

Transgenic East African Highland Banana Plants Are Protected against Radopholus similis through Host-Delivered RNAi

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Effect of dsRNA on Multiplication of Radopholus similis on Carrot Discs
2.2. RT-PCR Analysis of the Soaked Nematodes
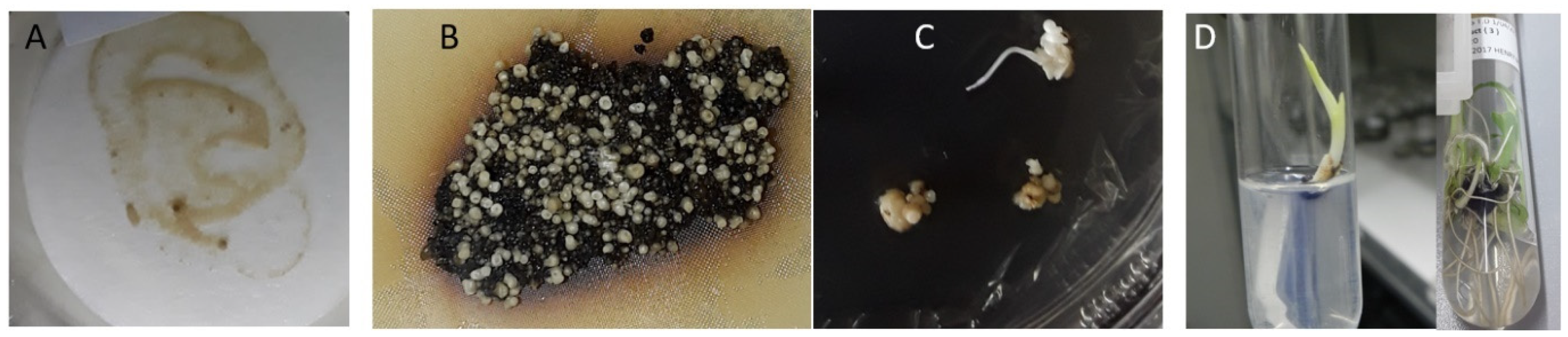
2.3. Generation of Transgenic Banana with Hairpin Constructs to R. similis Genes
2.4. Necrosis and Nematode Multiplication in dsRNA Plants
3. Discussion
4. Materials and Methods
4.1. Nematode Population
4.2. Culture of R. similis on Carrot Discs and Extraction of Nematodes
4.3. Target Gene Selection
4.4. RNA Isolation, cDNA Synthesis and Cloning Targets
4.4.1. RNA Isolation and cDNA Synthesis
4.4.2. Generation of Vectors for the Production of dsRNA
4.4.3. Generation of Vectors for Plant Transformation
4.5. RNAi of R. similis by Soaking in dsRNA
4.5.1. Synthesis and Isolation of dsRNA from Bacteria
4.5.2. Preparation of Mini Carrot Discs and Soaking Solution
4.5.3. Soaking Procedure
4.5.4. Analysis of R. similis Reproduction on Mini Carrot Discs
4.5.5. RT-PCR for Expression Analysis after Soaking
4.6. Generation and Testing of Transgenic Banana Plants with RNAi against R. similis
4.6.1. Generation of the Agrobacterium Strains and Culture
4.6.2. Transformation with Agrobacterium
4.6.3. PCR Analysis of Putatively Transformed Banana Plantlets
4.6.4. Preparation of Plants for Infection Tests
4.6.5. Infection Procedure
4.6.6. Assessing Damage and Nematode Reproduction
Assessing Root Damage
Extraction of Nematodes from Roots
Extraction of Nematodes from Soil
Characterization and Counting of Nematodes
Statistical Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pegg, K.; Moore, N.; Bentley, S. Fusarium wilt of banana in Australia: A review. Aust. J. Agric. Res. 1996, 47, 637–650. [Google Scholar]
- Marin, D.H.; Romero, R.A.; Guzman, M.; Sutton, T.B. Black Sigatoka: An increasing threat to banana cultivation. Plant Dis. 2003, 87, 208–222. [Google Scholar]
- Kalyebara, M.; Ragama, P.; Kikulwe, E.; Bagamba, F.; Nankinga, K.; Tushemereirwe, W. Economic importance of the banana bacterial wilt in Uganda. Afr. Crop Sci. J. 2006, 14, 93–103. [Google Scholar] [CrossRef]
- Daniells, J.; Geering, A.; Bryde, N.J.; Thomas, J.E. The effect of Banana streak virus on the growth and yield of dessert banana in tropical Australia. Ann. Appl. Biol. 2001, 139, 51–60. [Google Scholar] [CrossRef]
- Gold, C.; Kagezi, G.; Night, G.; Ragama, P. The effects of banana weevil, Cosmopolites sordidus, damage on highland banana growth, yield and stand duration in Uganda. Ann. Appl. Biol. 2004, 145, 263–269. [Google Scholar] [CrossRef]
- Davide, R. Overview of nematodes as a limiting factor in Musa production. In New Frontiers in Resistance Breeding for Nematode, Fusarium and Sigatoka; Frison, E.A., Horry, J.P., De Waele, D., Eds.; INIBAP: Montpellier, France, 1996; pp. 27–31. [Google Scholar]
- Kleynhans, K.; Berg, E.; Swart, A.; Marais, M.; Buckley, N. Plant Nematodes in South Africa; ARC-Plant Protection Research Institute: Pretoria, South Africa, 1996. [Google Scholar]
- Cobb, N.A. Nematodes, Mostly Australian and Fijian; F. Cunninghame & Company, Printers: Sydney, Australia, 1893. [Google Scholar]
- Thorne, G. On the classification of the Tylenchida, new order (Nematoda, Phasmidia). Proc. Helminthol. Soc. Wash. 1949, 16, 37–73. [Google Scholar]
- De Waele, D.; Davide, R.G. The Root-Knot Nematodes of Banana; INIBAP: Montpellier, France, 1998. [Google Scholar]
- Bridge, J.; Fogain, R.; Speijer, P.R. The root lesion nematodes of banana: Pratylenchus coffeae (Zimmermann, 1898) Filip. & Schu. Stek., 1941, Pratylenchus goodeyi Sher & Allen, 1953. 1997. Musa Pest Fact Sheet. Available online: https://hdl.handle.net/10568/105397 (accessed on 25 July 2023).
- McSorley, R.; Parrado, J. Nematological reviews: Helicotylenchus multicinctus on bananas: An international problem. Nematropica 1986, 16, 73–91. [Google Scholar]
- Barekye, A.; Kashaija, I.N.; Tushemereirwe, W.K.; Adipala, E. Comparison of damage levels caused by Radopholus similis and Helicotylenchus multicinctus on bananas in Uganda. Ann. Appl. Biol. 2000, 137, 273–278. [Google Scholar] [CrossRef]
- Sarah, J.; Pinochet, J.; Stanton, J. The Burrowing Nematode of Bananas, Radopholus Similis Cobb, 1913; Musa Pest Fact Sheet (INIBAP): Montpellier, France, 1996. [Google Scholar]
- Gowen, S.; Quénéhervé, P.; Fogain, R. Nematode parasites of bananas and plantains. Plant Parasit. Nematodes Subtrop. Trop. Agric. 2005, 2, 611–643. [Google Scholar]
- Leach, R. Blackhead Toppling Disease of Bananas. Nature 1958, 181, 204–205. [Google Scholar] [CrossRef]
- Price, N. The banana burrowing nematode, Radopholus similis (Cobb) Thorne, in the Lake Victoria region of East Africa: Its introduction, spread and impact. Nematology 2006, 8, 801–817. [Google Scholar]
- Tabora, P.; Shintani, M.; Elango, F. Banana researches in Costa Rica (Central America) with Effective Microorganisms. Available online: http://usi.earth.ac.cr/glas/sp/50000055.PDF (accessed on 25 July 2023).
- Daneel, M.; De Waele, D. Nematode pests of banana. In Nematology in South Africa: A View from the 21st Century; Fourie, H., Spaull, V.W., Jones, R.K., Daneel, M.S., De Waele, D., Eds.; Springer International: Cham, Switzerland, Chapter 16; pp. 359–371.
- Willmott, S.; Gooch, P.S.; Siddiqi, M.R.; Franklin, M.T. CIH Descriptions of Plant-Parasitic Nematodes; Commonwealth Agricultural Bureaux: Wallingford, UK, 1972. [Google Scholar]
- Fogain, R. Effect of Radopholus similis on plant growth and yield of plantains (Musa, AAB). Nematology 2000, 2, 129–133. [Google Scholar] [CrossRef]
- Sarah, J.L. Nematological review: Banana nematodes and their control in Africa. Nematropica 1989, 19, 199–216. [Google Scholar]
- Blake, C. Root rot of bananas caused by Radopholus similis (Cobb) and its control in New South Wales. Nematologica 1961, 6, 295–310. [Google Scholar] [CrossRef]
- Boncato, A.; Davide, R. Radopholus similis on Cavendish banana in Davao del Norte. 1. Host range and relative distribution and density. Philipp. Agric. 1980, 63, 111–119. [Google Scholar]
- Haegeman, A.; Elsen, A.; De Waele, D.; Gheysen, G. Emerging molecular knowledge on Radopholus similis, an important nematode pest of banana. Mol. Plant Pathol. 2010, 11, 315. [Google Scholar] [CrossRef]
- Mathew, R.; Opperman, C.H. The genome of the migratory nematode, Radopholus similis, reveals signatures of close association to the sedentary cyst nematodes. PLoS ONE 2019, 14, e0224391. [Google Scholar] [CrossRef] [PubMed]
- Fallas, G.; Sarah, J.L. Effect of temperature on the in vitro multiplication of seven Radopholus similis isolates from different banana producing zones of the world. Fundam. Appl. Nematol. 1995, 18, 445–449. [Google Scholar]
- Speijer, P.R.; De Waele, D. Screening of Musa Germplasm for Resistance and Tolerance to Nematodes; INIBAP. 1997. INIBAP Technical Guidelines. 1. Screening of Musa Germplasm for Resistance and Tolerance to Nematodes; INIBAP: Montpellier, France, 1997. [Google Scholar]
- Brooks, F. Symptoms and Signs. In Phytopathology News; American Phytopathological Society: St. Paul, MN, USA, 2021. [Google Scholar]
- Duncan, L.; Moens, M. Migratory endoparasitic nematodes. In Plant Nematology; CABI: Wallingford, UK, 2006; pp. 123–152. [Google Scholar]
- Speijer, P.R.; De Waele, D. Nematodes associated with East African highland cooking bananas and cv. Pisang Awak (Musa spp.) in Central Uganda. Nematology 2001, 3, 535–541. [Google Scholar]
- O’Bannon, J.H. Worldwide dissemination of Radopholus similis and its importance in crop production. J. Nematol. 1977, 9, 16–25. [Google Scholar]
- Bjornlund, V.; Bjornlund, H.; Van Rooyen, A.F. Why agricultural production in sub-Saharan Africa remains low compared to the rest of the world–a historical perspective. Int. J. Water Resour. Dev. 2020, 36, S20–S53. [Google Scholar] [CrossRef]
- Kalyebara, R.; Wood, S.; Abodi, P. The potential economic benefits of improved banana productivity in Uganda: An industry scale analysis. IFRI BRIEF 2005, 9. Available online: https://ussp.ifpri.info/files/2011/10/brief9_banana_meso_scale_economic_impacts.pdf (accessed on 25 July 2023).
- Risède, J.-M.; Chabrier, C.; Dorel, M.; Dambas, T.; Achard, R.; Quénéhervé, P. Integrated Management of Banana Nematodes: Lessons from a Case Study in the French West Indies. 2010. Available online: https://agritrop.cirad.fr/553878/1/document_553878.pdf (accessed on 25 July 2023).
- Ventura, W.; Watanabe, I.; Castillo, M.B.; De la Cruz, A. Involvement of nematodes in the soil sickness of a dryland rice-based cropping system. Soil Sci. Plant Nutr. 1981, 27, 305–315. [Google Scholar] [CrossRef]
- Edwards, D.I.; Wehunt, E.J. Host range of Radopholus similis from banana areas of Central America with indications of additional races. Plant Dis. Report. 1971, 55, 415–418. [Google Scholar]
- Nematodes Parasites of Citrus. In Plant and Insect Nematodes; Tarjan, A., O’bannon, J., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1984. [Google Scholar]
- Gowen, S.; Quénéhervé, P. Nematode parasites of bananas, plantains and Abaca. In Plant Parasitic Nematodes in Subtropical and Tropical Agriculture; CAB International: Wallingford, UK, 1990; pp. 431–460. [Google Scholar]
- Román, J.; Rivas, X.; Oramas, D.; Rodríguez, J. Further experiments on the chemical control of nematodes in plantains (Musa acuminata x M. Balbisiana, AAB). J. Agric. Univ. Puerto Rico 1977, 61, 192–199. [Google Scholar]
- Mekuria, W. The link between agricultural production and population dynamics in Ethiopia: A review. Adv. Plants Agric. Res. 2018, 8, 348–353. [Google Scholar]
- Hardaker, J.B. Guidelines for the Integration of Sustainable Agriculture and Rural Development into Agricultural Policies; Food & Agriculture Org.: Rome, Italy, 1997. [Google Scholar]
- Marin, D.H.; Sutton, T.B.; Barker, K.R. Dissemination of Bananas in Latin America and the Caribbean and Its Relationship to the Occurrence of Radopholus similis. Plant Dis. 1998, 82, 964–974. [Google Scholar] [PubMed]
- Bridge, J. Nematode Management in Sustainable and Subsistence Agriculture. Annu. Rev. Phytopathol. 1996, 34, 201–225. [Google Scholar] [CrossRef]
- Becker, D.K.; Dugdale, B.; Smith, M.K.; Harding, R.M.; Dale, J.L. Genetic transformation of Cavendish banana (Musa spp. AAA group) cv ‘Grand Nain’ via microprojectile bombardment. Plant Cell Rep. 2000, 19, 229–234. [Google Scholar] [CrossRef]
- Roderick, H.; Tripathi, L.; Babirye, A.; Wang, D.; Tripathi, J.; Urwin, P.E.; Atkinson, H.J. Generation of transgenic plantain (Musa spp.) with resistance to plant pathogenic nematodes. Mol. Plant Pathol. 2012, 13, 842–851. [Google Scholar] [CrossRef]
- Tripathi, L.; Tripathi, J.N.; Roderick, H.; Atkinson, H.J. Engineering Nematode Resistant Plantains for Sub-Saharan Africa 2013; International Society for Horticultural Science (ISHS): Leuven, Belgium, 2013. [Google Scholar]
- Yaqoob, A.; Shahid, A.A.; Samiullah, T.R.; Rao, A.Q.; Khan, M.A.U.; Tahir, S.; Mirza, S.A.; Husnain, T. Risk assessment of Bt crops on the non-target plant-associated insects and soil organisms. J. Sci. Food Agric. 2016, 96, 2613–2619. [Google Scholar] [PubMed]
- Casacuberta, J.M.; Devos, Y.; Du Jardin, P.; Ramon, M.; Vaucheret, H.; Nogué, F. Biotechnological uses of RNAi in plants: Risk assessment considerations. Trends Biotechnol. 2015, 33, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Induction and suppression of RNA silencing: Insights from viral infections. Nat. Rev. Genet. 2005, 6, 206–220. [Google Scholar] [PubMed]
- Sidahmed, A.M.; Wilkie, B. Endogenous antiviral mechanisms of RNA interference: A comparative biology perspective. Methods Mol. Biol. 2010, 623, 3–19. [Google Scholar]
- Dubelman, S.; Fischer, J.; Zapata, F.; Huizinga, K.; Jiang, C.; Uffman, J.; Levine, S.; Carson, D. Environmental Fate of Double-Stranded RNA in Agricultural Soils. PLoS ONE 2014, 9, e93155. [Google Scholar] [CrossRef]
- Fischer, J.R.; Zapata, F.; Dubelman, S.; Mueller, G.M.; Uffman, J.P.; Jiang, C.; Jensen, P.D.; Levine, S.L. Aquatic fate of a double-stranded RNA in a sediment–water system following an over-water application. Environ. Toxicol. Chem. 2017, 36, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.S.; Brower-Toland, B.; Jackson, A.L.; Kier, L.D. Safety assessment of food and feed from biotechnology-derived crops employing RNA-mediated gene regulation to achieve desired traits: A scientific review. Regul. Toxicol. Pharmacol. 2013, 66, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Fosu-Nyarko, J.; Jones, M.G.K. Attempt to Silence Genes of the RNAi Pathways of the Root-Knot Nematode, Meloidogyne incognita Results in Diverse Responses Including Increase and No Change in Expression of Some Genes. Front. Plant Sci. 2020, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.; Cox, P.; Shepherd, A. The chemical composition of the egg shells of the potato cyst-nematode, Heterodera rostochiensis Woll. Biochem. J. 1967, 104, 1056–1060. [Google Scholar] [CrossRef][Green Version]
- Fanelli, E.; Di Vito, M.; Jones, J.T.; De Giorgi, C. Analysis of chitin synthase function in a plant parasitic nematode, Meloidogyne artiellia, using RNAi. Gene 2005, 349, 87–95. [Google Scholar] [CrossRef]
- Kong, L.; Shi, X.; Chen, D.; Yang, N.; Yin, C.; Yang, J.; Wang, G.; Huang, W.; Peng, H.; Peng, D.; et al. Host-induced silencing of a nematode chitin synthase gene enhances resistance of soybeans to both pathogenic Heterodera glycines and Fusarium oxysporum. Plant Biotechnol. J. 2022, 20, 809–811. [Google Scholar] [CrossRef]
- Smardon, A.; Spoerke, J.M.; Stacey, S.C.; Klein, M.E.; Mackin, N.; Maine, E.M. EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr. Biol. 2000, 10, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Haegeman, A.; Jacob, J.; Vanholme, B.; Kyndt, T.; Gheysen, G. A family of GHF5 endo-1,4-beta-glucanases in the migratory plant-parasitic nematode Radopholus similis. Plant Pathol. 2008, 57, 581–590. [Google Scholar] [CrossRef]
- Peng, H.; Peng, D.; Long, H.; He, W.; Qiao, F.; Wang, G.; Huang, W. Characterisation and functional importance of β-1,4-endoglucanases from the potato rot nematode, Ditylenchus destructor. Nematology 2014, 16, 505–517. [Google Scholar] [CrossRef]
- Kranewitter, W.J.; Ylanne, J.; Gimona, M. UNC-87 Is an Actin-bundling Protein*. J. Biol. Chem. 2001, 276, 6306–6312. [Google Scholar] [CrossRef]
- Ono, K.; Obinata, T.; Yamashiro, S.; Liu, Z.; Ono, S. UNC-87 isoforms, Caenorhabditis elegans calponin-related proteins, interact with both actin and myosin and regulate actomyosin contractility. Mol. Biol. Cell 2015, 26, 1687–1698. [Google Scholar] [CrossRef]
- Kagawa, H.; Takuwa, K.; Sakube, Y. Mutations and expressions of the tropomyosin gene and the troponin C gene of Caenorhabditis elegans. Cell Struct. Funct. 1997, 22, 213–218. [Google Scholar] [CrossRef][Green Version]
- Joseph, S.; Gheysen, G.; Subramaniam, K. RNA interference in Pratylenchus coffeae: Knock down of Pc-pat-10 and Pc-unc-87 impedes migration. Mol. Biochem. Parasitol. 2012, 186, 51–59. [Google Scholar] [CrossRef]
- Nsengimana, J.; Bauters, L.; Haegeman, A.; Gheysen, G. Silencing of Mg-pat-10 and Mg-unc-87 in the plant parasitic nematode Meloidogyne graminicola using siRNAs. Agriculture 2013, 3, 567–578. [Google Scholar] [CrossRef]
- Cukras, A.R.; Southworth, D.R.; Brunelle, J.L.; Culver, G.M.; Green, R. Ribosomal proteins S12 and S13 function as control elements for translocation of the mRNA: tRNA complex. Mol. Cell 2003, 12, 321–328. [Google Scholar]
- Sonnichsen, B.; Koski, L.; Walsh, A.; Marschall, P.; Neumann, B.; Brehm, M.; Alleaume, A.-M.; Artelt, J.; Bettencourt, P.; Cassin, E. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature 2005, 434, 462–470. [Google Scholar] [PubMed]
- Liu, S.; Jaouannet, M.; Dempsey, D.M.A.; Imani, J.; Coustau, C.; Kogel, K.-H. RNA-based technologies for insect control in plant production. Biotechnol. Adv. 2020, 39, 107463. [Google Scholar]
- Whyard, S.; Singh, A.D.; Wong, S. Ingested double-stranded RNAs can act as species-specific insecticides. Insect Biochem. Mol. Biol. 2009, 39, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.A.; Bogaert, T.; Clinton, W.; Heck, G.R.; Feldmann, P.; Ilagan, O.; Johnson, S.; Plaetinck, G.; Munyikwa, T.; Pleau, M.; et al. Control of coleopteran insect pests through RNA interference. Nat. Biotechnol. 2007, 25, 1322–1326. [Google Scholar] [CrossRef]
- Mutti, N.S.; Park, Y.; Reese, J.C.; Reeck, G.R. RNAi knockdown of a salivary transcript leading to lethality in the pea aphid, Acyrthosiphon pisum. J. Insect Sci. 2006, 6, 38. [Google Scholar] [CrossRef]
- Tian, H.; Peng, H.; Yao, Q.; Chen, H.; Xie, Q.; Tang, B.; Zhang, W. Developmental control of a lepidopteran pest Spodoptera exigua by ingestion of bacteria expressing dsRNA of a non-midgut gene. PLoS ONE 2009, 4, e6225. [Google Scholar]
- Timmons, L.; Court, D.L.; Fire, A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 2001, 263, 103–112. [Google Scholar] [CrossRef]
- Huang, G.; Allen, R.; Davis, E.L.; Baum, T.J.; Hussey, R.S. Engineering broad root-knot resistance in transgenic plants by RNAi silencing of a conserved and essential root-knot nematode parasitism gene. Proc. Natl. Acad. Sci. USA 2006, 103, 14302–14306. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, K.; Xie, H.; Wang, D.-W.; Xu, C.-L.; Huang, X.; Wu, W.-J.; Li, D.-L. Cathepsin B cysteine proteinase is essential for the development and pathogenesis of the plant parasitic nematode Radopholus similis. Int. J. Biol. Sci. 2015, 11, 1073. [Google Scholar] [CrossRef]
- Li, Y.; Wang, K.; Lu, Q.; Du, J.; Wang, Z.; Wang, D.; Sun, B.; Li, H. Transgenic Nicotiana benthamiana plants expressing a hairpin RNAi construct of a nematode Rs-cps gene exhibit enhanced resistance to Radopholus similis. Sci. Rep. 2017, 7, 13126. [Google Scholar] [CrossRef]
- Li, Y.; Wang, K.; Xie, J.; Wang, Y.-T.; Wang, D.; Xu, C.-L.; Huang, X.; Wang, D.-S. A Nematode Calreticulin, Rs-CRT, Is a Key Effector in Reproduction and Pathogenicity of Radopholus similis. PLoS ONE 2015, 10, e0129351. [Google Scholar] [CrossRef]
- Jekayinoluwa, T.; Tripathi, J.N.; Dugdale, B.; Obiero, G.; Muge, E.; Dale, J.; Tripathi, L. Transgenic Expression of dsRNA Targeting the Pentalonia nigronervosa acetylcholinesterase Gene in Banana and Plantain Reduces Aphid Populations. Plants 2021, 10, 613. [Google Scholar] [CrossRef]
- Arguel, M.-J.; Jaouannet, M.; Magliano, M.; Abad, P.; Rosso, M.-N. siRNAs trigger efficient silencing of a parasitism gene in plant parasitic root-knot nematodes. Genes 2012, 3, 391–408. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Mitreva, M.; Vanholme, B.; Gheysen, G. Exploring the transcriptome of the burrowing nematode Radopholus similis. Mol. Genet. Genom. 2008, 280, 1–17. [Google Scholar] [CrossRef]
- Hooper, D. Extraction and processing of plant and soil nematodes. In Plant Parasitic Nematodes in Subtropical and Tropical Agriculture; CAB International: Wallingford, UK, 1990; pp. 45–68. [Google Scholar]
- Speijer, P.; Ssango, F. Evaluation of Musa host plant response using nematode densities and damage indices. Nematropica 1999, 29, 185–192. [Google Scholar]
- McSorley, R. Extraction of nematodes and sampling methods. In Principles and Practice of Nematode Control in Crops; Brown, R.H., Kerry, B.R., Eds.; Academic Press: Cambridge, MA, USA, 1987. [Google Scholar]
- Siddiqi, M.R. Tylenchida: Parasites of Plants and Insects; CABI: Wallingford, UK, 2000. [Google Scholar]
- Moody, E.H.; Lownsbery, B.F.; Ahmed, J.M. Culture of the Root-Lesion Nematode Pratylenchus vulnus on Carrot Disks. J. Nematol. 1973, 5, 225–226. [Google Scholar]
- Yuan, B.; Latek, R.; Hossbach, M.; Tuschl, T.; Lewitter, F. siRNA Selection Server: An automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004, 32, W130–W134. [Google Scholar] [CrossRef]
- Brown, R.B.; Audet, J. Current techniques for single-cell lysis. J. R. Soc. Interface 2008, 5, S131–S138. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. Extraction of nucleic acid fragments from gels. Anal. Biochem. 2002, 302, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.-Y.; Gomez-Sanchez, C.E. Universal TA cloning. Curr. Issues Mol. Biol. 2000, 2, 1–7. [Google Scholar] [PubMed]
- Lu, S. Rapid screening of recombinant plasmids. In E. coli Plasmid Vectors; Humana Press: Totowa, NJ, USA, 2003; pp. 169–174. [Google Scholar]
- Woodman, M.E. Direct PCR of intact bacteria (colony PCR). Curr. Protoc. Microbiol. 2008, 9, A-3D. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Subcloning of DNA fragments. Curr. Protoc. Mol. Biol. 1991, 13, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Taghian, D.G.; Nickoloff, J.A. Subcloning strategies and protocols. In Basic DNA and RNA Protocols; Humana Press: Totowa, NJ, USA, 1996; pp. 221–235. [Google Scholar]
- Spear, M.A. Efficient DNA subcloning through selective restriction endonuclease digestion. BioTechniques 2000, 28, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Ongvarrasopone, C.; Roshorm, Y.; Panyim, S. A simple and cost effective method to generate dsRNA for RNAi studies in invertebrates. ScienceAsia 2007, 33, 35–39. [Google Scholar] [CrossRef]
- Froger, A.; Hall, J.E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. 2007, 6, e253. [Google Scholar] [CrossRef]
- Bergkessel, M.; Guthrie, C. Colony PCR. Methods Enzymol. 2013, 529, 299–309. [Google Scholar]
- Travella, S.; Klimm, T.E.; Keller, B. RNA interference-based gene silencing as an efficient tool for functional genomics in hexaploid bread wheat. Plant Physiol. 2006, 142, 6–20. [Google Scholar] [CrossRef]
- Zeng, G. Sticky-end PCR: New method for subcloning. Biotechniques 1998, 25, 206–208. [Google Scholar] [CrossRef]
- Hengen, P.N. Methods and reagents: Recovering DNA from agarose gels. Trends Biochem. Sci. 1994, 19, 388–389. [Google Scholar] [CrossRef]
- Zeugin, J.A.; Hartley, J.L. Ethanol precipitation of DNA. Focus 1985, 7, 1–2. [Google Scholar]
- Joly, E. Purification of DNA fragments from agarose gels using glass beads. In Basic DNA and RNA Protocols; Humana Press: Totowa, NJ, USA, 1996; pp. 237–240. [Google Scholar]
- Williams, S.A.; Slatko, B.E.; McCarrey, J.R. Laboratory Investigations in Molecular Biology; Jones & Bartlett Learning: Burlington, MA, USA, 2007. [Google Scholar]
- Sarathi, M.; Simon, M.C.; Ahmed, V.I.; Kumar, S.R.; Hameed, A.S. Silencing VP28 gene of white spot syndrome virus of shrimp by bacterially expressed dsRNA. Mar. Biotechnol. 2008, 10, 198–206. [Google Scholar] [CrossRef]
- Mwaka, H.S.; Christiaens, O.; Bwesigye, P.N.; Kubiriba, J.; Tushemereirwe, W.K.; Gheysen, G.; Smagghe, G. First Evidence of Feeding-Induced RNAi in Banana Weevil via Exogenous Application of dsRNA. Insects 2022, 13, 40. [Google Scholar]
- Taning, C.N.T.; Christiaens, O.; Berkvens, N.; Casteels, H.; Maes, M.; Smagghe, G. Oral RNAi to control Drosophila suzukii: Laboratory testing against larval and adult stages. J. Pest Sci. 2016, 89, 803–814. [Google Scholar] [CrossRef]
- Nwokeoji, A.O.; Kilby, P.M.; Portwood, D.E.; Dickman, M.J. Accurate Quantification of Nucleic Acids Using Hypochromicity Measurements in Conjunction with UV Spectrophotometry. Anal. Chem. 2017, 89, 13567–13574. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.A.; Jones, M.G.; Fosu-Nyarko, J. Gene silencing in root lesion nematodes (Pratylenchus spp.) significantly reduces reproduction in a plant host. Exp. Parasitol. 2013, 133, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cahalan, M.D. Purifying plasmid DNA from bacterial colonies using the QIAGEN Miniprep Kit. J. Vis. Exp. 2007, 6, e247. [Google Scholar]
- Weigel, D.; Glazebrook, J. Transformation of agrobacterium using the freeze-thaw method. CSH Protoc. 2006, 7, pdb-prot4666. [Google Scholar] [CrossRef]
- Chen, H.; Nelson, R.; Sherwood, J. Enhanced recovery of transformants of Agrobacterium tumefaciens after freeze-thaw transformation and drug selection. Biotechniques 1994, 16, 664–668. [Google Scholar]
- Khanna, H.; Becker, D.; Kleidon, J.; Dale, J. Centrifugation assisted Agrobacterium tumefaciens-mediated transformation (CAAT) of embryogenic cell suspensions of banana (Musa spp. Cavendish AAA and Lady finger AAB). Mol. Breed. 2004, 14, 239–252. [Google Scholar] [CrossRef]
- Gao, N.; Shen, W.; Cao, Y.; Su, Y.; Shi, W. Influence of bacterial density during preculture on Agrobacterium-mediated transformation of tomato. Plant Cell Tissue Organ Cult. 2009, 98, 321–330. [Google Scholar] [CrossRef]
- MacWilliams, M.P.; Liao, M.K. Luria broth (LB) and Luria agar (LA) media and their uses protocol. Am. Soc. Microbiol. 2006, 2006, 1–4. [Google Scholar]
- Namanya, P.; Mutumba, G.; Magambo, S.; Tushemereirwe, W. Developing a cell suspension system for Musa-AAA-EA cv.‘Nakyetengu’: A critical step for genetic improvement of Matooke East African Highland bananas. In Vitro Cell. Dev. Biol. Plant 2014, 50, 442–450. [Google Scholar] [CrossRef]
- Sheikholeslam, S.N.; Weeks, D.P. Acetosyringone promotes high efficiency transformation of Arabidopsis thaliana explants by Agrobacterium tumefaciens. Plant Mol. Biol. 1987, 8, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Arinaitwe, G. Improved Agrobacterium-Mediated Transformation Methods for Banana and Plantain. Ph.D. Thesis, Catholic University of Leuven, Leuven, Belgium, 2008. [Google Scholar]
- Tripathi, L.; Mwaka, H.; Tripathi, J.N.; Tushemereirwe, W.K. Expression of sweet pepper Hrap gene in banana enhances resistance to Xanthomonas campestris pv. musacearum. Mol. Plant Pathol. 2010, 11, 721–731. [Google Scholar] [CrossRef]
- Gawel, N.; Jarret, R. A modified CTAB DNA extraction procedure for Musa and Ipomoea. Plant Mol. Biol. Report. 1991, 9, 262–266. [Google Scholar] [CrossRef]
- Dochez, C.; Speijer, P.R.; Hartman, J.; Vuylsteke, D.; De Waele, D. Screening Musa hybrids for resistance to Radopholus similis. InfoMusa 2000, 9, 3–4. [Google Scholar]
- Pinochet, J. A Method for Screening Bananas and Plantains to Lesion Forming Nematodes; INIBAP: Montpellier, France, 1988. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dsRNA Target | Mean | Mean Separation | SD | Relative Population Counts |
|---|---|---|---|---|
| Cs-Laccase-2 | 14,285 | a | 1287 | 1 |
| no dsRNA | 13,757 | a | 1154 | - |
| Rs-Chs-2 | 5170 | b | 530 | 0.36 |
| Rs-Pat-10 | 3292 | c | 676 | 0.23 |
| Rs-Ego-1 | 3021 | c | 477 | 0.21 |
| Rs-Unc-87 | 2927 | c | 502 | 0.21 |
| Rs-Eng1a | 2858 | c | 559 | 0.20 |
| Rs-Rps13 | 1833 | d | 464 | 0.13 |
| dsRNA Target Genes | Best Lines | Protection (%) |
|---|---|---|
| Rps13 | 86, 127, 98, 92, 87, 93, 54, 142, 68 | 92–85 |
| Pat-10 | 104, 80, 59, 84, 60, 103 | 92–85 |
| Unc-87 | 66, 140, 139 | 92–85 |
| Chs-2 | 24, 12, 44, 8 | 88–85 |
| Eng1a | 53, 58, 52 | 87–85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwaka, H.S.; Bauters, L.; Namaganda, J.; Marcou, S.; Bwesigye, P.N.; Kubiriba, J.; Smagghe, G.; Tushemereirwe, W.K.; Gheysen, G. Transgenic East African Highland Banana Plants Are Protected against Radopholus similis through Host-Delivered RNAi. Int. J. Mol. Sci. 2023, 24, 12126. https://doi.org/10.3390/ijms241512126
Mwaka HS, Bauters L, Namaganda J, Marcou S, Bwesigye PN, Kubiriba J, Smagghe G, Tushemereirwe WK, Gheysen G. Transgenic East African Highland Banana Plants Are Protected against Radopholus similis through Host-Delivered RNAi. International Journal of Molecular Sciences. 2023; 24(15):12126. https://doi.org/10.3390/ijms241512126
Chicago/Turabian StyleMwaka, Henry Shaykins, Lander Bauters, Josephine Namaganda, Shirley Marcou, Priver Namanya Bwesigye, Jerome Kubiriba, Guy Smagghe, Wilberforce Kateera Tushemereirwe, and Godelieve Gheysen. 2023. "Transgenic East African Highland Banana Plants Are Protected against Radopholus similis through Host-Delivered RNAi" International Journal of Molecular Sciences 24, no. 15: 12126. https://doi.org/10.3390/ijms241512126
APA StyleMwaka, H. S., Bauters, L., Namaganda, J., Marcou, S., Bwesigye, P. N., Kubiriba, J., Smagghe, G., Tushemereirwe, W. K., & Gheysen, G. (2023). Transgenic East African Highland Banana Plants Are Protected against Radopholus similis through Host-Delivered RNAi. International Journal of Molecular Sciences, 24(15), 12126. https://doi.org/10.3390/ijms241512126