
Dysregulated miRNA and mRNA Expression Affect Overlapping Pathways in a Huntington’s Disease Model

Abstract
1. Introduction
2. Results
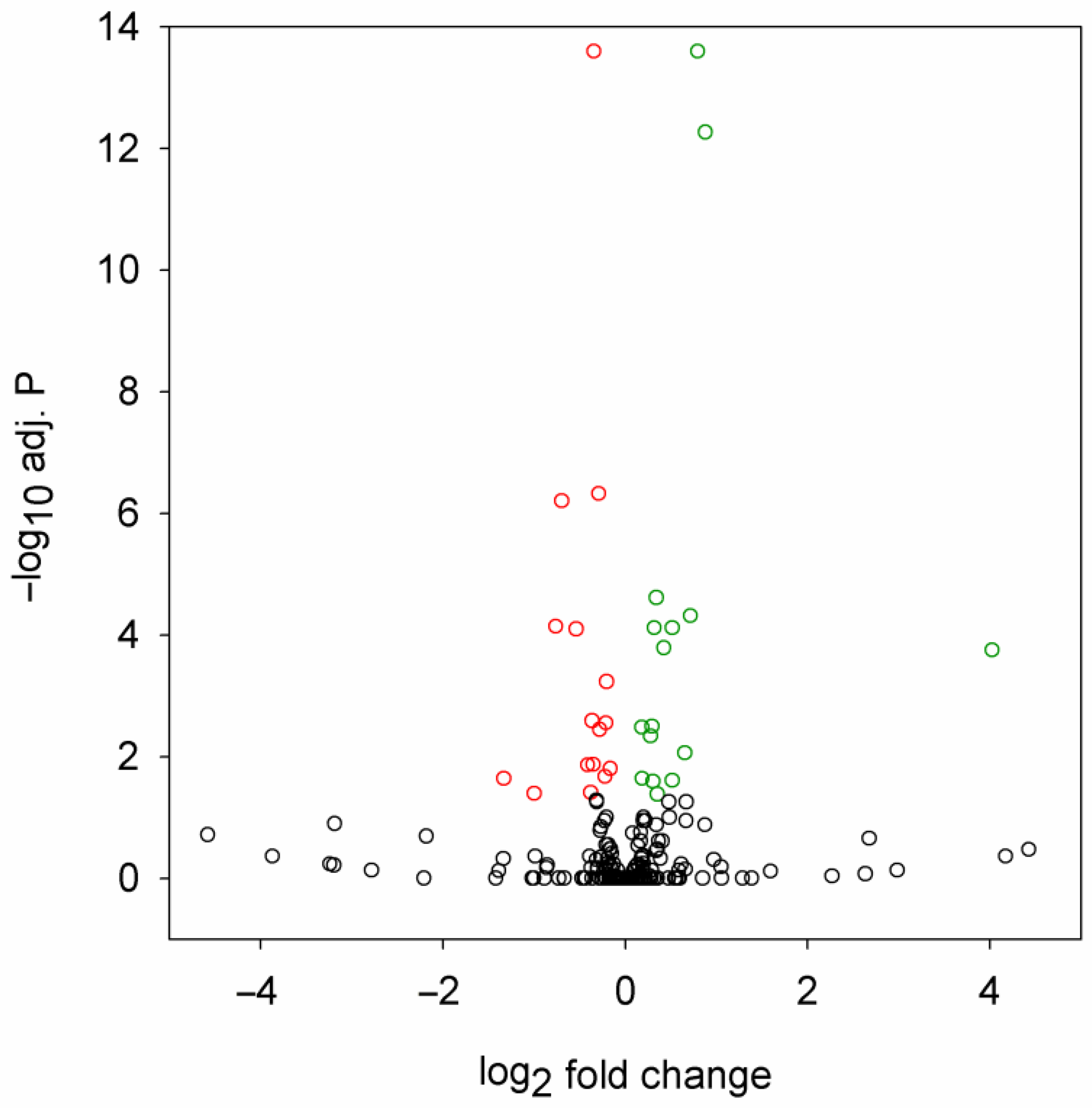
2.1. Mutant Huntingtin Induces Specific Changes in miRNA Expression
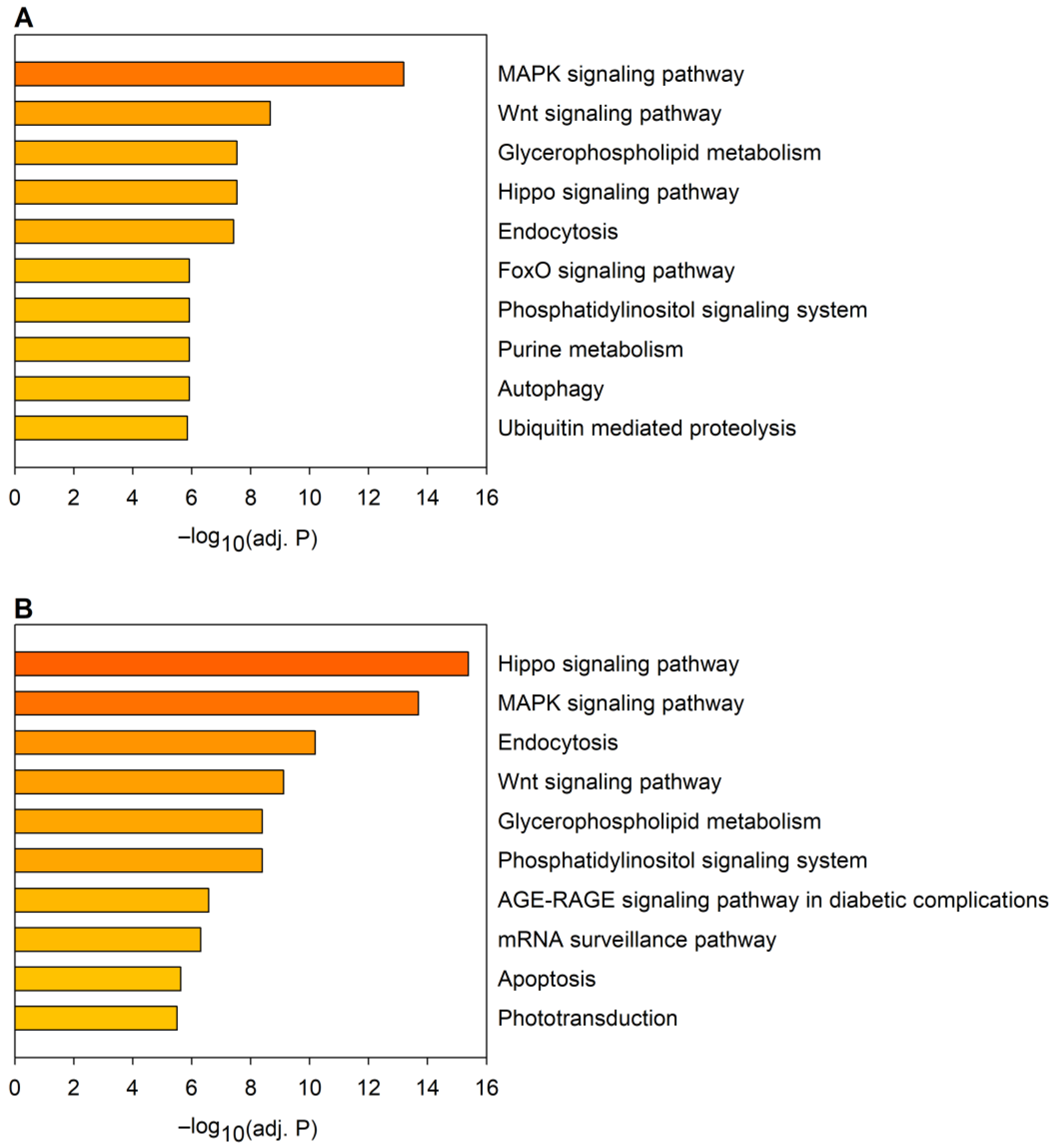
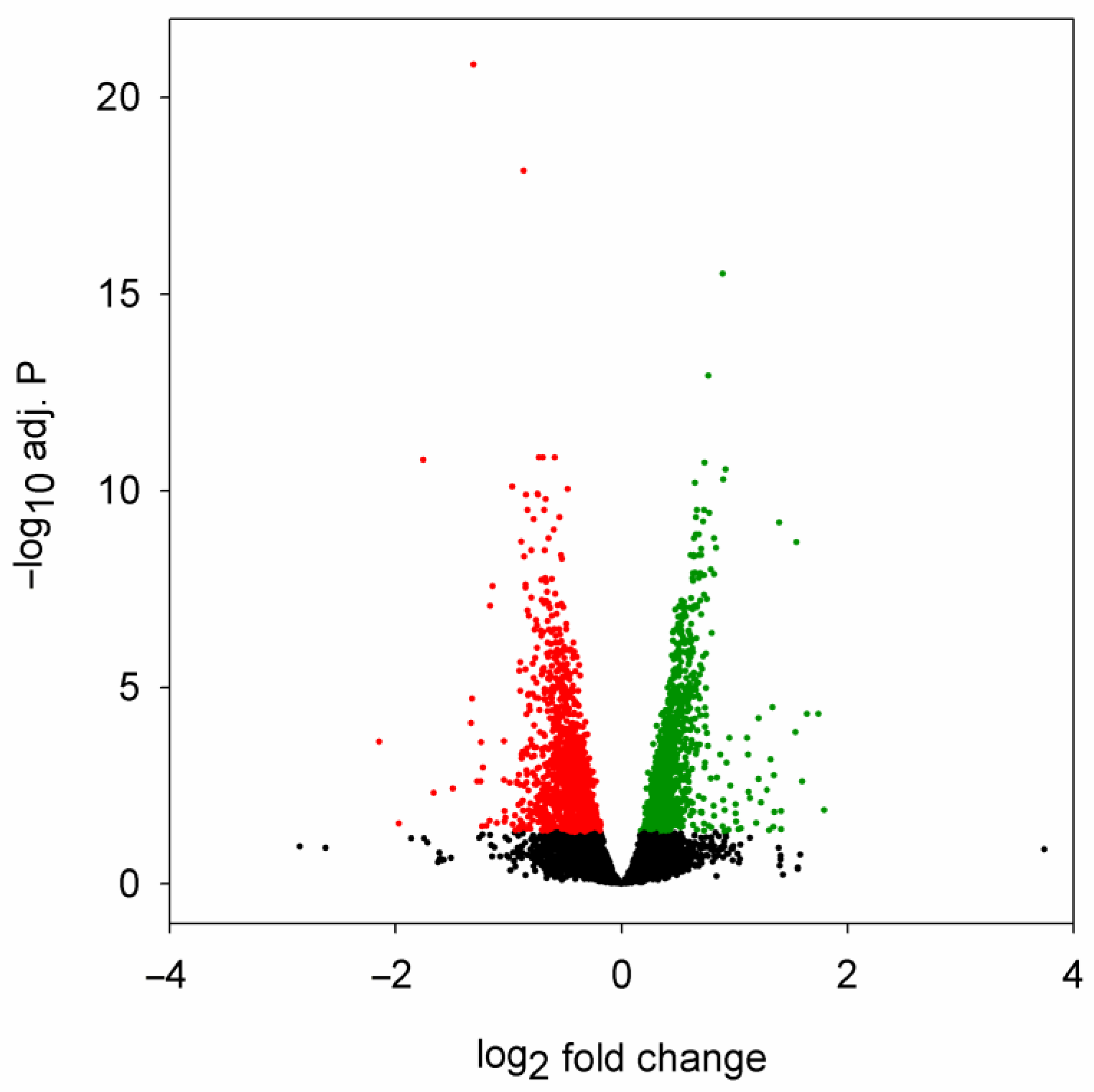
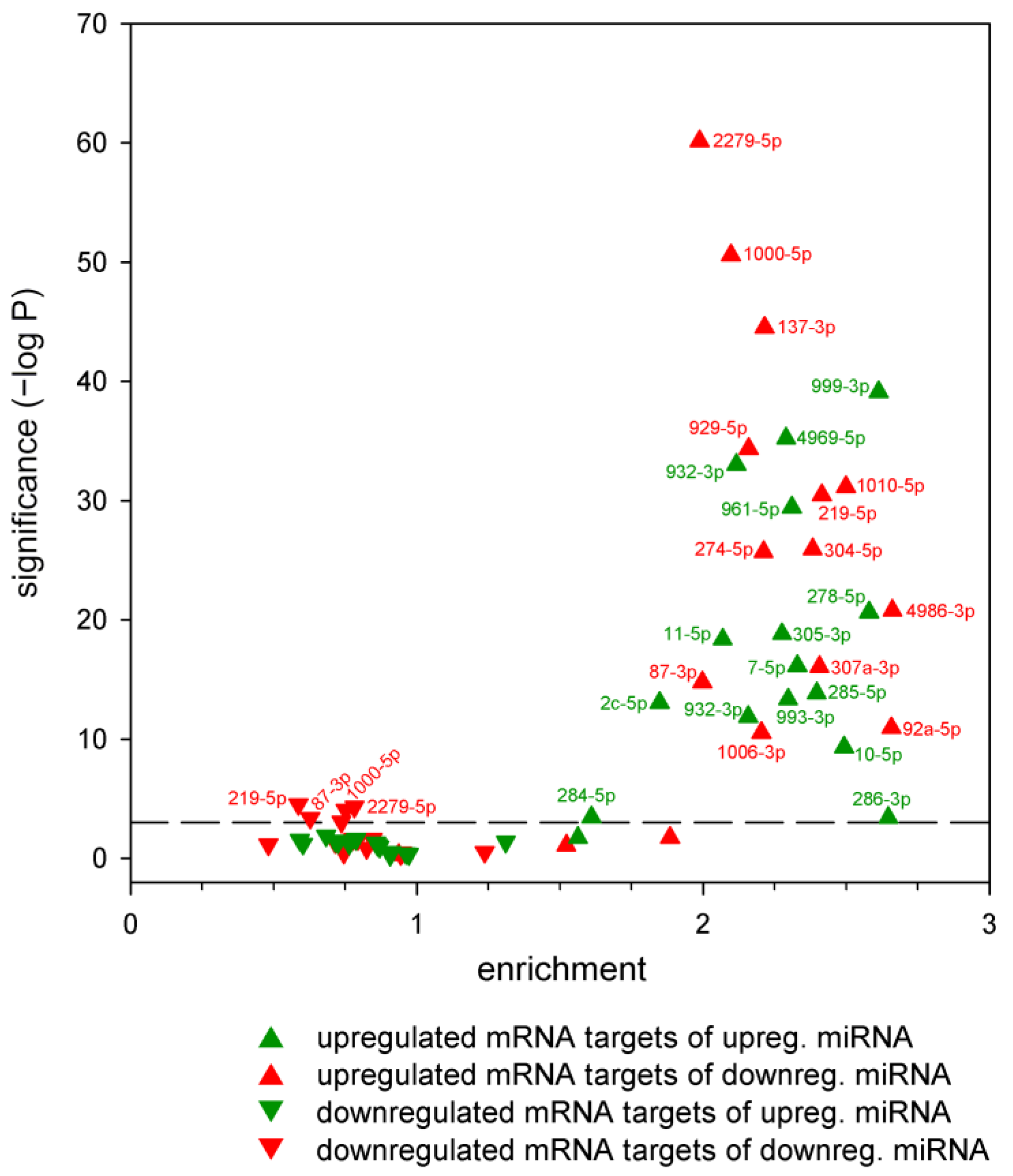
2.2. Correspondence between Dysregulated miRNA and mRNA Levels
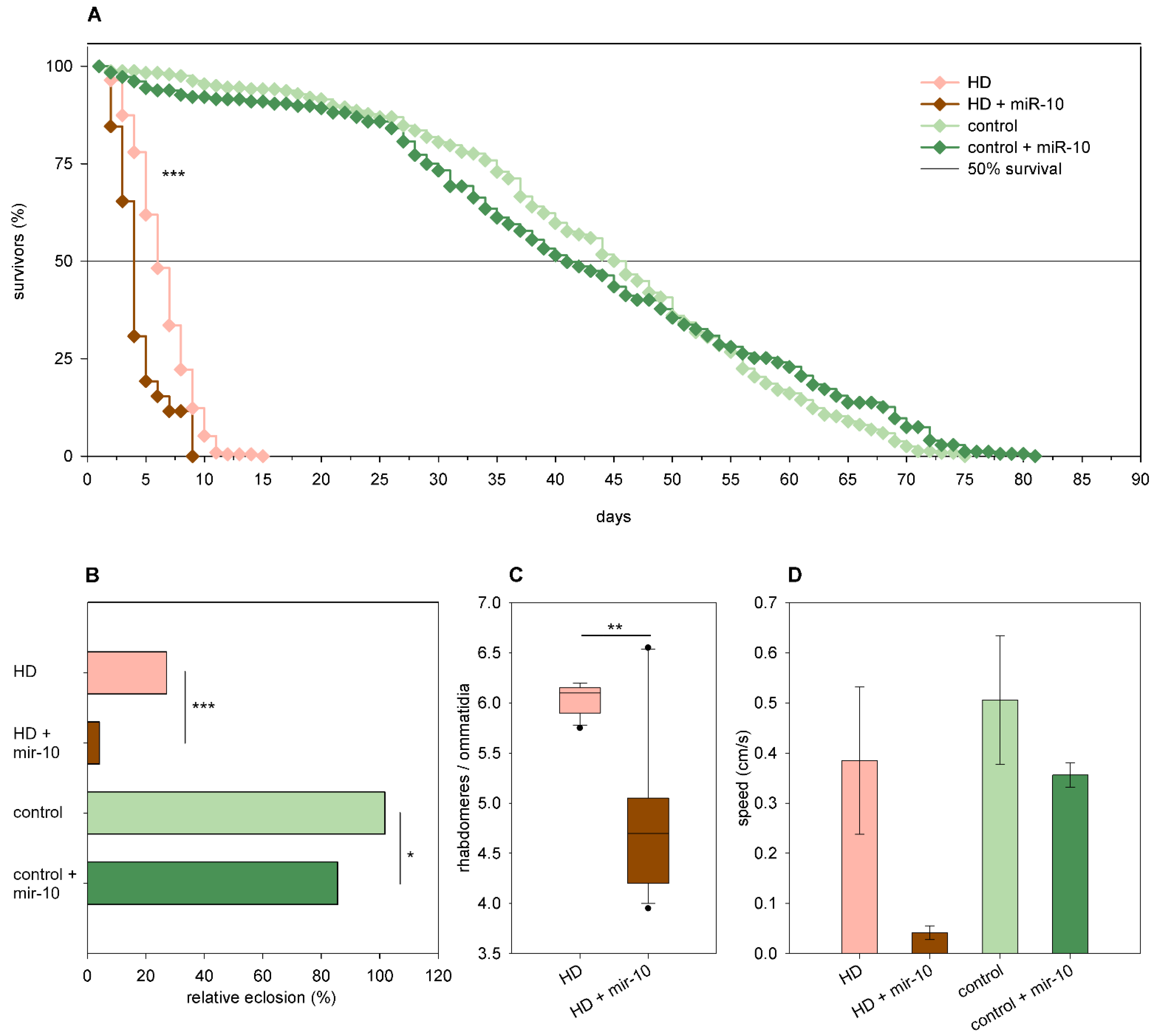
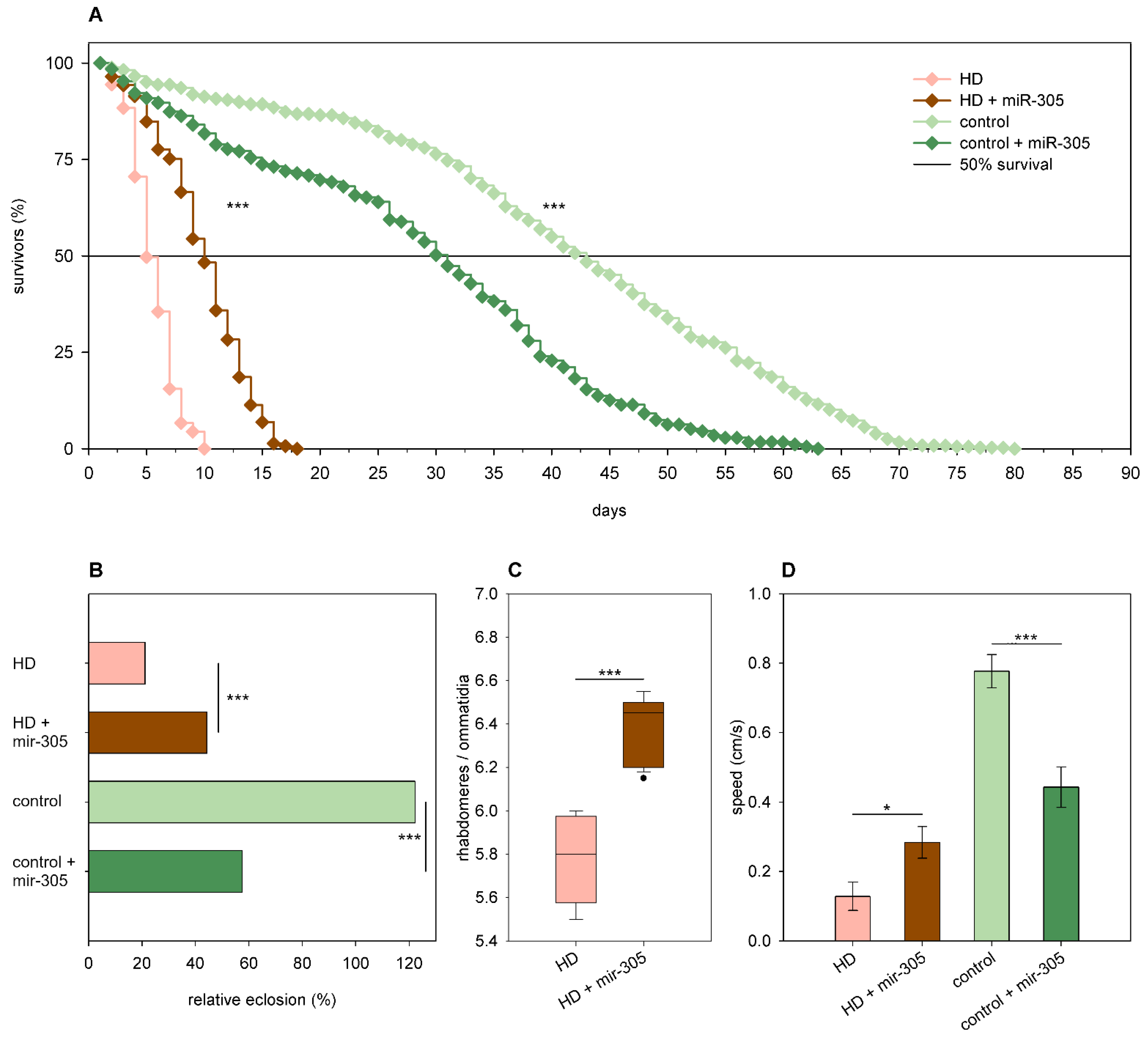
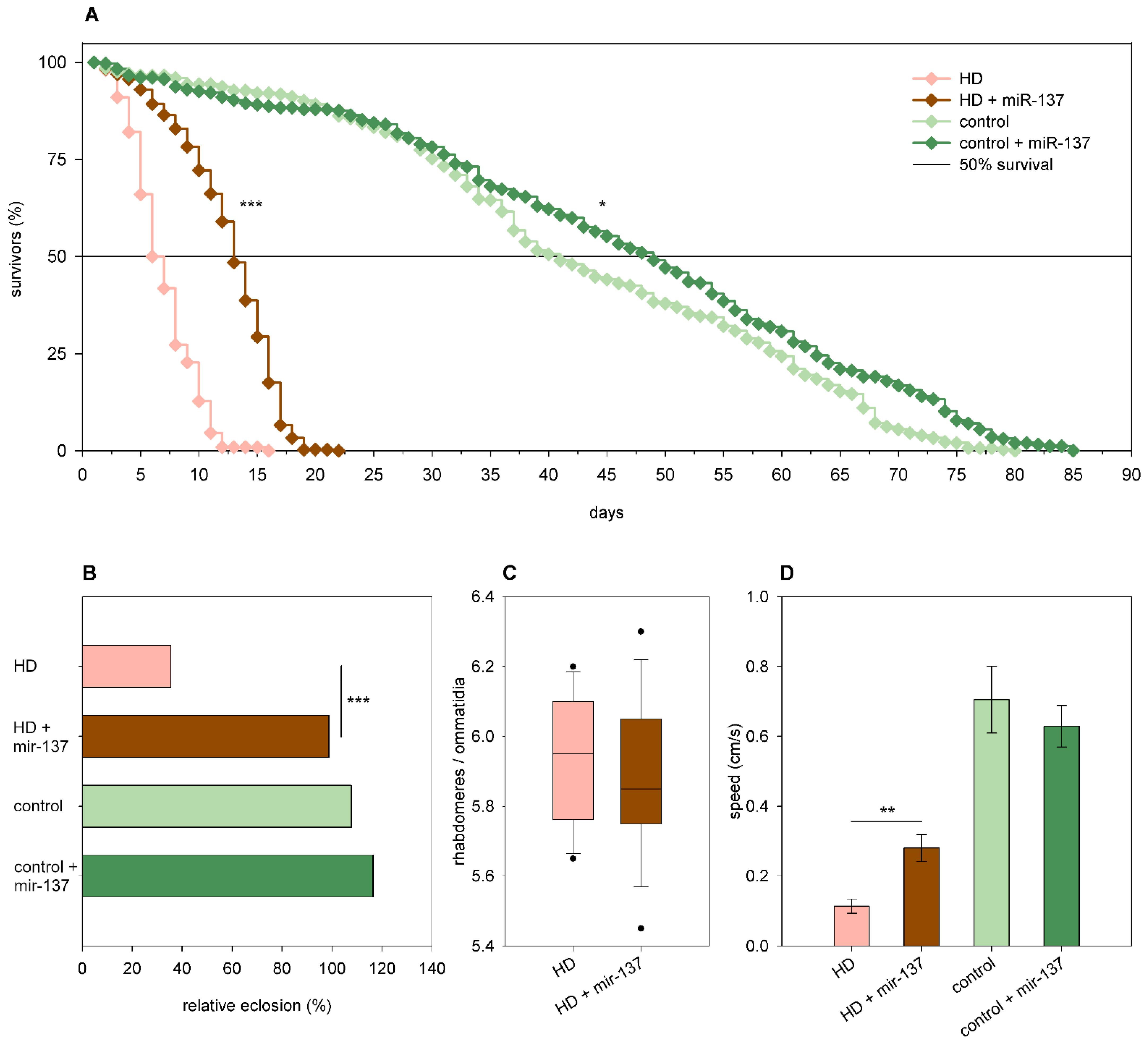
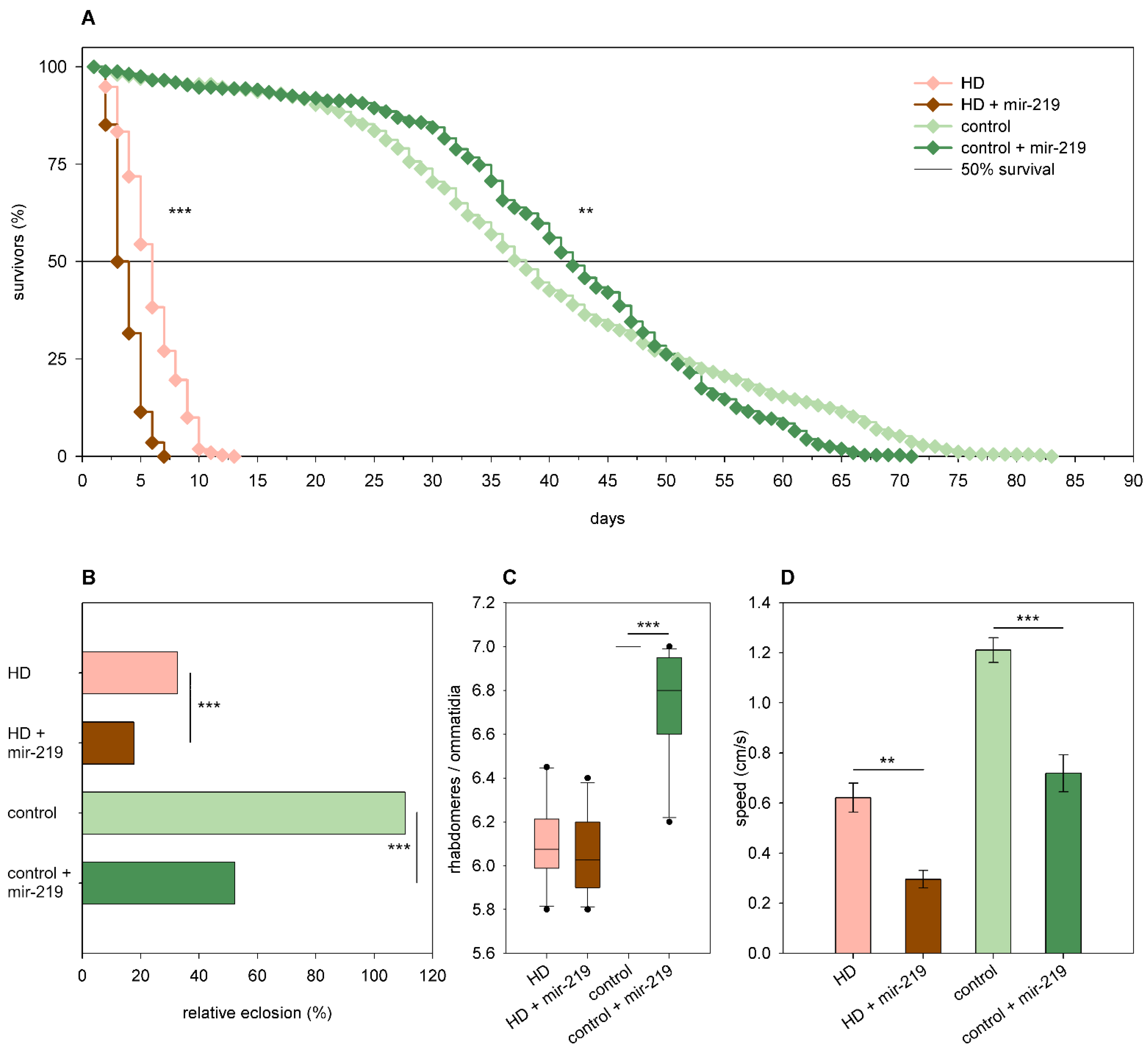
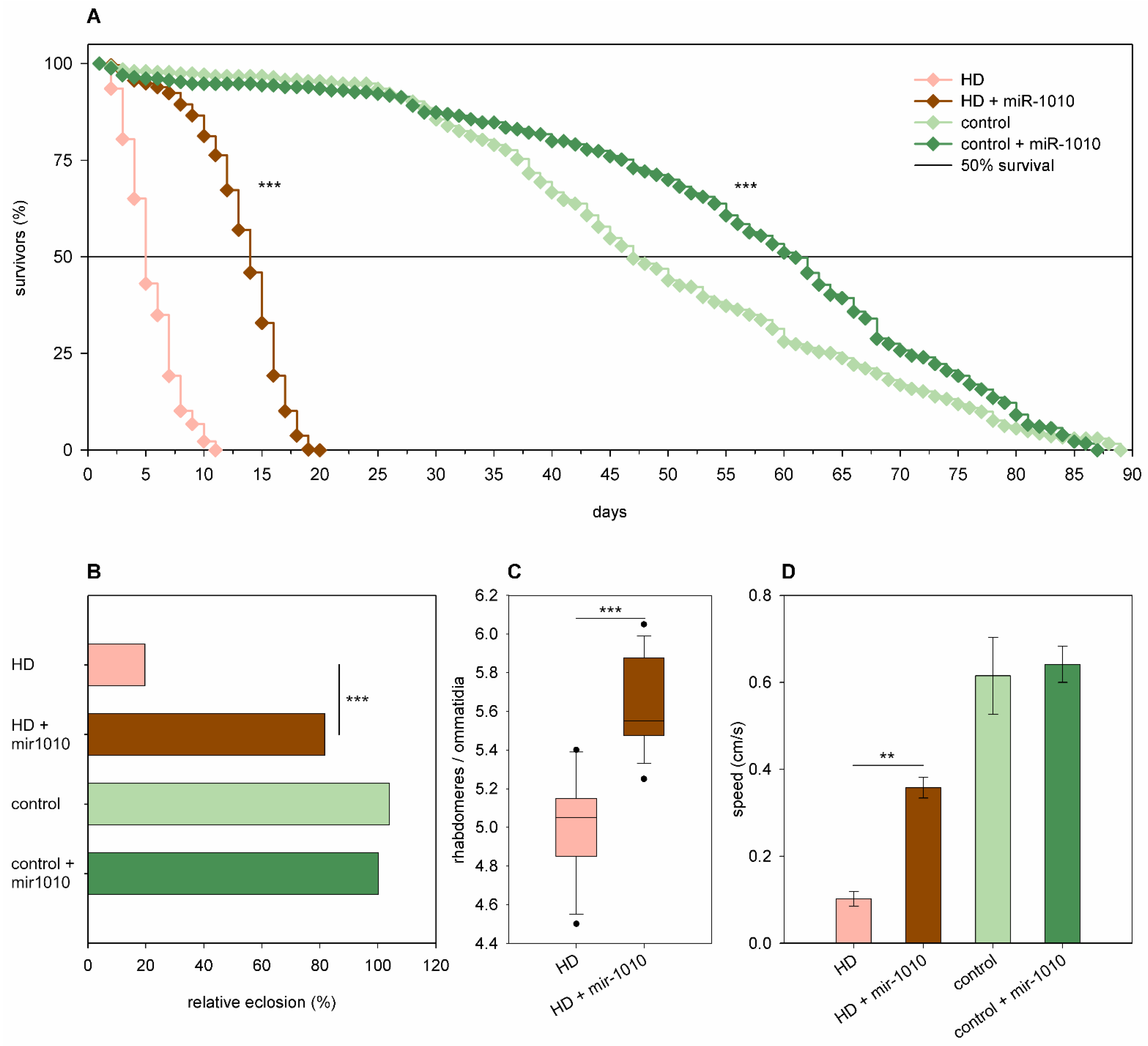
2.3. The Effects of miRNA Overexpression in mHtt-Expressing Drosophila
3. Discussion
3.1. Expression of Mutant Huntingtin Leads to miRNA Dysregulation
3.2. Overexpression of Specific miRNAs Alter mHtt-Induced Pathology
4. Materials and Methods
4.1. Drosophila Stocks and Crosses
4.2. RNA Preparation
4.3. Small-RNA Sequencing and RNA Sequencing
4.4. Secondary Sequence Analysis
4.5. Lifespan Analysis
4.6. Analysis of Neurodegeneration
4.7. Analysis of Motor Performance
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. Huntington’s Disease: Underlying Molecular Mechanisms and Emerging Concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Ortega, Z.; Lucas, J.J. Ubiquitin-Proteasome System Involvement in Huntington’s Disease. Front. Mol. Neurosci. 2014, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Bodai, L.; Marsh, J.L. A Novel Target for Huntington’s Disease: ERK at the Crossroads of Signaling. The ERK Signaling Pathway Is Implicated in Huntington’s Disease and Its Upregulation Ameliorates Pathology. BioEssays News Rev. Mol. Cell. Dev. Biol. 2012, 34, 142–148. [Google Scholar] [CrossRef]
- Dubinsky, J.M. Towards an Understanding of Energy Impairment in Huntington’s Disease Brain. J. Huntingt. Dis. 2017, 6, 267–302. [Google Scholar] [CrossRef]
- Valor, L.M. Transcription, Epigenetics and Ameliorative Strategies in Huntington’s Disease: A Genome-Wide Perspective. Mol. Neurobiol. 2015, 51, 406–423. [Google Scholar] [CrossRef]
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A MicroRNA-Based Gene Dysregulation Pathway in Huntington’s Disease. Neurobiol. Dis. 2008, 29, 438–445. [Google Scholar] [CrossRef]
- Martí, E.; Pantano, L.; Bañez-Coronel, M.; Llorens, F.; Miñones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A Myriad of MiRNA Variants in Control and Huntington’s Disease Brain Regions Detected by Massively Parallel Sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef]
- Michlewski, G.; Cáceres, J.F. Post-Transcriptional Control of MiRNA Biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef]
- Stavast, C.J.; Erkeland, S.J. The Non-Canonical Aspects of MicroRNAs: Many Roads to Gene Regulation. Cells 2019, 8, 1465. [Google Scholar] [CrossRef]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The Bifunctional MicroRNA MiR-9/MiR-9* Regulates REST and CoREST and Is Downregulated in Huntington’s Disease. J. Neurosci. 2008, 28, 14341–14346. [Google Scholar] [CrossRef]
- Jin, J.; Cheng, Y.; Zhang, Y.; Wood, W.; Peng, Q.; Hutchison, E.; Mattson, M.P.; Becker, K.G.; Duan, W. Interrogation of Brain MiRNA and MRNA Expression Profiles Reveals a Molecular Regulatory Network That Is Perturbed by Mutant Huntingtin. J. Neurochem. 2012, 123, 477–490. [Google Scholar] [CrossRef]
- Hoss, A.G.; Labadorf, A.; Latourelle, J.C.; Kartha, V.K.; Hadzi, T.C.; Gusella, J.F.; MacDonald, M.E.; Chen, J.-F.; Akbarian, S.; Weng, Z.; et al. MiR-10b-5p Expression in Huntington’s Disease Brain Relates to Age of Onset and the Extent of Striatal Involvement. BMC Med. Genom. 2015, 8, 10. [Google Scholar] [CrossRef]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical Aspects of MicroRNA Target Prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef]
- Sinha, M.; Ghose, J.; Bhattarcharyya, N.P. Micro RNA -214,-150,-146a and-125b Target Huntingtin Gene. RNA Biol. 2011, 8, 1005–1021. [Google Scholar] [CrossRef]
- Barbaro, B.A.; Lukacsovich, T.; Agrawal, N.; Burke, J.; Bornemann, D.J.; Purcell, J.M.; Worthge, S.A.; Caricasole, A.; Weiss, A.; Song, W.; et al. Comparative Study of Naturally Occurring Huntingtin Fragments in Drosophila Points to Exon 1 as the Most Pathogenic Species in Huntington’s Disease. Hum. Mol. Genet. 2015, 24, 913–925. [Google Scholar] [CrossRef]
- Ibáñez-Ventoso, C.; Vora, M.; Driscoll, M. Sequence Relationships among C. Elegans, D. Melanogaster and Human MicroRNAs Highlight the Extensive Conservation of MicroRNAs in Biology. PLoS ONE 2008, 3, e2818. [Google Scholar] [CrossRef]
- Naeli, P.; Winter, T.; Hackett, A.P.; Alboushi, L.; Jafarnejad, S.M. The Intricate Balance between MicroRNA-Induced MRNA Decay and Translational Repression. FEBS J. 2023, 290, 2508–2524. [Google Scholar] [CrossRef]
- Rosas-Arellano, A.; Estrada-Mondragón, A.; Piña, R.; Mantellero, C.A.; Castro, M.A. The Tiny Drosophila Melanogaster for the Biggest Answers in Huntington’s Disease. Int. J. Mol. Sci. 2018, 19, 2398. [Google Scholar] [CrossRef]
- Pouladi, M.A.; Morton, A.J.; Hayden, M.R. Choosing an Animal Model for the Study of Huntington’s Disease. Nat. Rev. Neurosci. 2013, 14, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Strand, A.; Peters, N.L.; Solano, S.M.; Hollingsworth, Z.R.; Menon, A.S.; Frey, A.S.; Spektor, B.S.; Penney, E.B.; Schilling, G.; et al. Decreased Expression of Striatal Signaling Genes in a Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2000, 9, 1259–1271. [Google Scholar] [CrossRef]
- Yildirim, F.; Ng, C.W.; Kappes, V.; Ehrenberger, T.; Rigby, S.K.; Stivanello, V.; Gipson, T.A.; Soltis, A.R.; Vanhoutte, P.; Caboche, J.; et al. Early Epigenomic and Transcriptional Changes Reveal Elk-1 Transcription Factor as a Therapeutic Target in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 24840–24851. [Google Scholar] [CrossRef] [PubMed]
- Agus, F.; Crespo, D.; Myers, R.H.; Labadorf, A. The Caudate Nucleus Undergoes Dramatic and Unique Transcriptional Changes in Human Prodromal Huntington’s Disease Brain. BMC Med. Genom. 2019, 12, 137. [Google Scholar] [CrossRef]
- Ueda, M.; Sato, T.; Ohkawa, Y.; Inoue, Y.H. Identification of MiR-305, a MicroRNA That Promotes Aging, and Its Target MRNAs in Drosophila. Genes Cells 2018, 23, 80–93. [Google Scholar] [CrossRef]
- Guo, S.; Yang, J.; Jiang, B.; Zhou, N.; Ding, H.; Zhou, G.; Wu, S.; Suo, A.; Wu, X.; Xie, W.; et al. MicroRNA Editing Patterns in Huntington’s Disease. Sci. Rep. 2022, 12, 3173. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, W.; Yi, Y.; Tong, Q. MiR-219-5p Inhibits Tau Phosphorylation by Targeting TTBK1 and GSK-3β in Alzheimer’s Disease. J. Cell. Biochem. 2019, 120, 9936–9946. [Google Scholar] [CrossRef]
- Denk, J.; Boelmans, K.; Siegismund, C.; Lassner, D.; Arlt, S.; Jahn, H. MicroRNA Profiling of CSF Reveals Potential Biomarkers to Detect Alzheimer’s Disease. PLoS ONE 2015, 10, e0126423. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Alaniz, M.E.; Renwick, N.; Cela, C.; Fulga, T.A.; Vactor, D.V.; Tuschl, T.; Clark, L.N.; Shelanski, M.L.; McCabe, B.D.; et al. Dysregulation of MicroRNA-219 Promotes Neurodegeneration through Post-Transcriptional Regulation of Tau. J. Clin. Investig. 2015, 125, 681–686. [Google Scholar] [CrossRef]
- Saraiva, C.; Esteves, M.; Bernardino, L. MicroRNA: Basic Concepts and Implications for Regeneration and Repair of Neurodegenerative Diseases. Biochem. Pharmacol. 2017, 141, 118–131. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c Regulates Serine Palmitoyltransferase and In Turn Amyloid β, Novel Targets in Sporadic Alzheimer’s Disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, B.; Chen, J.; Sui, Y.; Ren, L.; Li, J.; Zhang, H.; Guo, L.; Sun, X. Micro-RNA-137 Inhibits Tau Hyperphosphorylation in Alzheimer’s Disease and Targets the CACNA1C Gene in Transgenic Mice and Human Neuroblastoma SH-SY5Y Cells. Med. Sci. Monit. 2018, 24, 5635–5644. [Google Scholar] [CrossRef]
- Li, N.; Pan, X.; Zhang, J.; Ma, A.; Yang, S.; Ma, J.; Xie, A. Plasma Levels of MiR-137 and MiR-124 Are Associated with Parkinson’s Disease but Not with Parkinson’s Disease with Depression. Neurol. Sci. 2017, 38, 761–767. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, J.; Chen, L.; Jin, Y.; Zhang, G.; Lin, Z.; Du, S.; Fu, Z.; Chen, T.; Qin, Y.; et al. Serum Secreted MiR-137-Containing Exosomes Affects Oxidative Stress of Neurons by Regulating OXR1 in Parkinson’s Disease. Brain Res. 2019, 1722, 146331. [Google Scholar] [CrossRef]
- Kong, Y.; Liang, X.; Liu, L.; Zhang, D.; Wan, C.; Gan, Z.; Yuan, L. High Throughput Sequencing Identifies MicroRNAs Mediating α-Synuclein Toxicity by Targeting Neuroactive-Ligand Receptor Interaction Pathway in Early Stage of Drosophila Parkinson’s Disease Model. PLoS ONE 2015, 10, e0137432. [Google Scholar] [CrossRef]
- Kozlowska, E.; Krzyzosiak, W.J.; Koscianska, E. Regulation of Huntingtin Gene Expression by MiRNA-137,-214,-148a, and Their Respective IsomiRs. Int. J. Mol. Sci. 2013, 14, 16999–17016. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 August 2020).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Baras, A.S.; Mitchell, C.J.; Myers, J.R.; Gupta, S.; Weng, L.-C.; Ashton, J.M.; Cornish, T.C.; Pandey, A.; Halushka, M.K. MiRge-A Multiplexed Method of Processing Small RNA-Seq Data to Determine MicroRNA Entropy. PLoS ONE 2015, 10, e0143066. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Agarwal, V.; Subtelny, A.O.; Thiru, P.; Ulitsky, I.; Bartel, D.P. Predicting MicroRNA Targeting Efficacy in Drosophila. Genome Biol. 2018, 19, 152. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Diaz, J.E.L.; Flamholz, Z.N.; Keenan, A.B.; Lachmann, A.; Wojciechowicz, M.L.; Cagan, R.L.; Ma’ayan, A. ModEnrichr: A Suite of Gene Set Enrichment Analysis Tools for Model Organisms. Nucleic Acids Res. 2019, 47, W183–W190. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Smith, M.R.; Syed, A.; Lukacsovich, T.; Barbaro, B.A.; Purcell, J.; Bornemann, D.J.; Burke, J.; Marsh, J.L. Morphometric Analysis of Huntington’s Disease Neurodegeneration in Drosophila. In Tandem Repeats in Genes, Proteins, and Disease; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; pp. 41–57. ISBN 978-1-62703-437-1. [Google Scholar]
- Spierer, A.N.; Yoon, D.; Zhu, C.-T.; Rand, D.M. FreeClimber: Automated Quantification of Climbing Performance in Drosophila. J. Exp. Biol. 2021, 224, jeb229377. [Google Scholar] [CrossRef]
- Han, S.K.; Lee, D.; Lee, H.; Kim, D.; Son, H.G.; Yang, J.-S.; Lee, S.-J.V.; Kim, S. OASIS 2: Online Application for Survival Analysis 2 with Features for the Analysis of Maximal Lifespan and Healthspan in Aging Research. Oncotarget 2016, 7, 56147–56152. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Base Mean (CPM) 1 | log2(FC) 2 | Adjusted p Value 3 | Human Orthologs 4 |
|---|---|---|---|---|
| dme-miR-4986-3p | 6.02 | −1.33 | 2.29 × 10−2 | |
| dme-miR-2279-5p | 5.21 | −1 | 4.03 × 10−2 | |
| dme-miR-219-5p | 23.94 | −0.76 | 7.21 × 10−5 | hsa-miR-219-5p $# |
| dme-miR-1010-5p | 25.86 | −0.7 | 6.29 × 10−7 | hsa-miR-412 # |
| dme-miR-92a-5p | 39.12 | −0.54 | 8.01 × 10−5 | hsa-miR-25 $#, hsa-hsa-miR-92a $#, hsa-miR92b $#, hsa-miR-32 #, hsa-miR-363 #, hsa-miR-367 #, hsa-miR-885-5p # |
| dme-miR-307a-3p | 213.68 | −0.42 | 1.37 × 10−2 | |
| dme-miR-304-5p | 27.69 | −0.38 | 3.85 × 10−2 | hsa-miR-216a $# |
| dme-miR-929-5p | 117.03 | −0.36 | 2.60 × 10−3 | |
| dme-miR-274-5p | 1271.69 | −0.35 | 1.35 × 10−2 | hsa-miR-758 # |
| dme-miR-87-3p | 199.18 | −0.35 | 2.54 × 10−14 | |
| dme-miR-1006-3p | 198.74 | −0.29 | 4.75 × 10−7 | |
| dme-miR-137-3p | 934.73 | −0.28 | 3.60 × 10−3 | hsa-miR-137 $# |
| dme-miR-998-3p | 158.26 | −0.22 | 2.12 × 10−2 | hsa-miR-21* #, hsa-miR-29a #, hsa-miR-29b #, hsa-miR-29c #, hsa-miR-593* # |
| dme-miR-2b-3p | 2802.63 | −0.21 | 2.79 × 10−3 | hsa-miR-499-3p # |
| dme-miR-1000-5p | 936.13 | −0.21 | 5.86 × 10−4 | |
| dme-miR-13b-3p | 2027.42 | −0.16 | 1.57 × 10−2 | hsa-miR-499-3p # |
| dme-miR-11-5p | 256.11 | 0.18 | 3.29 × 10−3 | hsa-miR-27b $#, hsa-miR-27a #, hsa-miR-128 #, hsa-miR-499-3p #, hsa-miR-768-3p # |
| dme-miR-999-3p | 79,593.87 | 0.19 | 2.28 × 10−2 | |
| dme-miR-927-5p | 5481.4 | 0.28 | 4.59 × 10−3 | |
| dme-miR-932-3p | 89.88 | 0.29 | 3.17 × 10−3 | |
| dme-miR-993-3p | 306.58 | 0.3 | 2.56 × 10−2 | hsa-miR-100* $, hsa-mir-99a* #, hsa-miR-99b* #, hsa-miR-556-5p # |
| dme-miR-284-5p | 1428.7 | 0.31 | 7.61 × 10−5 | |
| dme-miR-285-5p | 309.28 | 0.34 | 2.44 × 10−5 | hsa-miR-29a $#, hsa-miR-29b $#, hsa-miR-29c $#, hsa-miR-21* #, hsa-miR-593* # |
| dme-miR-7-5p | 52,010.66 | 0.35 | 4.16 × 10−2 | hsa-miR-7 $#, hsa-miR-9* #, hsa-miR-548-3p #, hsa-miR-146a #, hsa-miR-146b-5p # |
| dme-miR-2c-5p | 214.24 | 0.42 | 1.62 × 10−4 | hsa-miR-499-3p # |
| dme-miR-10-5p | 1393.19 | 0.52 | 2.47 × 10−2 | hsa-miR-10a $#, hsa-miR-10b $#, hsa-miR-99a $, hsa-miR-100 $, hsa-miR-146b-3p # |
| dme-miR-278-5p | 618.26 | 0.52 | 7.61 × 10−5 | |
| dme-miR-6-3p | 70.38 | 0.65 | 8.73 × 10−3 | hsa-miR-27a #, hsa-miR-27b #, hsa-miR-128 #, hsa-miR-499-3p #, hsa-miR-768-3p # |
| dme-miR-305-3p | 487.46 | 0.71 | 4.85 × 10−5 | |
| dme-miR-286-3p | 16.1 | 0.79 | 2.54 × 10−14 | hsa-miR-134 # |
| dme-miR-4969-5p | 31.1 | 0.88 | 5.42 × 10−13 | |
| dme-miR-961-5p | 1.63 | 4.02 | 1.77 × 10−4 | hsa-miR-133a # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zsindely, N.; Nagy, G.; Siági, F.; Farkas, A.; Bodai, L. Dysregulated miRNA and mRNA Expression Affect Overlapping Pathways in a Huntington’s Disease Model. Int. J. Mol. Sci. 2023, 24, 11942. https://doi.org/10.3390/ijms241511942
Zsindely N, Nagy G, Siági F, Farkas A, Bodai L. Dysregulated miRNA and mRNA Expression Affect Overlapping Pathways in a Huntington’s Disease Model. International Journal of Molecular Sciences. 2023; 24(15):11942. https://doi.org/10.3390/ijms241511942
Chicago/Turabian StyleZsindely, Nóra, Gábor Nagy, Fruzsina Siági, Anita Farkas, and László Bodai. 2023. "Dysregulated miRNA and mRNA Expression Affect Overlapping Pathways in a Huntington’s Disease Model" International Journal of Molecular Sciences 24, no. 15: 11942. https://doi.org/10.3390/ijms241511942
APA StyleZsindely, N., Nagy, G., Siági, F., Farkas, A., & Bodai, L. (2023). Dysregulated miRNA and mRNA Expression Affect Overlapping Pathways in a Huntington’s Disease Model. International Journal of Molecular Sciences, 24(15), 11942. https://doi.org/10.3390/ijms241511942