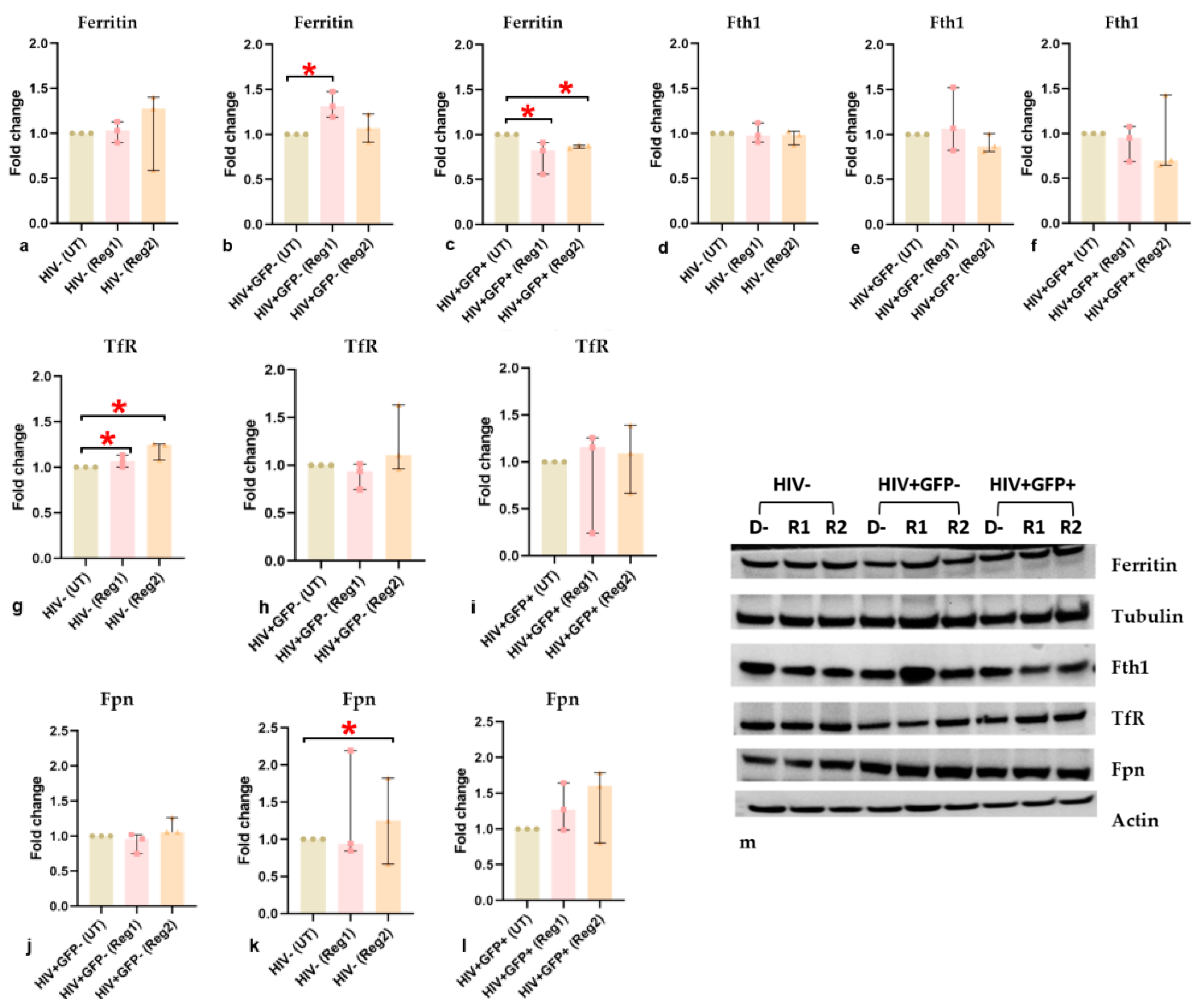
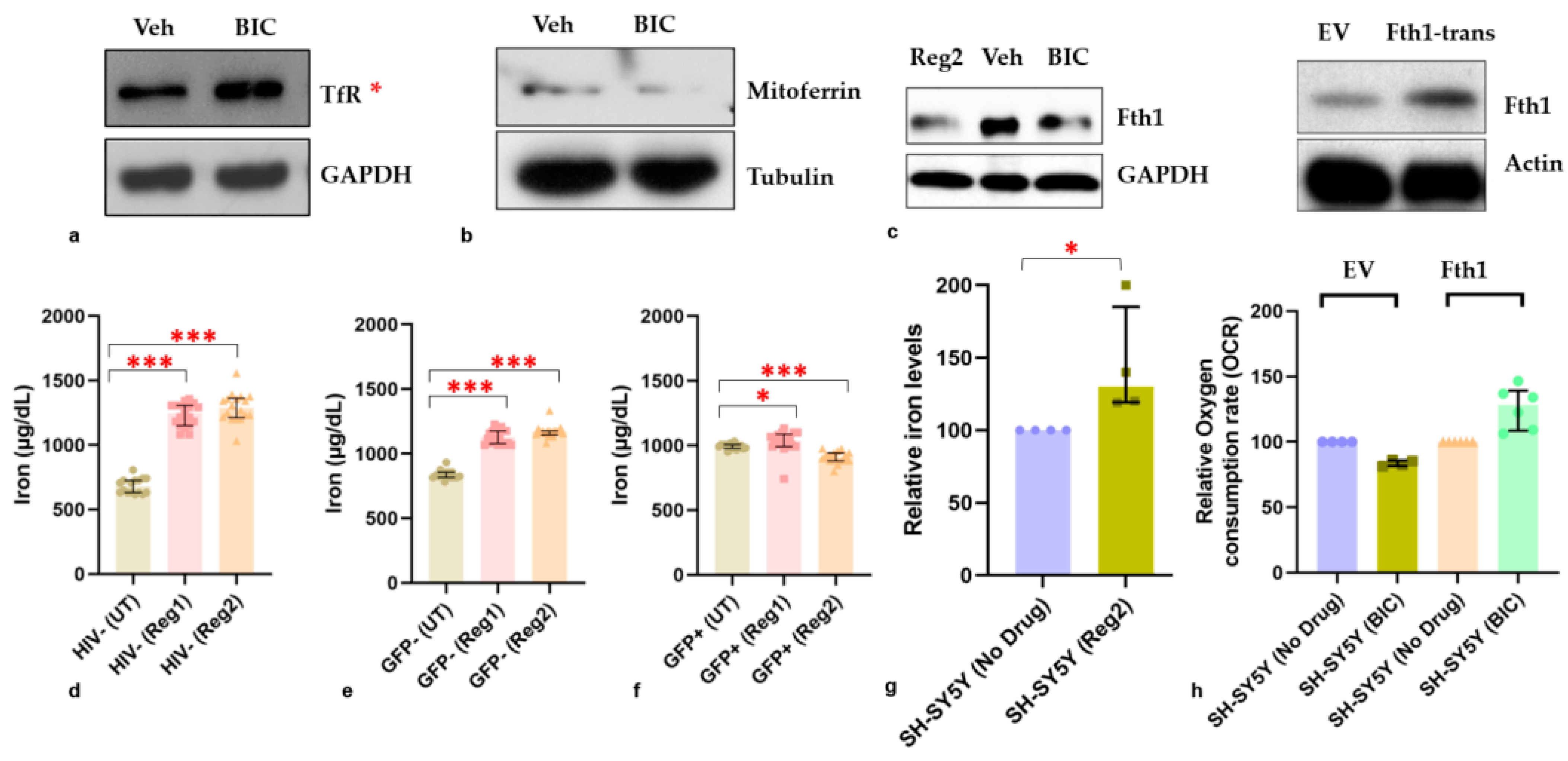
2.6. Effects of ARVs on Cellular Iron-Transport Phenotype and Iron Status
3. Discussion
4. Materials and Methods
4.1. Human Cell Lines and Cell Culture
4.2. Cell Culture in the Presence of ARVs
4.3. Real-Time Assays of Mitochondrial Respiratory Function in Intact Cells
4.4. Real-Time ATP Rate Assay (SH-SY5Y Cells)
4.5. Total and Mitochondrial Reactive Oxygen Species (ROS) Measurements
4.6. Protein Extraction and Quantification
4.7. Mitochondrial DNA (MtDNA) Copy Number Estimation and Morphologic Assessment
4.8. Transient Transfection of SH-SY5Y Cells and Other Pre-ARV-Treatment Interventions
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, S.; Dreyer, A.J.; Saylor, D.; Gisslen, M.; Winston, A.; Joska, J.A. Moving on from HAND: Why We Need New Criteria for Cognitive Impairment in Persons Living with Human Immunodeficiency Virus and a Proposed Way Forward. Clin. Infect. Dis. 2021, 73, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Falutz, J.; Branas, F.; Erlandson, K.M. Frailty: The current challenge for aging people with HIV. Curr. Opin. HIV AIDS 2021, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Masters, M.C.; Perez, J.; Wu, K.; Ellis, R.J.; Goodkin, K.; Koletar, S.L.; Andrade, A.; Yang, J.; Brown, T.T.; Palella, F.J.; et al. Baseline Neurocognitive Impairment (NCI) Is Associated with Incident Frailty but Baseline Frailty Does Not Predict Incident NCI in Older Persons with Human Immunodeficiency Virus (HIV). Clin. Infect. Dis. 2021, 73, 680–688. [Google Scholar] [CrossRef]
- Patton, S.M.; Wang, Q.; Hulgan, T.; Connor, J.R.; Jia, P.; Zhao, Z.; Letendre, S.L.; Ellis, R.J.; Bush, W.S.; Samuels, D.C.; et al. Cerebrospinal fluid (CSF) biomarkers of iron status are associated with CSF viral load, antiretroviral therapy, and demographic factors in HIV-infected adults. Fluids Barriers CNS 2017, 14, 11. [Google Scholar] [CrossRef]
- Volpe, K.; Samuels, D.; Kallianpur, A.; Ellis, R.; Franklin, D.; Letendre, S.; Heaton, R.K.; Hulgan, T. Mitochondrial DNA haplogroups and domain-specific neurocognitive performance in adults with HIV. J. Neurovirol. 2021, 27, 557–567. [Google Scholar] [CrossRef]
- Moulignier, A.; Costagliola, D. Metabolic Syndrome and Cardiovascular Disease Impacts on the Pathophysiology and Phenotype of HIV-Associated Neurocognitive Disorders. Curr. Top. Behav. Neurosci. 2021, 50, 367–399. [Google Scholar]
- Pfefferbaum, A.; Zahr, N.M.; Sassoon, S.A.; Kwon, D.; Pohl, K.M.; Sullivan, E.V. Accelerated and Premature Aging Characterizing Regional Cortical Volume Loss in Human Immunodeficiency Virus Infection: Contributions From Alcohol, Substance Use, and Hepatitis C Coinfection. Biol. Psychiatry. Cogn. Neurosci. Neuroimaging 2018, 3, 844–859. [Google Scholar] [CrossRef]
- Lanman, T.; Letendre, S.; Ma, Q.; Bang, A.; Ellis, R. CNS Neurotoxicity of Antiretrovirals. J. Neuroimmune. Pharmacol. 2021, 16, 130–143. [Google Scholar] [CrossRef]
- Ances, B.M.; Hammoud, D.A. Neuroimaging of HIV-associated neurocognitive disorders (HAND). Curr. Opin. HIV AIDS 2014, 9, 545–551. [Google Scholar] [CrossRef]
- Omeragic, A.; Kayode, O.; Hoque, M.T.; Bendayan, R. Potential pharmacological approaches for the treatment of HIV-1 associated neurocognitive disorders. Fluids Barriers CNS 2020, 17, 42. [Google Scholar] [CrossRef]
- Jordan, C.A.; Watkins, B.A.; Kufta, C.; Dubois-Dalcq, M. Infection of brain microglial cells by human immunodeficiency virus type 1 is CD4 dependent. J. Virol. 1991, 65, 736–742. [Google Scholar] [CrossRef]
- Kolson, D.L. Developments in Neuroprotection for HIV-Associated Neurocognitive Disorders (HAND). Curr. HIV/AIDS Rep. 2022, 19, 344–357. [Google Scholar] [CrossRef]
- Carroll, A.; Brew, B. HIV-associated neurocognitive disorders: Recent advances in pathogenesis, biomarkers, and treatment. F1000Research 2017, 6, 312. [Google Scholar] [CrossRef]
- Schier, C.J.; Marks, W.D.; Paris, J.J.; Barbour, A.J.; McLane, V.D.; Maragos, W.F.; McQuiston, A.R.; Knapp, P.E.; Hauser, K.F. Selective Vulnerability of Striatal D2 versus D1 Dopamine Receptor-Expressing Medium Spiny Neurons in HIV-1 Tat Transgenic Male Mice. J. Neurosci. 2017, 37, 5758–5769. [Google Scholar] [CrossRef]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Allen, C.N.S.; Arjona, S.P.; Santerre, M.; De Lucia, C.; Koch, W.J.; Sawaya, B.E. Metabolic Reprogramming in HIV-Associated Neurocognitive Disorders. Front. Cell. Neurosci. 2022, 16, 812887. [Google Scholar] [CrossRef]
- Cotto, B.; Natarajanseenivasan, K.; Langford, D. HIV-1 infection alters energy metabolism in the brain: Contributions to HIV-associated neurocognitive disorders. Prog. Neurobiol. 2019, 181, 101616. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Procopio, F.A.; Tarek, M.; Carlon-Andres, I.; Tang, H.Y.; Goldman, A.R.; Munshi, M.; Kumar Pal, V.; Forcato, M.; Sreeram, S.; et al. Glycolysis downregulation is a hallmark of HIV-1 latency and sensitizes infected cells to oxidative stress. EMBO Mol. Med. 2021, 13, e13901. [Google Scholar] [CrossRef]
- George, J.W.; Mattingly, J.E.; Roland, N.J.; Small, C.M.; Lamberty, B.G.; Fox, H.S.; Stauch, K.L. Physiologically Relevant Concentrations of Dolutegravir, Emtricitabine, and Efavirenz Induce Distinct Metabolic Alterations in HeLa Epithelial and BV2 Microglial Cells. Front. Immunol. 2021, 12, 639378. [Google Scholar] [CrossRef]
- Marra, C.M.; Zhao, Y.; Clifford, D.B.; Letendre, S.; Evans, S.; Henry, K.; Ellis, R.J.; Rodriguez, B.; Coombs, R.W.; Schifitto, G.; et al. Impact of combination antiretroviral therapy on cerebrospinal fluid HIV RNA and neurocognitive performance. AIDS 2009, 23, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.R.; Su, Z.; Margolis, D.M.; Krambrink, A.; Havlir, D.V.; Evans, S.; Skiest, D.J.; Team, A.S. Neurocognitive effects of treatment interruption in stable HIV-positive patients in an observational cohort. Neurology 2010, 74, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Gangwani, M.R.; Chaudhari, N.S.; Glazyrin, A.; Bhat, H.K.; Kumar, A. Neurotoxicity in the Post-HAART Era: Caution for the Antiretroviral Therapeutics. Neurotox. Res. 2016, 30, 677–697. [Google Scholar] [CrossRef] [PubMed]
- Rathore, U. Mitochondrial dysfunction in people with HIV receiving contemporary antiretroviral therapy. AIDS 2022, 36, 2063–2064. [Google Scholar] [CrossRef]
- Fields, J.A.; Ellis, R.J. HIV in the cART era and the mitochondrial: Immune interface in the CNS. Int. Rev. Neurobiol. 2019, 145, 29–65. [Google Scholar]
- Rensvold, J.W.; Ong, S.E.; Jeevananthan, A.; Carr, S.A.; Mootha, V.K.; Pagliarini, D.J. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis. Cell Rep. 2013, 3, 237–245. [Google Scholar] [CrossRef]
- Okwuegbuna, O.K.; Kaur, H.; Jennifer, I.; Bush, W.S.; Bharti, A.; Umlauf, A.; Ellis, R.J.; Franklin, D.R.; Heaton, R.K.; McCutchan, J.A.; et al. Anemia and Erythrocyte Indices Are Associated with Neurocognitive Performance Across Multiple Ability Domains in Adults with HIV. J. Acquir. Immune Defic. Syndr. 2023, 92, 414–421. [Google Scholar] [CrossRef]
- Armitage, A.E.; Stacey, A.R.; Giannoulatou, E.; Marshall, E.; Sturges, P.; Chatha, K.; Smith, N.M.; Huang, X.; Xu, X.; Pasricha, S.R.; et al. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proc. Natl. Acad. Sci. USA 2014, 111, 12187–12192. [Google Scholar] [CrossRef]
- Sotolongo, K.; Ghiso, J.; Rostagno, A. Nrf2 activation through the PI3K/GSK-3 axis protects neuronal cells from Abeta-mediated oxidative and metabolic damage. Alzheimer’s Res. Ther. 2020, 12, 13. [Google Scholar] [CrossRef]
- Kim, J.; Han, J.; Jang, Y.; Kim, S.J.; Lee, M.J.; Ryu, M.J.; Kweon, G.R.; Heo, J.Y. High-capacity glycolytic and mitochondrial oxidative metabolisms mediate the growth ability of glioblastoma. Int. J. Oncol. 2015, 47, 1009–1016. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Webb, M.; Sideris, D.P. Intimate Relations-Mitochondria and Ageing. Int. J. Mol. Sci. 2020, 21, 7580. [Google Scholar] [CrossRef]
- Goping, I.S.; Millar, D.G.; Shore, G.C. Identification of the human mitochondrial protein import receptor, huMas20p. Complementation of delta mas20 in yeast. FEBS Lett. 1995, 373, 45–50. [Google Scholar] [CrossRef]
- Dickson, K.B.; Zhou, J. Role of reactive oxygen species and iron in host defense against infection. Front. Biosci. 2020, 25, 1600–1616. [Google Scholar]
- Sandoval-Acuna, C.; Torrealba, N.; Tomkova, V.; Jadhav, S.B.; Blazkova, K.; Merta, L.; Lettlova, S.; Adamcova, M.K.; Rosel, D.; Brabek, J.; et al. Targeting Mitochondrial Iron Metabolism Suppresses Tumor Growth and Metastasis by Inducing Mitochondrial Dysfunction and Mitophagy. Cancer Res. 2021, 81, 2289–2303. [Google Scholar] [CrossRef]
- Bereczki, E.; Branca, R.M.; Francis, P.T.; Pereira, J.B.; Baek, J.H.; Hortobagyi, T.; Winblad, B.; Ballard, C.; Lehtio, J.; Aarsland, D. Synaptic markers of cognitive decline in neurodegenerative diseases: A proteomic approach. Brain 2018, 141, 582–595. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef]
- Yuan, N.Y.; Kaul, M. Beneficial and Adverse Effects of cART Affect Neurocognitive Function in HIV-1 Infection: Balancing Viral Suppression against Neuronal Stress and Injury. J. Neuroimmune Pharmacol. 2021, 16, 90–112. [Google Scholar] [CrossRef]
- Klaus, S.; Ost, M. Mitochondrial uncoupling and longevity—A role for mitokines? Exp. Gerontol. 2020, 130, 110796. [Google Scholar] [CrossRef]
- Sfera, A.; Gradini, R.; Cummings, M.; Diaz, E.; Price, A.I.; Osorio, C. Rusty Microglia: Trainers of Innate Immunity in Alzheimer’s Disease. Front. Neurol. 2018, 9, 1062. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.A.; Kerskens, C.; Lynch, M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Sosa, J.C.; Gardeck, A.M.; Baez, A.S.; Lee, C.H.; Wessling-Resnick, M. Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 2018, 293, 7853–7863. [Google Scholar] [CrossRef]
- Holland, R.; McIntosh, A.L.; Finucane, O.M.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.A.; Gun’ko, Y.K.; Lynch, M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Valdebenito, S.; D’Amico, D.; Prideaux, B.; Eugenin, E.A. HIV infection of astrocytes compromises inter-organelle interactions and inositol phosphate metabolism: A potential mechanism of bystander damage and viral reservoir survival. Prog. Neurobiol. 2021, 206, 102157. [Google Scholar] [CrossRef] [PubMed]
- Deme, P.; Rubin, L.H.; Yu, D.; Xu, Y.; Nakigozi, G.; Nakasujja, N.; Anok, A.; Kisakye, A.; Quinn, T.C.; Reynolds, S.J.; et al. Immunometabolic Reprogramming in Response to HIV Infection Is Not Fully Normalized by Suppressive Antiretroviral Therapy. Viruses 2022, 14, 1313. [Google Scholar] [CrossRef]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-mediated microglial activation: Role of mitochondrial dysfunction and defective mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free. Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Dietrich, C.; Hofmann, T.G. Ferroptosis Meets Cell-Cell Contacts. Cells 2021, 10, 2462. [Google Scholar] [CrossRef]
- Drakesmith, H.; Chen, N.; Ledermann, H.; Screaton, G.; Townsend, A.; Xu, X.N. HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. Proc. Natl. Acad. Sci. USA 2005, 102, 11017–11022. [Google Scholar] [CrossRef]
- Huang, H.; Zhou, Z.H.; Adhikari, R.; Yamada, K.M.; Dhawan, S. Defective iron homeostasis in human immunodeficiency virus type-1 latency. Curr. Trends Immunol. 2016, 17, 125–131. [Google Scholar]
- Arosio, P.; Carmona, F.; Gozzelino, R.; Maccarinelli, F.; Poli, M. The importance of eukaryotic ferritins in iron handling and cytoprotection. Biochem. J. 2015, 472, 1–15. [Google Scholar] [CrossRef]
- Shankaran, P.; Madlenakova, M.; Hajkova, V.; Jilich, D.; Svobodova, I.; Horinek, A.; Fujikura, Y.; Melkova, Z. Effects of heme degradation products on reactivation of latent HIV-1. Acta Virol. 2017, 61, 86–96. [Google Scholar] [CrossRef][Green Version]
- Shytaj, I.L.; Lucic, B.; Forcato, M.; Penzo, C.; Billingsley, J.; Laketa, V.; Bosinger, S.; Stanic, M.; Gregoretti, F.; Antonelli, L.; et al. Alterations of redox and iron metabolism accompany the development of HIV latency. EMBO J. 2020, 39, e102209. [Google Scholar] [CrossRef]
- Kaur, H.; Bush, W.S.; Letendre, S.L.; Ellis, R.J.; Heaton, R.K.; Patton, S.M.; Connor, J.R.; Samuels, D.C.; Franklin, D.R.; Hulgan, T.; et al. Higher CSF Ferritin Heavy-Chain (Fth1) and Transferrin Predict Better Neurocognitive Performance in People with HIV. Mol. Neurobiol. 2021, 58, 4842–4855. [Google Scholar] [CrossRef]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R., Jr.; Ballinger, S.; Dell’Italia, L.; Zhang, J.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef]
- Yauger, Y.J.; Bermudez, S.; Moritz, K.E.; Glaser, E.; Stoica, B.; Byrnes, K.R. Iron accentuated reactive oxygen species release by NADPH oxidase in activated microglia contributes to oxidative stress in vitro. J. Neuroinflammation 2019, 16, 41. [Google Scholar] [CrossRef]
- Mak, I.T.; Kramer, J.H.; Iantorno, M.; Chmielinska, J.J.; Weglicki, W.B.; Spurney, C.F. Deficiency in gp91Phox (NOX2) Protects against Oxidative Stress and Cardiac Dysfunction in Iron Overloaded Mice. Hearts 2020, 1, 117–125. [Google Scholar] [CrossRef]
- Lopez-Suarez, L.; Awabdh, S.A.; Coumoul, X.; Chauvet, C. The SH-SY5Y human neuroblastoma cell line, a relevant in vitro cell model for investigating neurotoxicology in human: Focus on organic pollutants. Neurotoxicology 2022, 92, 131–155. [Google Scholar] [CrossRef]
- Gonzalez, H.; Podany, A.; Al-Harthi, L.; Wallace, J. The far-reaching HAND of cART: cART effects on astrocytes. J. Neuroimmune Pharmacol. 2021, 16, 144–158. [Google Scholar] [CrossRef]
- Robertson, K.; Liner, J.; Meeker, R.B. Antiretroviral neurotoxicity. J. Neurovirol. 2012, 18, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Funes, H.A.; Apostolova, N.; Alegre, F.; Blas-Garcia, A.; Alvarez, A.; Marti-Cabrera, M.; Esplugues, J.V. Neuronal bioenergetics and acute mitochondrial dysfunction: A clue to understanding the central nervous system side effects of efavirenz. J. Infect. Dis. 2014, 210, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Ciavatta, V.T.; Bichler, E.K.; Speigel, I.A.; Elder, C.C.; Teng, S.L.; Tyor, W.R.; Garcia, P.S. In vitro and Ex vivo Neurotoxic Effects of Efavirenz are Greater than Those of Other Common Antiretrovirals. Neurochem. Res. 2017, 42, 3220–3232. [Google Scholar] [CrossRef]
- Blas-Garcia, A.; Polo, M.; Alegre, F.; Funes, H.A.; Martinez, E.; Apostolova, N.; Esplugues, J.V. Lack of mitochondrial toxicity of darunavir, raltegravir and rilpivirine in neurons and hepatocytes: A comparison with efavirenz. J. Antimicrob. Chemother. 2014, 69, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.E5. [Google Scholar] [CrossRef]
- Ambikan, A.T.; Svensson-Akusjarvi, S.; Krishnan, S.; Sperk, M.; Nowak, P.; Vesterbacka, J.; Sonnerborg, A.; Benfeitas, R.; Neogi, U. Genome-scale metabolic models for natural and long-term drug-induced viral control in HIV infection. Life Sci. Alliance 2022, 5, e202201405. [Google Scholar] [CrossRef]
- Mikaeloff, F.; Svensson Akusjarvi, S.; Ikomey, G.M.; Krishnan, S.; Sperk, M.; Gupta, S.; Magdaleno, G.D.V.; Escos, A.; Lyonga, E.; Okomo, M.C.; et al. Trans cohort metabolic reprogramming towards glutaminolysis in long-term successfully treated HIV-infection. Commun. Biol. 2022, 5, 27. [Google Scholar] [CrossRef]
- Singh, K.; Sarafianos, S.G.; Sonnerborg, A. Long-Acting Anti-HIV Drugs Targeting HIV-1 Reverse Transcriptase and Integrase. Pharmaceuticals 2019, 12, 62. [Google Scholar] [CrossRef]
- Ajaykumar, A.; Zhu, M.; Kakkar, F.; Brophy, J.; Bitnun, A.; Alimenti, A.; Soudeyns, H.; Saberi, S.; Albert, A.Y.K.; Money, D.M.; et al. Elevated Blood Mitochondrial DNA in Early Life Among Uninfected Children Exposed to Human Immunodeficiency Virus and Combination Antiretroviral Therapy in utero. J. Infect. Dis. 2021, 223, 621–631. [Google Scholar] [CrossRef]
- Schnoll, J.G.; Temsamrit, B.; Zhang, D.; Song, H.; Ming, G.L.; Christian, K.M. Evaluating Neurodevelopmental Consequences of Perinatal Exposure to Antiretroviral Drugs: Current Challenges and New Approaches. J. Neuroimmune Pharmacol. 2021, 16, 113–129. [Google Scholar] [CrossRef]
- Shen, J.; Liberty, A.; Shiau, S.; Strehlau, R.; Pierson, S.; Patel, F.; Wang, L.; Burke, M.; Violari, A.; Coovadia, A.; et al. Mitochondrial Impairment in Well-Suppressed Children with Perinatal HIV-Infection on Antiretroviral Therapy. AIDS Res. Hum. Retroviruses 2020, 36, 27–38. [Google Scholar] [CrossRef]
- Garcia-Otero, L.; Lopez, M.; Guitart-Mampel, M.; Moren, C.; Gonce, A.; Esteve, C.; Salazar, L.; Gomez, O.; Martinez, J.M.; Torres, B.; et al. Cardiac and mitochondrial function in HIV-uninfected fetuses exposed to antiretroviral treatment. PLoS ONE 2019, 14, e0213279. [Google Scholar] [CrossRef]
- Horvath, S.; Lin, D.T.S.; Kobor, M.S.; Zoller, J.A.; Said, J.W.; Morgello, S.; Singer, E.; Yong, W.H.; Jamieson, B.D.; Levine, A.J. HIV, pathology and epigenetic age acceleration in different human tissues. Geroscience 2022, 44, 1609–1620. [Google Scholar] [CrossRef]
- Chen, Y.F.; Stampley, J.E.; Irving, B.A.; Dugas, T.R. Chronic Nucleoside Reverse Transcriptase Inhibitors Disrupt Mitochondrial Homeostasis and Promote Premature Endothelial Senescence. Toxicol. Sci. 2019, 172, 445–456. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Jay, T.R.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017, 23, 47–66. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Milne, S.; Das, B.; Dobrowolski, C.; Rojas, R.; Karn, J. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 2017, 14, 9. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249. [Google Scholar] [CrossRef]
- Ye, F.; Alvarez-Carbonell, D.; Nguyen, K.; Leskov, K.; Garcia-Mesa, Y.; Sreeram, S.; Valadkhan, S.; Karn, J. Recruitment of the CoREST transcription repressor complexes by Nerve Growth factor IB-like receptor (Nurr1/NR4A2) mediates silencing of HIV in microglial cells. PLoS Pathog. 2022, 18, e1010110. [Google Scholar] [CrossRef]
- Varatharajan, L.; Thomas, S.A. The transport of anti-HIV drugs across blood-CNS interfaces: Summary of current knowledge and recommendations for further research. Antivir. Res. 2009, 82, A99–A109. [Google Scholar] [CrossRef]
- Yilmaz, A.; Price, R.W.; Gisslen, M. Antiretroviral drug treatment of CNS HIV-1 infection. J. Antimicrob. Chemother. 2012, 67, 299–311. [Google Scholar] [CrossRef]
- Ma, Q.; Ocque, A.J.; Morse, G.D.; Sanders, C.; Burgi, A.; Little, S.J.; Letendre, S.L. Switching to Tenofovir Alafenamide in Elvitegravir-Based Regimens: Pharmacokinetics and Antiviral Activity in Cerebrospinal Fluid. Clin. Infect. Dis. 2020, 71, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.; Bumpus, N.N.; Ma, Q.; Ellis, R.J.; Soontornniyomkij, V.; Fields, J.A.; Bharti, A.; Achim, C.L.; Moore, D.J.; Letendre, S.L. Antiretroviral drug concentrations in brain tissue of adult decedents. AIDS 2020, 34, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Ohuchi, M.; Yamane, N.; Aikawa, H.; Gatanaga, H.; Oka, S.; Hamada, A. High-performance liquid chromatography-tandem mass spectrometry for simultaneous determination of raltegravir, dolutegravir and elvitegravir concentrations in human plasma and cerebrospinal fluid samples. Biomed. Chromatogr. 2018, 32, e4058. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, A.; Pinnetti, C.; De Nicolo, A.; Scarvaglieri, E.; Gisslen, M.; Tempestilli, M.; D’Avolio, A.; Fedele, V.; Di Perri, G.; Antinori, A.; et al. Cerebrospinal fluid abacavir concentrations in HIV-positive patients following once-daily administration. Br. J. Clin. Pharmacol. 2018, 84, 1380–1383. [Google Scholar] [CrossRef]
- Imaz, A.; Niubo, J.; Amara, A.; Khoo, S.; Ferrer, E.; Tiraboschi, J.M.; Acerete, L.; Garcia, B.; Vila, A.; Podzamczer, D. Cerebrospinal fluid drug concentrations and viral suppression in HIV-1-infected patients receiving ritonavir-boosted atazanavir plus lamivudine dual antiretroviral therapy (Spanish HIV/AIDS Research Network, PreEC/RIS 39). J. Neurovirol. 2018, 24, 391–397. [Google Scholar] [CrossRef]
- Gele, T.; Cheret, A.; Castro Gordon, A.; Nkam, L.; Furlan, V.; Pallier, C.; Becker, P.H.; Catalan, P.; Goujard, C.; Taburet, A.M.; et al. Cerebrospinal fluid exposure to bictegravir/emtricitabine/tenofovir in HIV-1-infected patients with CNS impairment. J. Antimicrob. Chemother. 2021, 76, 3280–3285. [Google Scholar] [CrossRef]
- Rigo-Bonnin, R.; Tiraboschi, J.M.; Alvarez-Alvarez, M.; Perez-Fernandez, G.A.; Sanjuas-Iglesias, M.; Scevola, S.; Niubo, J.; Videla, S.; Podzamczer, D. Measurement of total and unbound bictegravir concentrations in plasma and cerebrospinal fluid by UHPLC-MS/MS. J. Pharm. Biomed. Anal. 2020, 185, 113250. [Google Scholar] [CrossRef]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Huttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Huttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, H.; Minchella, P.; Alvarez-Carbonell, D.; Purandare, N.; Nagampalli, V.K.; Blankenberg, D.; Hulgan, T.; Gerschenson, M.; Karn, J.; Aras, S.; et al. Contemporary Antiretroviral Therapy Dysregulates Iron Transport and Augments Mitochondrial Dysfunction in HIV-Infected Human Microglia and Neural-Lineage Cells. Int. J. Mol. Sci. 2023, 24, 12242. https://doi.org/10.3390/ijms241512242
Kaur H, Minchella P, Alvarez-Carbonell D, Purandare N, Nagampalli VK, Blankenberg D, Hulgan T, Gerschenson M, Karn J, Aras S, et al. Contemporary Antiretroviral Therapy Dysregulates Iron Transport and Augments Mitochondrial Dysfunction in HIV-Infected Human Microglia and Neural-Lineage Cells. International Journal of Molecular Sciences. 2023; 24(15):12242. https://doi.org/10.3390/ijms241512242
Chicago/Turabian StyleKaur, Harpreet, Paige Minchella, David Alvarez-Carbonell, Neeraja Purandare, Vijay K. Nagampalli, Daniel Blankenberg, Todd Hulgan, Mariana Gerschenson, Jonathan Karn, Siddhesh Aras, and et al. 2023. "Contemporary Antiretroviral Therapy Dysregulates Iron Transport and Augments Mitochondrial Dysfunction in HIV-Infected Human Microglia and Neural-Lineage Cells" International Journal of Molecular Sciences 24, no. 15: 12242. https://doi.org/10.3390/ijms241512242
APA StyleKaur, H., Minchella, P., Alvarez-Carbonell, D., Purandare, N., Nagampalli, V. K., Blankenberg, D., Hulgan, T., Gerschenson, M., Karn, J., Aras, S., & Kallianpur, A. R. (2023). Contemporary Antiretroviral Therapy Dysregulates Iron Transport and Augments Mitochondrial Dysfunction in HIV-Infected Human Microglia and Neural-Lineage Cells. International Journal of Molecular Sciences, 24(15), 12242. https://doi.org/10.3390/ijms241512242