
Blood–Brain Barrier Dysfunction and Aβ42/40 Ratio Dose-Dependent Modulation with the ApoE Genotype within the ATN Framework


Abstract
1. Introduction
2. Results
2.1. Overview of Participants within the ATN Framework and Between-Group Comparisons
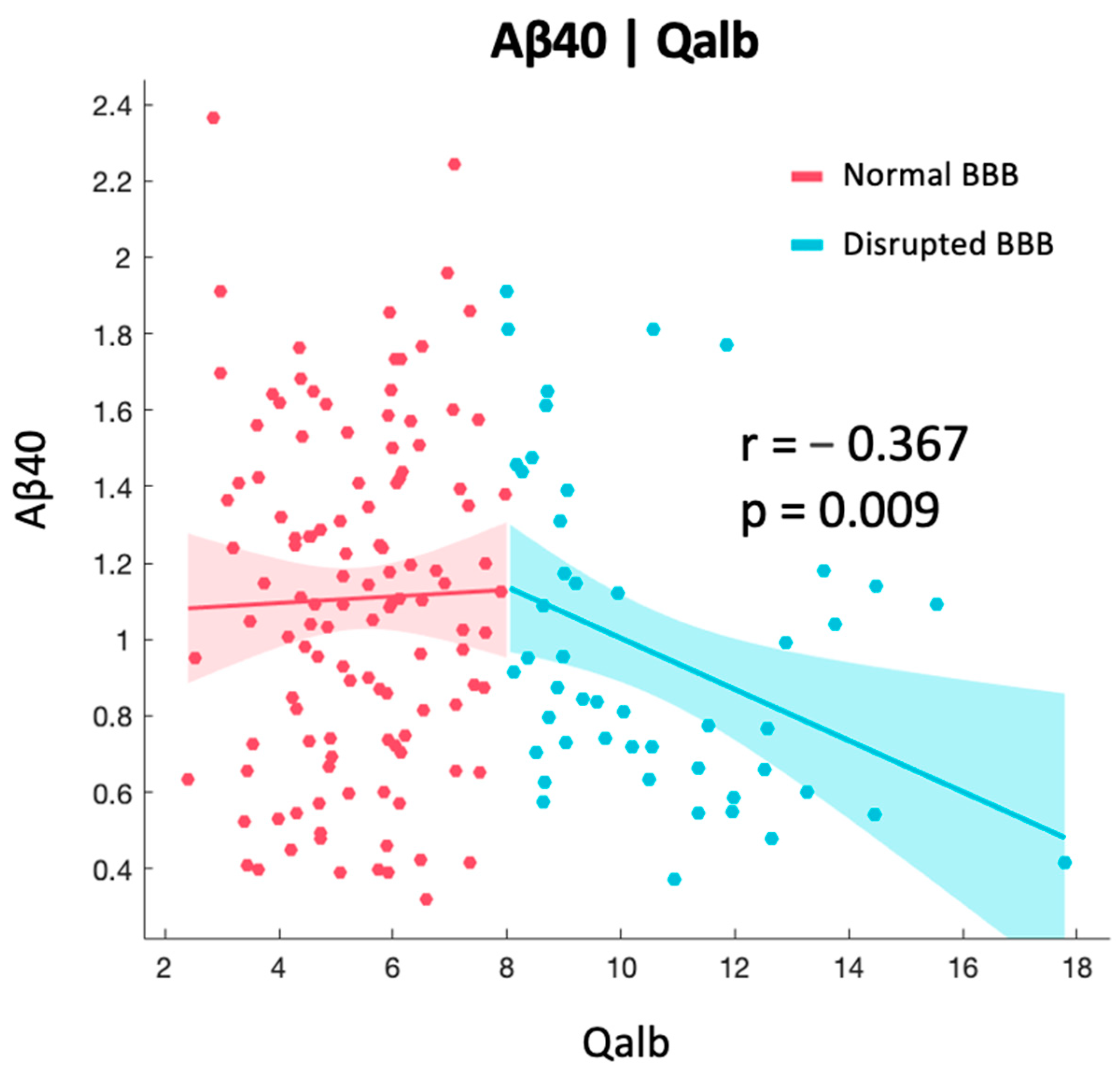
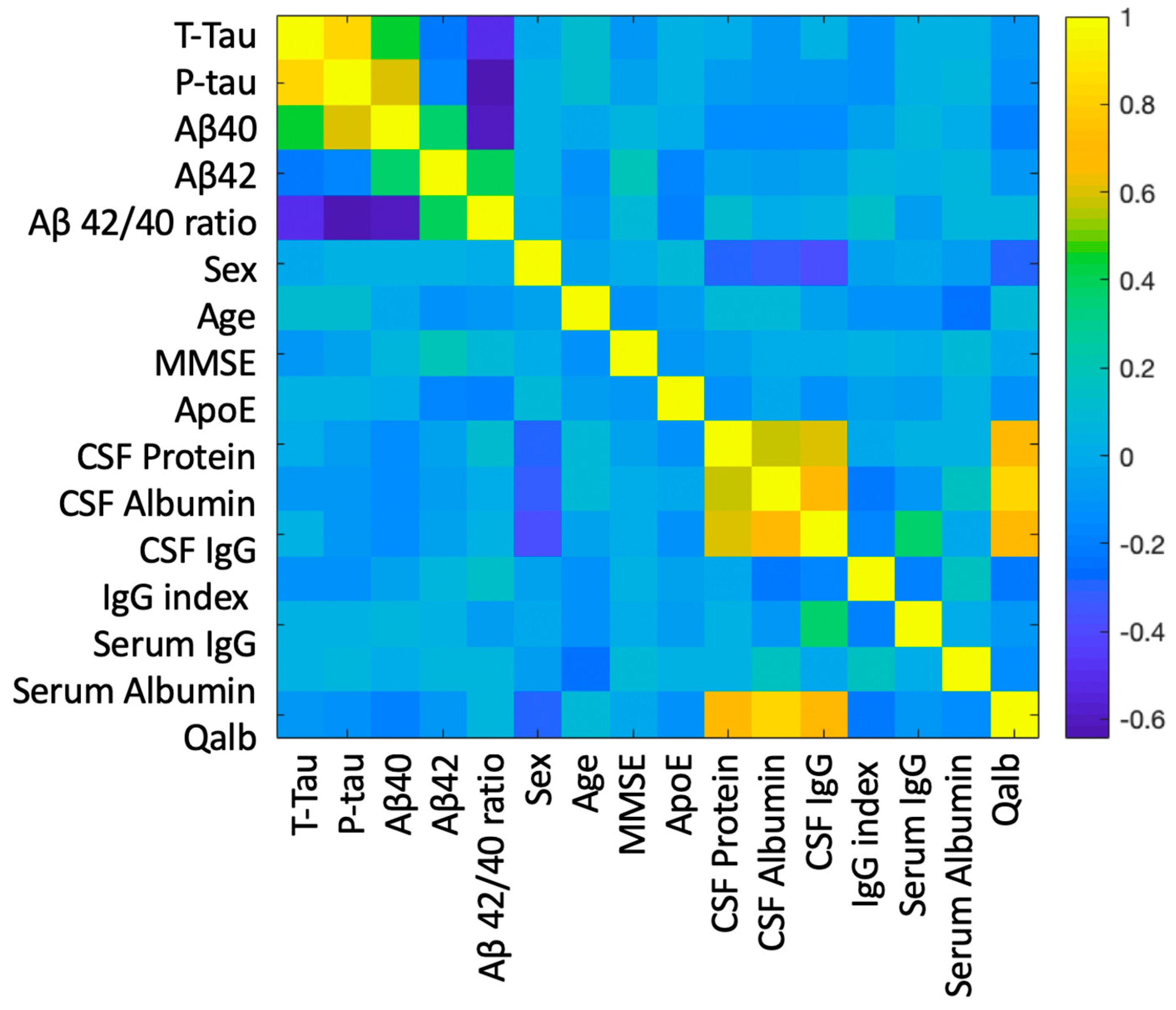
2.2. Correlation between BBB and AD-Specific Biomarkers
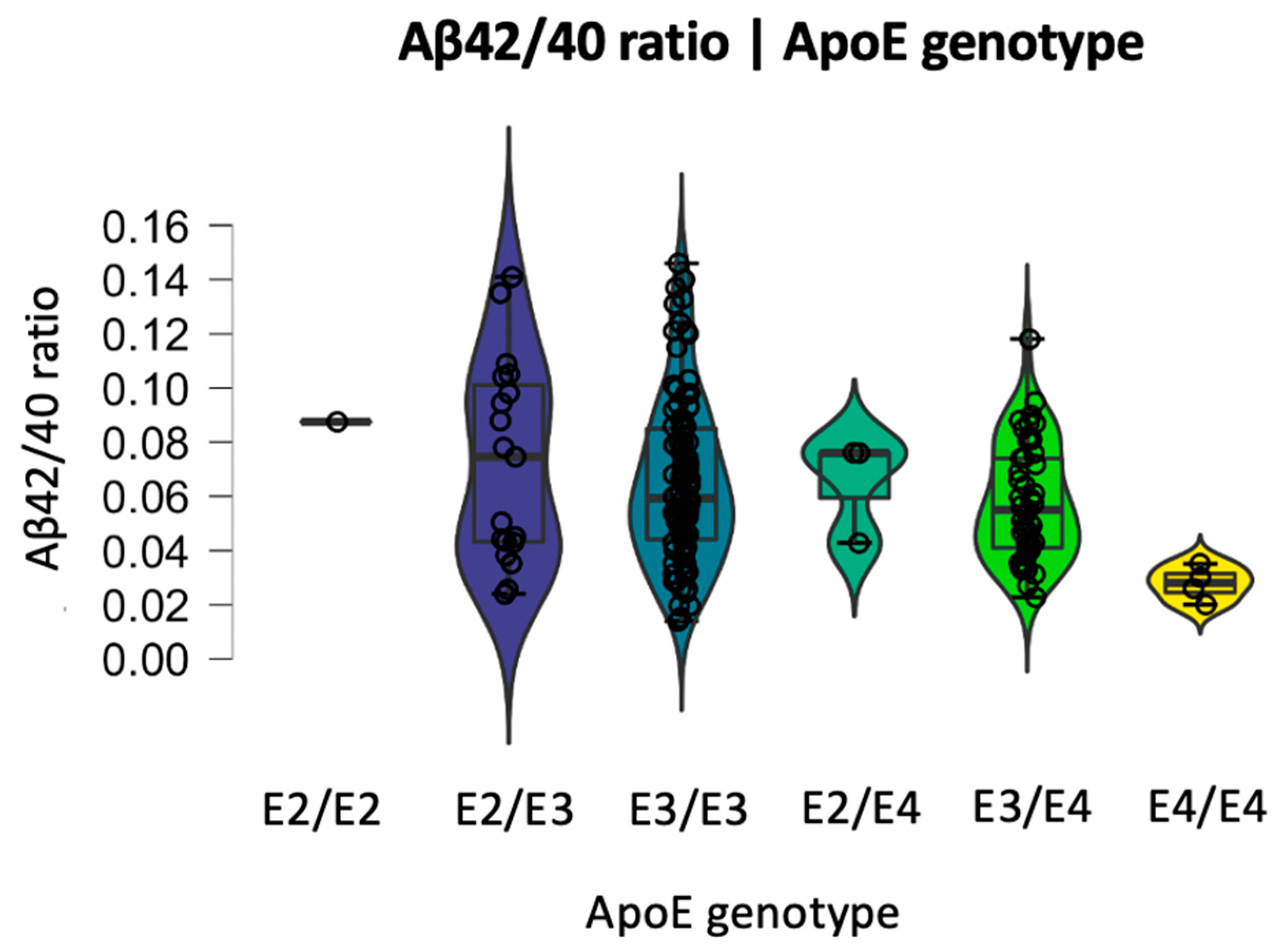
2.3. Relationship between BBB Biomarkers, Demographics, and Cognition
2.4. Principal Component Analysis
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Head, E.; Schmitt, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J.; et al. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Blacker, D.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Johnson, K.A.; Frosch, M.P.; Sperling, R.A.; et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann. Neurol. 2014, 75, 597–601. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef]
- Adamowicz, D.H.; Roy, S.; Salmon, D.P.; Galasko, D.R.; Hansen, L.A.; Masliah, E.; Gage, F.H. Hippocampal α-Synuclein in Dementia with Lewy Bodies Contributes to Memory Impairment and Is Consistent with Spread of Pathology. J. Neurosci. 2016, 37, 1675–1684. [Google Scholar] [CrossRef]
- Rodriguez, R.D.; Grinberg, L.T. Argyrophilic grain disease: An underestimated tauopathy. Dement. Neuropsychol. 2015, 9, 2–8. [Google Scholar] [CrossRef]
- Nelson, P.T.; Trojanowski, J.Q.; Abner, E.L.; Al-Janabi, O.M.; Jicha, G.A.; Schmitt, F.A.; Smith, C.D.; Fardo, D.W.; Wang, W.-X.; Kryscio, R.J.; et al. “New Old Pathologies”: AD, PART, and Cerebral Age-Related TDP-43 With Sclerosis (CARTS). J. Neuropathol. Exp. Neurol. 2016, 75, 482–498. [Google Scholar] [CrossRef]
- Duits, F.H.; Teunissen, C.E.; Bouwman, F.H.; Visser, P.; Mattsson, N.; Zetterberg, H.; Blennow, K.; Hansson, O.; Minthon, L.; Andreasen, N.; et al. The cerebrospinal fluid “Alzheimer profile”: Easily said, but what does it mean? Alzheimer’s Dement. 2014, 10, 713–723.e2. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Lelental, N.; Spitzer, P.; Maler, J.M.; Kornhuber, J. Amyloid-β 42/40 Cerebrospinal Fluid Concentration Ratio in the Diagnostics of Alzheimer’s Disease: Validation of Two Novel Assays. J. Alzheimer’s Dis. 2014, 43, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Matsubara, E.; Kanai, M.; Watanabe, M.; Nakamura, T.; Tomidokoro, Y.; Shizuka, M.; Wakabayashi, K.; Igeta, Y.; Ikeda, Y.; et al. Combination assay of CSF Tau, Aβ1-40 and Aβ1-42(43) as a biochemical marker of Alzheimer’s disease. J. Neurol. Sci. 1998, 158, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Hüll, M.; Hampel, H.; Kessler, H.; Frölich, L.; Schröder, J.; Peters, O.; Jessen, F.; et al. Amyloid β peptide ratio 42/40 but not Aβ42 correlates with phospho-Tau in patients with low- and high-CSF Aβ40 load. J. Neurochem. 2006, 101, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Andreasson, U.; Londos, E.; Minthon, L.; Blennow, K. Prediction of Alzheimer’s Disease Using the CSF Aβ42/Aβ40 Ratio in Patients with Mild Cognitive Impairment. Dement. Geriatr. Cogn. Disord. 2007, 23, 316–320. [Google Scholar] [CrossRef]
- Dorey, A.; Perret-Liaudet, A.; Tholance, Y.; Fourier, A.; Quadrio, I. Cerebrospinal Fluid Aβ40 Improves the Interpretation of Aβ42 Concentration for Diagnosing Alzheimer’s Disease. Front. Neurol. 2015, 6, 247. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, R.; Mizuno, T.; Mizuno, T.; Mori, S.; Nakajima, K.; Fushiki, S.; Yanagisawa, K. Age-Dependent Change in the Levels of Aβ40 and Aβ42 in Cerebrospinal Fluid from Control Subjects, and a Decrease in the Ratio of Aβ42 to Aβ40 Level in Cerebrospinal Fluid from Alzheimer’s Disease Patients. Eur. Neurol. 2000, 43, 155–160. [Google Scholar] [CrossRef]
- Lewczuk, P.; Matzen, A.; Blennow, K.; Parnetti, L.; Molinuevo, J.L.; Eusebi, P.; Kornhuber, J.; Morris, J.C.; Fagan, A.M. Cerebrospinal Fluid Aβ42/40 Corresponds Better than Aβ42 to Amyloid PET in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 55, 813–822. [Google Scholar] [CrossRef]
- Slaets, S.; Le Bastard, N.; Martin, J.-J.; Sleegers, K.; Van Broeckhoven, C.; De Deyn, P.P.; Engelborghs, S. Cerebrospinal Fluid Aβ1-40 Improves Differential Dementia Diagnosis in Patients with Intermediate P-tau181P Levels. J. Alzheimer’s Dis. 2013, 36, 759–767. [Google Scholar] [CrossRef]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schiossmacher, M.; Whaley, J.; Swindlehurst, C.; et al. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 1992, 359, 325–327. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Karikari, T.K.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Moscoso, A.; Grothe, M.J.; Ashton, N.J.; Karikari, T.K.; Rodriguez, J.L.; Snellman, A.; Suárez-Calvet, M.; Zetterberg, H.; Blennow, K.; Schöll, M.; et al. Time course of phosphorylated-tau181 in blood across the Alzheimer’s disease spectrum. Brain 2020, 144, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Husain, M. Blood tests to screen for Alzheimer’s disease. Brain 2021, 144, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Elovaara, I.; Icen, A.; Palo, J.; Erkinjuntti, T. CSF in Alzheimer’s disease: Studies on blood-brain barrier function and intrathecal protein synthesis. J. Neurol. Sci. 1985, 70, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Burgmans, S.; van de Haar, H.J.; Verhey, F.R.J.; Backes, W.H. Amyloid-β Interacts with Blood-Brain Barrier Function in Dementia: A Systematic Review. J. Alzheimer’s Dis. 2013, 35, 859–873. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Zimmermann, R.; Beck, G.; Knispel, S.; Maler, J.M.; Weih, M.; Wiltfang, J.; Kornhuber, J.; Lewczuk, P. Intrathecal IgG Synthesis in Patients with Alterations in the Neurochemical Dementia Diagnostics. J. Alzheimer’s Dis. 2010, 19, 1199–1203. [Google Scholar] [CrossRef]
- Blennow, K.; Wallin, A.; Fredman, P.; Karlsson, I.; Gottfries, C.G.; Svennerholm, L. Blood-brain barrier disturbance in patients with Alzheimer’s disease is related to vascular factors. Acta Neurol. Scand. 1990, 81, 323–326. [Google Scholar] [CrossRef]
- Hampel, H.; Kötter, H.U.; Padberg, F.; A Körschenhausen, D.; Möller, H.J. Oligoclonal Bands and Blood-Cerebrospinal-Fluid Barrier Dysfunction in a Subset of Patients with Alzheimer Disease: Comparison with Vascular Dementia, Major Depression, and Multiple Sclerosis. Alzheimer Dis. Assoc. Disord. 1999, 13, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Yanase, D.; Noguchi-Shinohara, M.; Ono, K.; Yoshita, M.; Yamada, M. Blood-Brain Barrier Permeability Correlates with Medial Temporal Lobe Atrophy but Not with Amyloid-β Protein Transport across the Blood-Brain Barrier in Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2007, 23, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Algotsson, A.; Winblad, B. The integrity of the blood?brain barrier in Alzheimer?s disease. Acta Neurol. Scand. 2007, 115, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Castellazzi, M.; Morotti, A.; Tamborino, C.; Alessi, F.; Pilotto, S.; Baldi, E.; Caniatti, L.M.; Trentini, A.; Casetta, I.; Granieri, E.; et al. Increased age and male sex are independently associated with higher frequency of blood–cerebrospinal fluid barrier dysfunction using the albumin quotient. Fluids Barriers CNS 2020, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Skillbäck, T.; Blennow, K.; Zetterberg, H.; Shams, S.; Machado, A.; Pereira, J.; Lindberg, O.; Mielke, M.M.; Zettergren, A.; Ryden, L.; et al. Sex differences in CSF biomarkers for neurodegeneration and blood-brain barrier integrity. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2021, 13, e12141. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Fredman, P.; Wallin, A.; Gottfries, C.-G.; Karlsson, I.; Långstrom, G.; Skoog, I.; Svennerholm, L.; Wikkelsö, C. Protein Analysis in Cerebrospinal Fluid. Eur. Neurol. 1993, 33, 129–133. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Halliday, M.R.; Pomara, N.; Sagare, A.P.; Mack, W.J.; Frangione, B.; Zlokovic, B.V. Relationship Between Cyclophilin A Levels and Matrix Metalloproteinase 9 Activity in Cerebrospinal Fluid of Cognitively Normal Apolipoprotein E4 Carriers and Blood-Brain Barrier Breakdown. JAMA Neurol. 2013, 70, 1198–1200. [Google Scholar] [CrossRef]
- Janelidze, S.; Hertze, J.; Nägga, K.; Nilsson, K.; Nilsson, C.; Wennström, M.; van Westen, D.; Blennow, K.; Zetterberg, H.; Hansson, O. Increased blood-brain barrier permeability is associated with dementia and diabetes but not amyloid pathology or APOE genotype. Neurobiol. Aging 2017, 51, 104–112. [Google Scholar] [CrossRef]
- Jesse, S.; Brettschneider, J.; Süssmuth, S.D.; Landwehrmeyer, B.G.; Arnim, C.A.F.; Ludolph, A.C.; Tumani, H.; Otto, M. Summary of cerebrospinal fluid routine parameters in neurodegenerative diseases. J. Neurol. 2010, 258, 1034–1041. [Google Scholar] [CrossRef]
- Seyfert, S.; Faulstich, A.; Marx, P. What determines the CSF concentrations of albumin and plasma-derived IgG? J. Neurol. Sci. 2004, 219, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Stanyon, H.F.; Viles, J.H. Human Serum Albumin Can Regulate Amyloid-β Peptide Fiber Growth in the Brain Interstitium. J. Biol. Chem. 2012, 287, 28163–28168. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, C. Aβ40 Protects Non-toxic Aβ42 Monomer from Aggregation. J. Mol. Biol. 2007, 369, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Boada, M.; López, O.; Núñez, L.; Szczepiorkowski, Z.M.; Torres, M.; Grifols, C.; Páez, A. Plasma exchange for Alzheimer’s disease Management by Albumin Replacement (AMBAR) trial: Study design and progress. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Boada, M.; López, O.L.; Olazarán, J.; Núñez, L.; Pfeffer, M.; Puente, O.; Piñol-Ripoll, G.; Gámez, J.E.; Anaya, F.; Kiprov, D.; et al. Neuropsychological, neuropsychiatric, and quality-of-life assessments in Alzheimer’s disease patients treated with plasma exchange with albumin replacement from the randomized AMBAR study. Alzheimer’s Dement. 2021, 18, 1314–1324. [Google Scholar] [CrossRef]
- Sunderland, T.; Mirza, N.; Putnam, K.T.; Linker, G.; Bhupali, D.; Durham, R.; Soares, H.; Kimmel, L.; Friedman, D.; Bergeson, J.; et al. Cerebrospinal fluid β-amyloid1–42 and tau in control subjects at risk for Alzheimer’s disease: The effect of APOE ε4 allele. Biol. Psychiatry 2004, 56, 670–676. [Google Scholar] [CrossRef]
- Radanovic, M.; Oshiro, C.A.; Freitas, T.Q.; Talib, L.L.; Forlenza, O.V. Correlation between CSF biomarkers of Alzheimer’s disease and global cognition in a psychogeriatric clinic cohort. Rev. Bras. Psiquiatr. 2019, 41, 479–484. [Google Scholar] [CrossRef]
- Small, G.W.; Rosenthal, M.; Tourtellotte, W.W. Central Nervous System IgG Synthesis Rates in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 1994, 8, 29–37. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Yanase, D.; Noguchi-Shinohara, M.; Ono, K.; Yoshita, M.; Yamada, M. Cerebrospinal Fluid/Serum IgG Index Is Correlated with Medial Temporal Lobe Atrophy in Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2007, 25, 144–147. [Google Scholar] [CrossRef]
- Chiaravalloti, A.; Fiorentini, A.; Francesco, U.; Martorana, A.; Koch, G.; Belli, L.; Toniolo, S.; Di Pietro, B.; Motta, C.; Schillaci, O. Is cerebral glucose metabolism related to blood–brain barrier dysfunction and intrathecal IgG synthesis in Alzheimer disease? Medicine 2016, 95, e4206. [Google Scholar] [CrossRef]
- Ballerini, S.; Bellincampi, L.; Bernardini, S.; Casciani, S.; Motti, C.; Cortese, C.; Federici, G. Apolipoprotein E genotyping: A comparative study between restriction endonuclease mapping and allelic discrimination with the LightCycler. Clin. Chim. Acta 2001, 317, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Reiber, H.; Felgenhauer, K. Protein transfer at the blood cerebrospinal fluid barrier and the quantitation of the humoral immune response within the central nervous system. Clin. Chim. Acta 1987, 163, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal ATN (n = 57) | Alzheimer’s Pathologic Change (A+T-N-) (n = 21) | Alzheimer’s Disease (A+T+N-, A+T+N+) (n = 70) | Non-Alzheimer’s Pathologic Change (A-T+N-, A-T+N+) (n = 24) | p-Value | |
|---|---|---|---|---|---|
| Age, years | 68.3 ± 8.7 | 69.48 ± 7.9 | 71.9 ± 6.7 | 70.5 ± 8.2 | 0.085 |
| Sex (M/F ratio) | 0.49 | 0.29 | 0.51 | 0.41 | 0.290 |
| MMSE | 23.71 ± 4.7 | 24.5 ± 5.7 | 22.8 ± 5.2 | 25.1 ± 3.2 | 0.397 |
| ApoE status | 2.3 | 2.5 | 2.7 | 2.3 | 0.285 |
| CSF total-tau | 198.6 ± 65.1 | 247.8 ± 65.8 | 702.8 ± 397.0 | 511.0 ± 383.7 | <0.001 |
| CSF p-tau | 28.1 ± 8.7 | 37.7 ± 10.2 | 85.6 ± 36.8 | 59.7 ± 16.1 | <0.001 |
| CSF Aβ42 | 656.9 ± 223.3 | 519.2 ± 218.3 | 498.9 ± 197.4 | 872.6 ± 255.3 | <0.001 |
| CSF Aβ40 | 7287 ± 2863 | 10,280 ± 4148 | 13,270 ± 4312 | 11,694 ± 3937 | <0.001 |
| Aβ42/ 40 ratio | 0.09 ± 0.02 | 0.051 ± 0.01 | 0.039 ± 0.01 | 0.077 ± 0.02 | <0.001 |
| Normal ATN (n = 57) | Alzheimer’s Disease Pathologic Change (n = 21) | Alzheimer’s Disease (n = 70) | Non-Alzheimer’s Pathologic Change (n = 24) | p-Value | |
|---|---|---|---|---|---|
| CSF Protein | 51.1 ± 23.6 | 47.7 ± 12.1 | 48.9 ± 16.3 | 49.5 ± 26.1 | 0.085 |
| Serum Albumin | 4103 ± 448 | 4030 ± 642 | 4042 ± 443 | 4204 ± 360 | 0.897 |
| CSF Albumin | 29.5 ± 14.3 | 28.2 ± 9.5 | 28.0 ± 12.8 | 31.9 ± 18.5 | 0.465 |
| Qalb | 7.6 ± 4.4 | 7.2 ± 3.0 | 6.9 ± 3.0 | 7.6 ± 4.6 | 0.690 |
| Serum IgG | 944.7 ± 234 | 970.3 ± 256 | 960.0 ± 236 | 931.3 ± 196 | 0.931 |
| CSF IgG | 3.7 ± 3.1 | 3.2 ± 1.3 | 3.1 ± 1.4 | 3.4 ± 1.5 | 0.454 |
| IgG Index | 0.50 ± 0.1 | 0.48 ± 0.1 | 0.47 ± 0.0 | 0.49 ± 0.1 | 0.106 |
| RC1 | RC2 | RC3 | RC4 | RC5 | |
|---|---|---|---|---|---|
| T-tau | 0.832 | ||||
| P-tau | 0.926 | ||||
| Aβ40 | 0.813 | 0.448 | |||
| Aβ42 | 0.851 | ||||
| Aβ42/40 ratio | −0.768 | ||||
| MMSE | 0.497 | ||||
| ApoE status | −0.564 | ||||
| Sex | −0.499 | ||||
| Age | −0.701 | ||||
| Serum Albumin | 0.781 | ||||
| Serum IgG | 0.889 | ||||
| CSF Albumin | 0.936 | ||||
| CSF IgG | 0.836 | ||||
| CSF Protein | 0.899 | ||||
| IgG Index | −0.528 | ||||
| Qalb | 0.944 |
| RC1 | RC2 | RC3 | RC4 | RC5 | |
|---|---|---|---|---|---|
| RC1 | 1 | −0.751 | −0.919 | 0.483 | |
| RC2 | −0.751 | 1 | 0.680 | ||
| RC3 | 1 | ||||
| RC4 | −0.919 | 0.680 | 1 | −0.470 | |
| RC5 | 0.483 | −0.470 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toniolo, S.; Di Lorenzo, F.; Bernardini, S.; Mercuri, N.B.; Sancesario, G.M. Blood–Brain Barrier Dysfunction and Aβ42/40 Ratio Dose-Dependent Modulation with the ApoE Genotype within the ATN Framework. Int. J. Mol. Sci. 2023, 24, 12151. https://doi.org/10.3390/ijms241512151
Toniolo S, Di Lorenzo F, Bernardini S, Mercuri NB, Sancesario GM. Blood–Brain Barrier Dysfunction and Aβ42/40 Ratio Dose-Dependent Modulation with the ApoE Genotype within the ATN Framework. International Journal of Molecular Sciences. 2023; 24(15):12151. https://doi.org/10.3390/ijms241512151
Chicago/Turabian StyleToniolo, Sofia, Francesco Di Lorenzo, Sergio Bernardini, Nicola Biagio Mercuri, and Giulia Maria Sancesario. 2023. "Blood–Brain Barrier Dysfunction and Aβ42/40 Ratio Dose-Dependent Modulation with the ApoE Genotype within the ATN Framework" International Journal of Molecular Sciences 24, no. 15: 12151. https://doi.org/10.3390/ijms241512151
APA StyleToniolo, S., Di Lorenzo, F., Bernardini, S., Mercuri, N. B., & Sancesario, G. M. (2023). Blood–Brain Barrier Dysfunction and Aβ42/40 Ratio Dose-Dependent Modulation with the ApoE Genotype within the ATN Framework. International Journal of Molecular Sciences, 24(15), 12151. https://doi.org/10.3390/ijms241512151