

Inflammatory Bowel Diseases: An Updated Overview on the Heat Shock Protein Involvement



 ,
,  , and
, and
Abstract
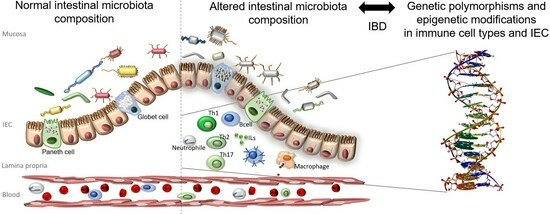
1. Introduction
2. Heat Shock Protein Expression in Healthy Gastrointestinal Mucosa and IBD Mucosa
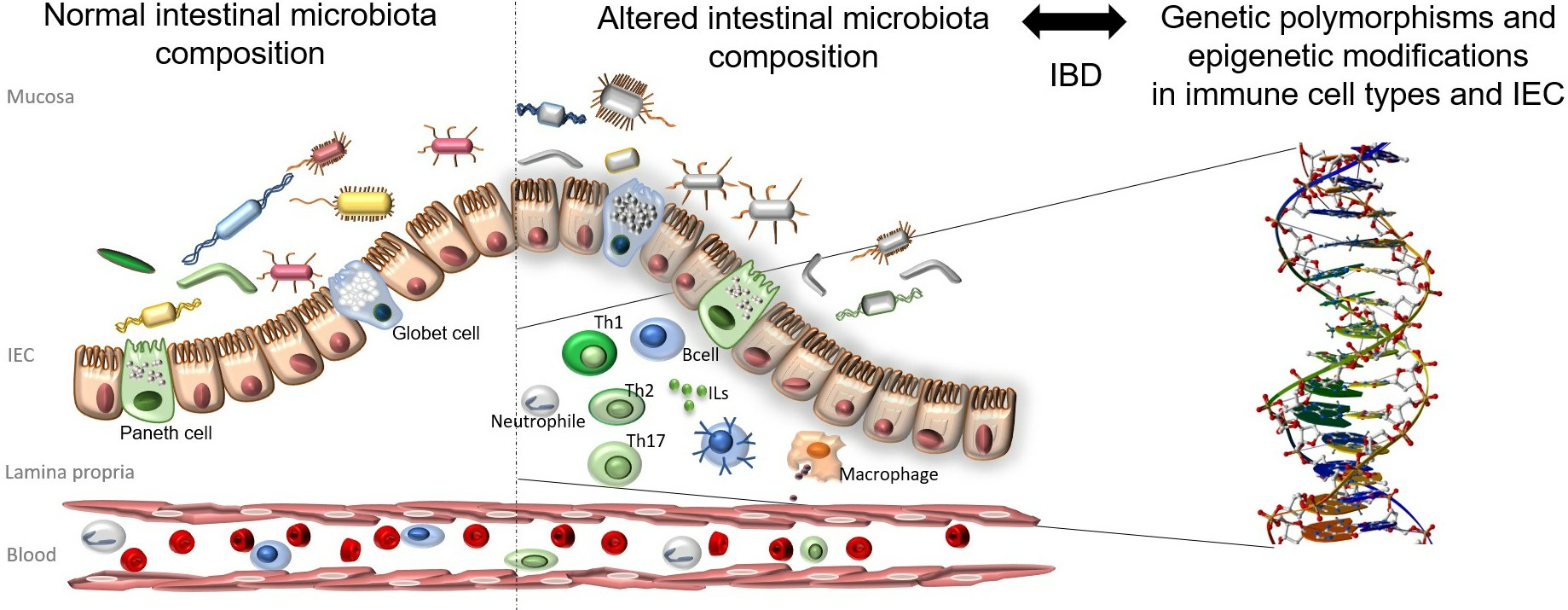
3. Genetic Alterations in IBD- and HSP-Related Genes
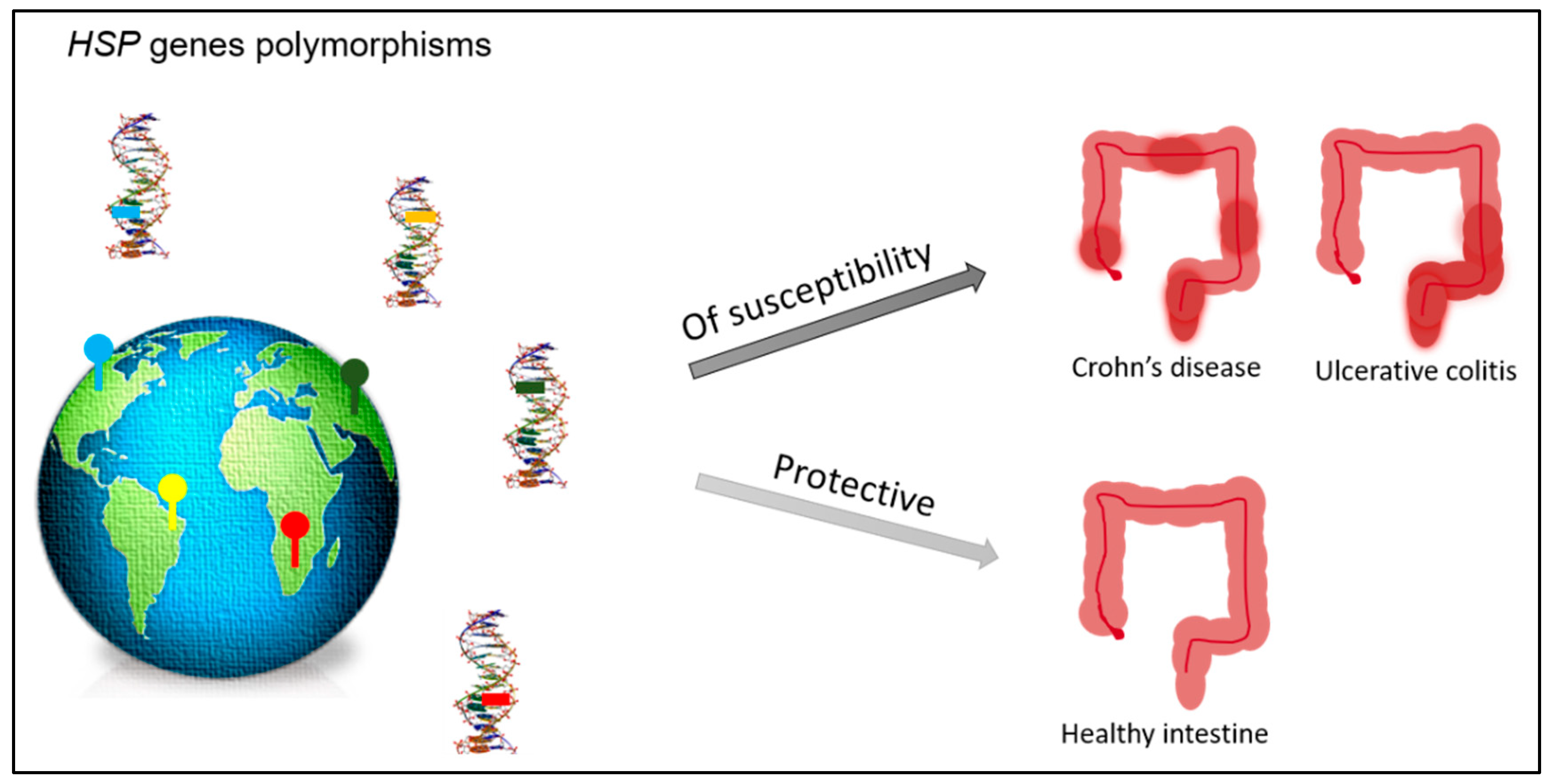
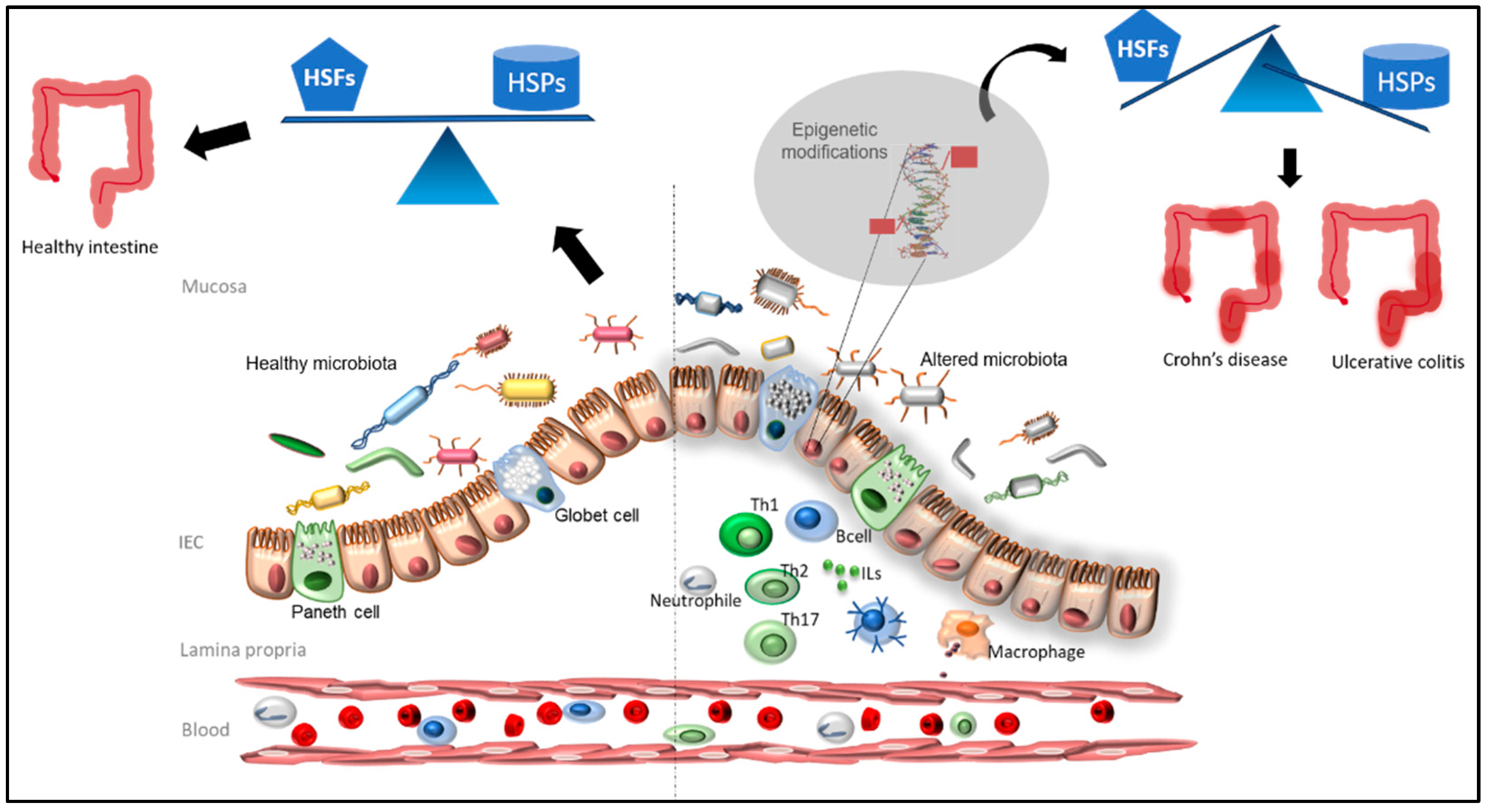
4. Epigenetic Alterations in IBD- and HSP-Related Proteins
5. Immunology and Inflammation in IBD and HSP Involvement
6. Involvement of HSPs in the Progression of IBD to Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chu, H. Host gene-microbiome interactions: Molecular mechanisms in inflammatory bowel disease. Genome Med. 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, M.; Carini, F.; Leone, A.; Damiani, P.; Jurjus, A.; Geagea, A.; Jurjus, R.; Assi Bou, T.; Trovato, E.; Rappa, F.; et al. Inflammatory bowel disease and colorectal cancer, nutraceutical aspects. Euromediterr. Biomed. J. 2016, 11, 123–129. [Google Scholar]
- Carini, F.; Gagliardo, C.; Mazzola, M.; Lo Presti, E.; Scaglione, M.; Jurjus, A.; Geagea, A.; Zerbe, R.; Leone, A.; Rappa, F.; et al. Immunological aspects of Crohn’s disease: A regulatory function of tim-3/galectin-9 in lyth1. Euromediterr. Biomed. J. 2017, 12, 66–68. [Google Scholar]
- El Menyiy, N.; El Allam, A.; Aboulaghras, S.; Jaouadi, I.; Bakrim, S.; El Omari, N.; Shariati, M.A.; Miftakhutdinov, A.; Wilairatana, P.; Mubarak, M.S.; et al. Inflammatory auto-immune diseases of the intestine and their management by natural bioactive compounds. Biomed. Pharmacother. 2022, 151, 113158. [Google Scholar] [CrossRef] [PubMed]
- Jodeleit, H.; Milchram, L.; Soldo, R.; Beikircher, G.; Schönthaler, S.; Al-Amodi, O.; Wolf, E.; Beigel, F.; Weinhäusel, A.; Siebeck, M.; et al. Autoantibodies as diagnostic markers and potential drivers of inflammation in ulcerative colitis. PLoS ONE 2020, 15, e0228615. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.; Lakatos, P.L.; Papp, M.; Jacobsen, S.; Nemes, E.; Polgar, M.; Solyom, E.; Bodi, P.; Horvath, A.; Muller, K.E.; et al. Pancreatic Autoantibodies and Autoantibodies Against Goblet Cells in Pediatric Patients With Inflammatory Bowel Disease. J. Pediatr. Gastr. Nutr. 2012, 55, 429–435. [Google Scholar] [CrossRef]
- Onuma, E.K.; Amenta, P.S.; Ramaswamy, K.; Lin, J.J.; Das, K.M. Autoimmunity in ulcerative colitis (UC): A predominant colonic mucosal B cell response against human tropomyosin isoform 5. Clin. Exp. Immunol. 2000, 121, 466–471. [Google Scholar] [CrossRef]
- Wen, Z.; Fiocchi, C. Inflammatory bowel disease: Autoimmune or immune-mediated pathogenesis? Clin. Dev. Immunol. 2004, 11, 195–204. [Google Scholar]
- Matricon, J.; Barnich, N.; Ardid, D. Immunopathogenesis of inflammatory bowel disease. Self/Nonself 2010, 1, 299–309. [Google Scholar] [CrossRef]
- Mazzola, M.; Carini, F.; Leone, A.; Damiani, P.; Messina, M.; Jurjus, A.; Geagea, G.; Jurjus, R.; Tomaselo, G. Ibd, malignancy and oral microbiota: Analysis of the literature. Int. J. Clin. Dent. 2016, 9, 273–278. [Google Scholar]
- Cappello, F.; Mazzola, M.; Jurjus, A.; Zeenny, M.N.; Jurjus, R.; Carini, F.; Leone, A.; Bonaventura, G.; Tomasello, G.; Bucchieri, F.; et al. Hsp60 as a Novel Target in IBD Management: A Prospect. Front. Pharmacol. 2019, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell. Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef]
- Macario, A.J.; de Macario, E.C. Molecular mechanisms in chaperonopathies: Clues to understanding the histopathological abnormalities and developing novel therapies. J. Pathol. 2020, 250, 9–18. [Google Scholar] [CrossRef]
- Gong, M.; Zhang, F.; Miao, Y.; Niu, J. Advances of Heat Shock Family in Ulcerative Colitis. Front. Pharmacol. 2022, 13, 869930. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E.; Cappello, F. The Chaperonopathies. Diseases with Defective Molecular Chaperones; Springer: Berlin/Heidelberg, Germany, 2013; ISBN 978-94-007-4666-4. [Google Scholar]
- Scalia, F.; Vitale, A.M.; Santonocito, R.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. The Neurochaperonopathies: Anomalies of the Chaperone System with Pathogenic Effects in Neurodegenerative and Neuromuscular Disorders. Appl. Sci. 2021, 11, 898. [Google Scholar] [CrossRef]
- Barbatis, C.; Tsopanomichalou, M. Heat shock proteins in inflammatory bowel disease. Ann. Gastroenterol. 2009, 22, 244–247. [Google Scholar]
- Dudeja, V.; Vickers, S.M.; Saluja, A.K. The role of heat shock proteins in gastrointestinal diseases. Gut 2009, 58, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.L.; Conway de Macario, E. Stress and molecular chaperones in disease. Int. J. Clin. Lab. Res. 2000, 30, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Nakamura, K.; Yoshii, A.; Yokoi, Y.; Kikuchi, M.; Shinozaki, R.; Nakamura, S.; Ohira, S.; Sugimoto, R.; Ayabe, T. Paneth cell alpha-defensin misfolding correlates with dysbiosis and ileitis in Crohn’s disease model mice. Life Sci. Alliance 2020, 3, e201900592. [Google Scholar] [CrossRef] [PubMed]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, J.Y.; Lee, H.C.; Ahn, J.B.; Kim, J.H.; Park, I.S.; Cheon, J.H.; Kim, D.H. Downregulation of Heat Shock Protein 72 Contributes to Fibrostenosis in Crohn’s Disease. Gut Liver, 2023; online ahead of print. [Google Scholar] [CrossRef]
- Ren, H.; Musch, M.W.; Kojima, K.; Boone, D.; Ma, A.; Chang, E.B. Short-chain fatty acids induce intestinal epithelial heat shock protein 25 expression in rats and IEC 18 cells. Gastroenterology 2001, 121, 631–639. [Google Scholar] [CrossRef]
- Miao, J.; Niu, J.; Wang, K.; Xiao, Y.; Du, Y.; Zhou, L.; Duan, L.; Li, S.; Yang, G.; Chen, L.; et al. Heat shock factor 2 levels are associated with the severity of ulcerative colitis. PLoS ONE 2014, 9, e88822. [Google Scholar] [CrossRef]
- Tanaka, K.; Tsutsumi, S.; Arai, Y.; Hoshino, T.; Suzuki, K.; Takaki, E.; Ito, T.; Takeuchi, K.; Nakai, A.; Mizushima, T. Genetic evidence for a protective role of heat shock factor 1 against irritant-induced gastric lesions. Mol. Pharmacol. 2007, 71, 985–993. [Google Scholar] [CrossRef]
- Tanaka, K.; Namba, T.; Arai, Y.; Fujimoto, M.; Adachi, H.; Sobue, G.; Takeuchi, K.; Nakai, A.; Mizushima, T. Genetic evidence for a protective role for heat shock factor 1 and heat shock protein 70 against colitis. J. Biol. Chem. 2007, 282, 23240–23252. [Google Scholar] [CrossRef]
- Hirakawa, T.; Rokutan, K.; Nikawa, T.; Kishi, K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology 1996, 111, 345–357. [Google Scholar] [CrossRef]
- Konturek, J.W.; Fischer, H.; Konturek, P.C.; Huber, V.; Boknik, P.; Luess, H.; Neumann, J.; Brzozowski, T.; Schmitz, W.; Hahn, E.G.; et al. Heat shock protein 70 (HSP70) in gastric adaptation to aspirin in Helicobacter pylori infection. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2001, 52, 153–164. [Google Scholar]
- Liu, W.L.; Chen, S.J.; Chen, Y.; Sun, L.M.; Zhang, W.; Zeng, Y.M.; Zhou, T.H.; Si, J.M. Protective effects of heat shock protein 70 induced by geranylgeranylacetone in atrophic gastritis in rats. Acta Pharmacol. Sin. 2007, 28, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Musch, M.W.; Ren, H.; Boone, D.L.; Hendrickson, B.A.; Ma, A.; Chang, E.B. Enteric flora and lymphocyte-derived cytokines determine expression of heat shock proteins in mouse colonic epithelial cells. Gastroenterology 2003, 124, 1395–1407. [Google Scholar] [CrossRef]
- Asea, A.; Rehli, M.; Kabingu, K.; Boch, J.A.; Baré, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef]
- Rodolico, V.; Tomasello, G.; Zerilli, M.; Martorana, A.; Pitruzzella, A.; Gammazza, A.M.; David, S.; Zummo, G.; Damiani, P.; Accomando, S.; et al. Hsp60 and Hsp10 increase in colon mucosa of Crohn’s disease and ulcerative colitis. Cell Stress Chaperones 2010, 15, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Ludwig, D.; Fellermann, K.; Stange, E.F. Intestinal expression of human heat shock protein 90 in patients with Crohn’s disease and ulcerative colitis. Dig. Dis. Sci. 1998, 43, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, G.; Sciumé, C.; Rappa, F.; Rodolico, V.; Zerilli, M.; Martorana, A.; Cicero, G.; De Luca, R.; Damiani, P.; Accardo, F.M.; et al. Hsp10, Hsp70, and Hsp90 immunohistochemical levels change in ulcerative colitis after therapy. Eur. J. Histochem. EJH 2011, 55, e38. [Google Scholar] [CrossRef]
- Honzawa, Y.; Nakase, H.; Shiokawa, M.; Yoshino, T.; Imaeda, H.; Matsuura, M.; Kodama, Y.; Ikeuchi, H.; Andoh, A.; Sakai, Y.; et al. Involvement of interleukin-17A-induced expression of heat shock protein 47 in intestinal fibrosis in Crohn’s disease. Gut 2014, 63, 1902–1912. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kennedy, N.A.; Nimmo, E.R.; Satsangi, J. Beyond gene discovery in inflammatory bowel disease: The emerging role of epigenetics. Gastroenterology 2013, 145, 293–308. [Google Scholar] [CrossRef]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef]
- Annese, V. Genetics and epigenetics of IBD. Pharmacol. Res. 2020, 159, 104892. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Ha, C. Epidemiology and Pathogenesis of Ulcerative Colitis. Gastroenterol. Clin. N. Am. 2020, 49, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Diaz Pena, R.; Valdes, E.; Cofre, C.; Castro-Santos, P. Th17 response and autophagy–main pathways implicated in the development of inflammatory bowel disease by genome-wide association studies. Rev. Esp. Enferm. Dig. 2015, 107, 559–565. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tahara, T.; Shibata, T.; Arisawa, T.; Nakamura, M.; Yoshioka, D.; Okubo, M.; Maruyama, N.; Kamano, T.; Kamiya, Y.; Fujita, H.; et al. The BB genotype of heat-shock protein (HSP) 70-2 gene is associated with gastric pre-malignant condition in H. pylori-infected older patients. Anticancer Res. 2009, 29, 3453–3458. [Google Scholar]
- Chen, J.; Ren, J.; Gu, G.; Wang, G.; Wu, X.; Yan, D.; Liu, S.; Li, J. Crohn’s disease and polymorphism of heat shock protein gene HSP70-2 in the Chinese population. J. Gastroenterol. Hepatol. 2013, 28, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Zouiten-Mekki, L.; Karoui, S.; Kharrat, M.; Fekih, M.; Matri, S.; Boubaker, J.; Filali, A.; Chaabouni, H. Crohn’s disease and polymorphism of heat shock protein gene HSP70-2 in the Tunisian population. Eur. J. Gastroenterol. Hepatol. 2007, 19, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.Y.; Kim, N.; Kim, J.S.; Lim, S.H.; Jung, H.C.; Song, I.S. Heat shock protein gene 70-2 polymorphism is differentially associated with the clinical phenotypes of ulcerative colitis and Crohn’s disease. J. Gastroenterol. Hepatol. 2007, 22, 1032–1038. [Google Scholar] [CrossRef]
- Lu, W.G.; Zou, Y.F.; Feng, X.L.; Yuan, F.L.; Gu, Y.L.; Li, X.; Li, C.W.; Jin, C.; Li, J.P. Association of NOD1 (CARD4) insertion/deletion polymorphism with susceptibility to IBD: A meta-analysis. World J. Gastroenterol. 2010, 16, 4348–4356. [Google Scholar] [CrossRef]
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 2011, 60, 1580–1588. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Dietrich, G.; Hugot, J.P.; Barreau, F. Nod2: The intestinal gate keeper. PLoS Pathog. 2017, 13, e1006177. [Google Scholar] [CrossRef]
- Cavanaugh, J. NOD2: Ethnic and geographic differences. World. J. Gastroenterol. 2006, 12, 3673–3677. [Google Scholar] [CrossRef]
- Mohanan, V.; Grimes, C.L. The molecular chaperone HSP70 binds to and stabilizes NOD2, an important protein involved in Crohn disease. J. Biol. Chem. 2014, 289, 18987–18998. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.F.; Xu, J.H.; Gu, Y.Y.; Pan, F.M.; Tao, J.H.; Wang, D.G.; Xu, S.Q.; Xiao, H.; Chen, P.L.; Liu, S.; et al. Single nucleotide polymorphisms of HSP90AA1 gene influence response of SLE patients to glucocorticoids treatment. Springerplus 2016, 5, 222. [Google Scholar] [CrossRef]
- Yin, Y.; Wan, J.; Yu, J.; Wu, K. Molecular Pathogenesis of Colitis-associated Colorectal Cancer: Immunity, Genetics, and Intestinal Microecology. Inflamm. Bowel Dis. 2023, izad081. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Sun, D.; Chen, Y.; Fang, J.Y. Influence of the microbiota on epigenetics in colorectal cancer. Natl. Sci. Rev. 2019, 6, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Woo, V.; Alenghat, T. Epigenetic regulation by gut microbiota. Gut Microbes 2022, 14, 2022407. [Google Scholar] [CrossRef] [PubMed]
- Miro-Blanch, J.; Yanes, O. Epigenetic Regulation at the Interplay Between Gut Microbiota and Host Metabolism. Front. Genet. 2019, 10, 638. [Google Scholar] [CrossRef]
- Shock, T.; Badang, L.; Ferguson, B.; Martinez-Guryn, K. The interplay between diet, gut microbes, and host epigenetics in health and disease. J. Nutr. Biochem. 2021, 95, 108631. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kennedy, N.A.; Adams, A.T.; Kalla, R.; Heath, S.; O’Leary, K.R.; Drummond, H.; IBD BIOM consortium; IBD CHARACTER consortium; Wilson, D.C.; et al. Integrative epigenome-wide analysis demonstrates that DNA methylation may mediate genetic risk in inflammatory bowel disease. Nat. Commun. 2016, 7, 13507. [Google Scholar] [CrossRef]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury. PLoS ONE 2013, 8, e66814. [Google Scholar] [CrossRef] [PubMed]
- Vieujean, S.; Caron, B.; Haghnejad, V.; Jouzeau, J.Y.; Netter, P.; Heba, A.C.; Ndiaye, N.C.; Moulin, D.; Barreto, G.; Danese, S.; et al. Impact of the Exposome on the Epigenome in Inflammatory Bowel Disease Patients and Animal Models. Int. J. Mol. Sci. 2022, 23, 7611. [Google Scholar] [CrossRef]
- Kalla, R.; Adams, A.T.; Nowak, J.K.; Bergemalm, D.; Vatn, S.; Ventham, N.T.; Kennedy, N.A.; Ricanek, P.; Lindstrom, J.; IBD-Character Consortium; et al. Analysis of Systemic Epigenetic Alterations in Inflammatory Bowel Disease: Defining Geographical, Genetic and Immune-Inflammatory influences on the Circulating Methylome. J. Crohn’s Colitis 2023, 17, 170–184. [Google Scholar] [CrossRef]
- Hou, H.W.; Wang, J.M.; Wang, D.; Wu, R.; Ji, Z.L. Triptolide exerts protective effects against fibrosis following ileocolonic anastomosis by mechanisms involving the miR-16-1/HSP70 pathway in IL-10-deficient mice. Int. J. Mol. Med. 2017, 40, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yang, M.F.; Liang, Y.J.; Xu, J.; Xu, H.M.; Nie, Y.Q.; Wang, L.S.; Yao, J.; Li, D.F. Immunology of Inflammatory Bowel Disease: Molecular Mechanisms and Therapeutics. J. Inflamm. Res. 2022, 15, 1825–1844. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.Y.; Boivin, M.A.; Ye, D.; Pedram, A.; Said, H.M. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: Role of myosin light-chain kinase protein expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G422–G430. [Google Scholar] [CrossRef]
- Hoter, A.; Naim, H.Y. The Functions and Therapeutic Potential of Heat Shock Proteins in Inflammatory Bowel Disease-An Update. Int. J. Mol. Sci. 2019, 20, 5331. [Google Scholar] [CrossRef]
- Kurumi, H.; Takata, T.; Kanda, T.; Sugihara, T.; Kakugawa, T.; Yokota, S.I.; Morisaki, T.; Akashi, T.; Isomoto, H. Investigating the role of heat shock protein 47 in fibrosis in Crohn’s disease. Sci. Rep. 2022, 12, 10966. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.M.; Kim, S.H.; Kim, E.H. The molecular mechanism of transforming growth factor-beta signaling for intestinal fibrosis: A mini-review. Front. Pharmacol. 2019, 10, 162. [Google Scholar] [CrossRef]
- Kitamura, H.; Yamamoto, S.; Nakase, H.; Matsuura, M.; Honzawa, Y.; Matsumura, K.; Takeda, Y.; Uza, N.; Nagata, K.; Chiba, T. Role of heat shock protein 47 in intestinal fibrosis of experimental colitis. Biochem. Biophys. Res. Commun. 2011, 404, 599–604. [Google Scholar] [CrossRef]
- Dulle, J.E.; Fort, P.E. Crystallins and neuroinflammation: The glial side of the story. Biochim. Biophys. Acta 2016, 1860 Pt B, 278–286. [Google Scholar] [CrossRef]
- Scalia, F.; Barone, R.; Rappa, F.; Marino Gammazza, A.; Lo Celso, F.; Lo Bosco, G.; Barone, G.; Antona, V.; Vadalà, M.; Vitale, A.M.; et al. Muscle Histopathological Abnormalities in a Patient With a CCT5 Mutation Predicted to Affect the Apical Domain of the Chaperonin Subunit. Front. Mol. Biosci. 2022, 9, 887336. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.S.; Liang, P.Z.; Lu, S.Z.; Chen, R.; Yin, Y.Q.; Zhou, J.W. Extracellular αB-crystallin modulates the inflammatory responses. Biochem. Biophys. Res. Commun. 2019, 508, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Guo, Y.; Huang, Z.; Zhao, H.; Zhou, M.; Huang, Y.; Wen, D.; Song, J.; Zhu, Z.; Sun, M.; et al. Small heat shock protein CRYAB inhibits intestinal mucosal inflammatory responses and protects barrier integrity through suppressing IKKβ activity. Mucosal Immunol. 2019, 12, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Woznicki, J.A.; Saini, N.; Flood, P.; Rajaram, S.; Lee, C.M.; Stamou, P.; Skowyra, A.; Bustamante-Garrido, M.; Regazzoni, K.; Crawford, N.; et al. TNF-alpha synergises with IFN-gamma to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis. 2021, 12, 864. [Google Scholar] [CrossRef] [PubMed]
- Ousman, S.S.; Tomooka, B.H.; van Noort, J.M.; Wawrousek, E.F.; O’Connor, K.C.; Hafler, D.A.; Sobel, R.A.; Robinson, W.H.; Steinman, L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 2007, 448, 474–479. [Google Scholar] [CrossRef]
- Sato, S.; Oka, M.; Noguchi, Y.; Soda, H.; Tsurutani, J.; Nakamura, Y.; Kitazaki, T.; Mizuta, Y.; Takeshima, F.; Murase, K.; et al. Autoimmunity to heat shock protein 40 in ulcerative colitis. J. Int. Med. Res. 2004, 32, 141–148, Erratum in J. Int. Med. Res. 2004, 32, 341. [Google Scholar] [CrossRef]
- Stevens, T.R.; Winrow, V.R.; Blake, D.R.; Rampton, D.S. Circulating antibodies to heat-shock protein 60 in Crohn’s disease and ulcerative colitis. Clin. Exp. Immunol. 1992, 90, 271–274. [Google Scholar] [CrossRef]
- Chatila, W.K.; Walch, H.; Hechtman, J.F.; Moyer, S.M.; Sgambati, V.; Faleck, D.M.; Srivastava, A.; Tang, L.; Benhamida, J.; Ismailgeci, D.; et al. Integrated clinical and genomic analysis identifies driver events and molecular evolution of colitis-associated cancers. Nat. Commun. 2023, 14, 110. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Ruan, Q.; Han, S.; Jiang, W.G.; Boulton, M.E.; Chen, Z.J.; Law, B.K.; Cai, J. alphaB-crystallin, an effector of unfolded protein response, confers anti-VEGF resistance to breast cancer via maintenance of intracrine VEGF in endothelial cells. Mol. Cancer Res. 2011, 9, 1632–1643. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Lai, Y.; Xu, P.; Yang, Z. HspB5 correlates with poor prognosis in colorectal cancer and prompts epithelial-mesenchymal transition through ERK signaling. PLoS ONE 2017, 12, e0182588. [Google Scholar] [CrossRef]
- Li, J.; Song, P.; Jiang, T.; Dai, D.; Wang, H.; Sun, J.; Zhu, L.; Xu, W.; Feng, L.; Shin, V.Y.; et al. Heat Shock Factor 1 Epigenetically Stimulates Glutaminase-1-Dependent mTOR Activation to Promote Colorectal Carcinogenesis. Mol. Ther. 2018, 26, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Levi-Galibov, O.; Lavon, H.; Wassermann-Dozorets, R.; Pevsner-Fischer, M.; Mayer, S.; Wershof, E.; Stein, Y.; Brown, L.E.; Zhang, W.; Friedman, G.; et al. Heat Shock Factor 1-dependent extracellular matrix remodeling mediates the transition from chronic intestinal inflammation to colon cancer. Nat. Commun. 2020, 11, 6245. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Role | IBD | CD | UC |
|---|---|---|---|
| IL23/TH17 pathway | IL23R, JAK2, TYK2, ICOSLG, TNFSF15 | STAT3 | IL21 |
| Autophagy | CUL2 | ATG16L1, IRGM, NOD2, LRRK2 | PARK7, DAP |
| T-cell regulation | TNFSF8, IL12B, IL23, PRDM1, ICOSLG | NDFIP1, TAGAP, IL2R | tNFRSF9, PIM3, IL/R, TNFSF8, IGNG, IL23 |
| Innate mucosal defense | CARD 9, RER | NOD2, ITLN1 | SLC11A1, FGR2a/B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalia, F.; Carini, F.; David, S.; Giammanco, M.; Mazzola, M.; Rappa, F.; Bressan, N.I.; Maida, G.; Tomasello, G. Inflammatory Bowel Diseases: An Updated Overview on the Heat Shock Protein Involvement. Int. J. Mol. Sci. 2023, 24, 12129. https://doi.org/10.3390/ijms241512129
Scalia F, Carini F, David S, Giammanco M, Mazzola M, Rappa F, Bressan NI, Maida G, Tomasello G. Inflammatory Bowel Diseases: An Updated Overview on the Heat Shock Protein Involvement. International Journal of Molecular Sciences. 2023; 24(15):12129. https://doi.org/10.3390/ijms241512129
Chicago/Turabian StyleScalia, Federica, Francesco Carini, Sabrina David, Marco Giammanco, Margherita Mazzola, Francesca Rappa, Noemi Irma Bressan, Giorgio Maida, and Giovanni Tomasello. 2023. "Inflammatory Bowel Diseases: An Updated Overview on the Heat Shock Protein Involvement" International Journal of Molecular Sciences 24, no. 15: 12129. https://doi.org/10.3390/ijms241512129
APA StyleScalia, F., Carini, F., David, S., Giammanco, M., Mazzola, M., Rappa, F., Bressan, N. I., Maida, G., & Tomasello, G. (2023). Inflammatory Bowel Diseases: An Updated Overview on the Heat Shock Protein Involvement. International Journal of Molecular Sciences, 24(15), 12129. https://doi.org/10.3390/ijms241512129