

C∙∙∙O and Si∙∙∙O Tetrel Bonds: Substituent Effects and Transfer of the SiF3 Group




Abstract
1. Introduction
2. Theoretical Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Ramis, R.; Frontera, A. Computational Study of Anion Recognition Based on Tetrel and Hydrogen Bonding Interaction by Calix [4] Pyrrole Derivatives. Comput. Theor. Chem. 2014, 1038, 67–70. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel Bond–σ-Hole Bond as a Preliminary Stage of the SN2 Reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Southern, S.A.; Bryce, D.L. NMR Investigations of Noncovalent Carbon Tetrel Bonds. Computational Assessment and Initial Experimental Observation. J. Phys. Chem. A 2015, 119, 11891–11899. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, Q.Z.; Cheng, J.B.; Li, W.Z.; Li, H.B. Tetrel Bond of Pseudohalide Anions with XH3F (X = C, Si, Ge, and Sn) and its Role in SN2 Reaction. J. Chem. Phys. 2016, 145, 224310. [Google Scholar] [CrossRef]
- Scheiner, S. Systematic Elucidation of Factors That Influence the Strength of Tetrel Bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef]
- Scheiner, S. The Ditetrel Bond: Noncovalent Bond between Neutral Tetrel Atoms. Phys. Chem. Chem. Phys. 2020, 22, 16606–16614. [Google Scholar] [CrossRef]
- Scheiner, S. Relative Strengths of a Pnicogen and a Tetrel Bond and Their Mutual Effects upon One Another. J. Phys. Chem. A 2021, 125, 2631–2641. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-Hole Concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- de Paul N. Nziko, V.; Scheiner, S. Comparison of π-Hole Tetrel Bonding with σ-Hole Halogen Bonds in Complexes of XCN (X = F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. The X–C···Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘Carbon Bond’ and Hydrophobic Interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef] [PubMed]
- Mundlapati, V.R.; Sahoo, D.K.; Bhaumik, S.; Chandrakar, A.; Jena, S.; Biswal, H.S. Noncovalent Carbon-Bonding Interactions in Proteins. Angew. Chem. Int. Ed. 2018, 57, 16496–16500. [Google Scholar] [CrossRef] [PubMed]
- García-Llinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R–CF3···O Tetrel Bonding Interactions in Biological Systems. J. Phys. Chem. A 2017, 121, 5371–5376. [Google Scholar] [CrossRef]
- Karim, A.; Schulz, N.; Andersson, H.; Nekoueishahraki, B.; Carlsson, A.C.C.; Sarabi, D.; Valkonen, A.; Rissanen, K.; Gräfenstein, J.; Keller, S.; et al. Carbon’s Three-Center, Four-Electron Tetrel Bond, Treated Experimentally. J. Am. Chem. Soc. 2018, 140, 17571–17579. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. Microwave Spectroscopic and Atoms in Molecules Theoretical Investigations on the Ar···Propargyl Alcohol Complex: Ar···H-O, Ar···π, and Ar···C Interactions. ChemPhysChem 2013, 14, 754–763. [Google Scholar] [CrossRef]
- Thomas, S.P.; Pavan, M.S.; Row, T.N.G. Experimental Evidence for ‘Carbon Bonding’ in the Solid State from Charge Density Analysis. Chem. Commun. 2014, 50, 49–51. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Non-Covalent sp3 Carbon Bonding with ArCF3 is Analogous to CH–π Interactions. Chem. Commun. 2014, 50, 12626–12629. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.Z.; Scheiner, S. Comparison of Tetrel Bonds in Neutral and Protonated Complexes of PyridineTF3 and FuranTF3 (T = C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef]
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, G.; Gong, H. Copper-Catalyzed Reductive Trifluoromethylation of Alkyl Iodides with Togni’s Reagent. Org. Lett. 2018, 20, 4677–4680. [Google Scholar] [CrossRef]
- Zhang, M.; Lin, J.H.; Xiao, J.C. A Readily Available Trifluoromethylation Reagent and Its Difunctionalization of Alkenes. Org. Lett. 2021, 23, 6079–6083. [Google Scholar] [CrossRef] [PubMed]
- Kalim, J.; Duhail, T.; Le, T.N.; Vanthuyne, N.; Anselmi, E.; Togni, A.; Magnier, E. Merging Hypervalent Iodine and Sulfoximine Chemistry: A New Electrophilic Trifluoromethylation Reagent. Chem. Sci. 2019, 10, 10516–10523. [Google Scholar] [CrossRef]
- Hof, F.; Scofield, D.M.; Schweizer, W.B.; Diederich, F. A Weak Attractive Interaction between Organic Fluorine and an Amide Group. Angew. Chem. Int. Ed. 2004, 43, 5056–5059. [Google Scholar] [CrossRef]
- Wu, Q.Z.; Xie, X.Y.; Li, Q.Z.; Scheiner, S. Enhancement of Tetrel Bond Involving Tetrazole-TtR3 (Tt = C, Si; R = H, F). Promotion of SiR3 Transfer by a Triel Bond. Phys. Chem. Chem. Phys. 2022, 24, 25895–25903. [Google Scholar] [CrossRef] [PubMed]
- Voronkov, A.M.G.; Grebneva, E.A.; Trofimova, O.M.; Chernov, N.F.; Albanov, A.I.; Chipanina, N.N. The Unusual Reaction of Phenyltrifluorosilane with 2-Aminoethanol and its N-Methyl Derivatives. Dokl. Chem. 2006, 409, 139–141. [Google Scholar] [CrossRef]
- Tamao, K.; Hayashi, T.; Ito, Y. Anion Complexation by Bidentate Lewis Acidic Hosts, Ortho-bis(fluorosilyl)benzenes. J. Organomet. Chem. 1996, 506, 85–91. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Trofimova, O.M.; Chernov, N.F.; Albanov, A.I.; Chipanina, N.N.; Grebneva, E.A. Reaction of Phenyltrifluorosilane with 8-Hydroxy- or 8-Mercapto-Quinoline and their Derivatives as a Route to New Heterocyclic Compounds of Pentacoordinate Silicon. Appl. Organomet. Chem. 2005, 19, 538–541. [Google Scholar] [CrossRef]
- Nakash, M.; Gut, D.; Goldvaser, M. Formation of Hypervalent Complexes of Trifluorosilanes with Pyridine and with 4-Methoxypyridine, through Intermolecular Silicon···Nitrogen Interactions. Inorg. Chem. 2005, 44, 1023–1030. [Google Scholar] [CrossRef]
- Kobelt, C.; Burschka, C.; Bertermann, R.; Guerra, C.F.; Bickelhaupt, F.M.; Tacke, R. Synthesis and Structural Characterisation of Neutral Pentacoordinate Silicon(iv) Complexes with a Tridentate Dianionic N,N,S Chelate Ligand. Dalton Trans. 2012, 41, 2148–2162. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.; Panisch, R.; Bowden, A.; Bassindale, A.R.; Taylor, P.G.; Korlyukov, A.A.; Arkhipov, D.E.; Male, L.; Callear, S.; Coles, S.J.; et al. Pentacoordinate Silicon Complexes with Dynamic Motion Resembling a Pendulum on the SN2 Reaction Pathway. Dalton Trans. 2013, 42, 10971–10981. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wu, Q.Z.; Li, Q.Z.; Scheiner, S. Promotion of TH3 (T = Si and Ge) Group Transfer within a Tetrel Bond by a Cation–π Interaction. Phys. Chem. Chem. Phys. 2022, 24, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xie, X.Y.; Li, Q.Z.; Scheiner, S. Enhancement of the Tetrel Bond by the Effects of Substituents, Cooperativity, and Electric Field: Transition from Noncovalent to Covalent Bond. ChemPhysChem 2021, 22, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-Hole Strengths and Interactions of F3MX Molecules (M = C, Si, Ge and X = F, Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef]
- Scheiner, S. Highly Selective Halide Receptors Based on Chalcogen, Pnicogen, and Tetrel Bonds. Chem. Eur. J. 2016, 22, 18850–18858. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative Analysis of Molecular Surfaces: Areas, Volumes, Electrostatic Potentials and Average Local Ionization Energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules; A Quantum Theory; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F.A. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Su, P.F.; Li, H. Energy Decomposition Analysis of Covalent Bonds and Intermolecular Interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Arnold, W.D.; Oldfield, E. The Chemical Nature of Hydrogen Bonding in Proteins via NMR: J-Couplings, Chemical Shifts, and AIM Theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Zhuo, H.Y.; Jiang, L.X.; Li, Q.Z.; Li, W.Z.; Cheng, J.B. Is There an Attractive Interaction between Two Methyl Groups? Chem. Phys. Lett. 2014, 608, 90–94. [Google Scholar] [CrossRef]
- Daolio, A.; Scilabra, P.; Terraneo, G.; Resnati, G. C(sp3) Atoms as Tetrel Bond Donors: A Crystallographic Survey. Coord. Chem. Rev. 2020, 413, 213265. [Google Scholar] [CrossRef]
- Southern, S.A.; West, M.S.; Bradshaw, M.J.Z.; Bryce, D.L. Experimental 13C and 1H Solid-State NMR Response in Weakly Tetrel-Bonded Methyl Groups. J. Phys. Chem. C 2021, 125, 2111–2123. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Asadollahi, S.; Mousavian, P. Anionic Tetrel Bonds: An ab Initio Study. Chem. Phys. Lett. 2018, 691, 394–400. [Google Scholar] [CrossRef]
- Xu, H.L.; Cheng, J.B.; Li, H.B.; Yang, X.; Li, Q.Z. Tetrel bonds between PhSiF3/PhTH3 (T = Si, Ge, Sn) and H3ZO (Z = N, P, As): A Pentacoordinate Silicon (IV) Complex. Int. J. Quantum Chem. 2018, 118, e25660. [Google Scholar] [CrossRef]
- Xu, H.L.; Cheng, J.B.; Yu, X.F.; Li, Q.Z. Abnormal Tetrel Bonds between Formamidine and TH3F: Substituent Effects. ChemistrySelect 2018, 3, 2842–2849. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Implications of Monomer Deformation for Tetrel and Pnicogen Bonds. Phys. Chem. Chem. Phys. 2018, 20, 8832–8841. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M. On the Opposite Tends of Correlations between Interaction Energies and Electrostatic Potentials of Chlorinated and Methylated Amine Complexes Stabilized by Halogen Bond. Theor. Chem. Acc. 2017, 136, 125. [Google Scholar] [CrossRef]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of Halogen Bonding in Solution: Substituent, Structural, and Solvent Effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef]
- An, X.L.; Yang, X.; Li, Q.Z. Tetrel Bonds between Phenyltrifluorosilane and Dimethyl Sulfoxide: Influence of Basis Sets, Substitution and Competition. Molecules 2021, 26, 7231. [Google Scholar] [CrossRef]
- Wei, Y.X.; Li, H.B.; Cheng, J.B.; Li, W.Z.; Li, Q.Z. Prominent Enhancing Effects of Substituents on the Strength of π···σ-Hole Tetrel Bond. Int. J. Quantum Chem. 2017, 117, e25448. [Google Scholar] [CrossRef]
- Wei, Y.X.; Cheng, J.B.; Li, W.Z.; Li, Q.Z. Regulation of Coin Metal Substituents and Cooperativity on the Strength and Nature of Tetrel Bonds. RSC Adv. 2017, 7, 46321–46328. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, Q.Z.; Li, W.Z.; Cheng, J.B. Carbene Tetrel-Bonded Complexes. Struct. Chem. 2017, 28, 823–831. [Google Scholar] [CrossRef]






{kind=link}
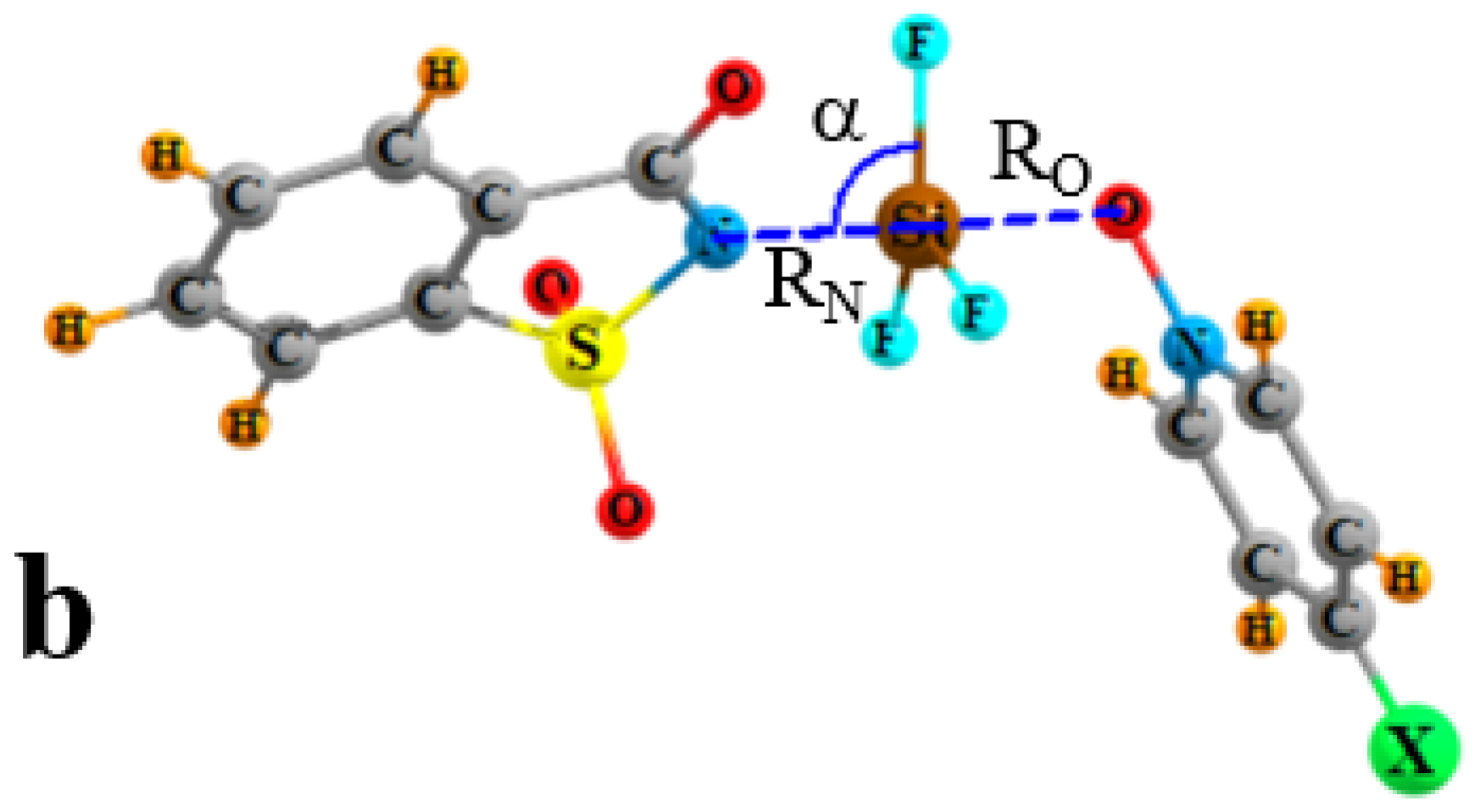{kind=link}
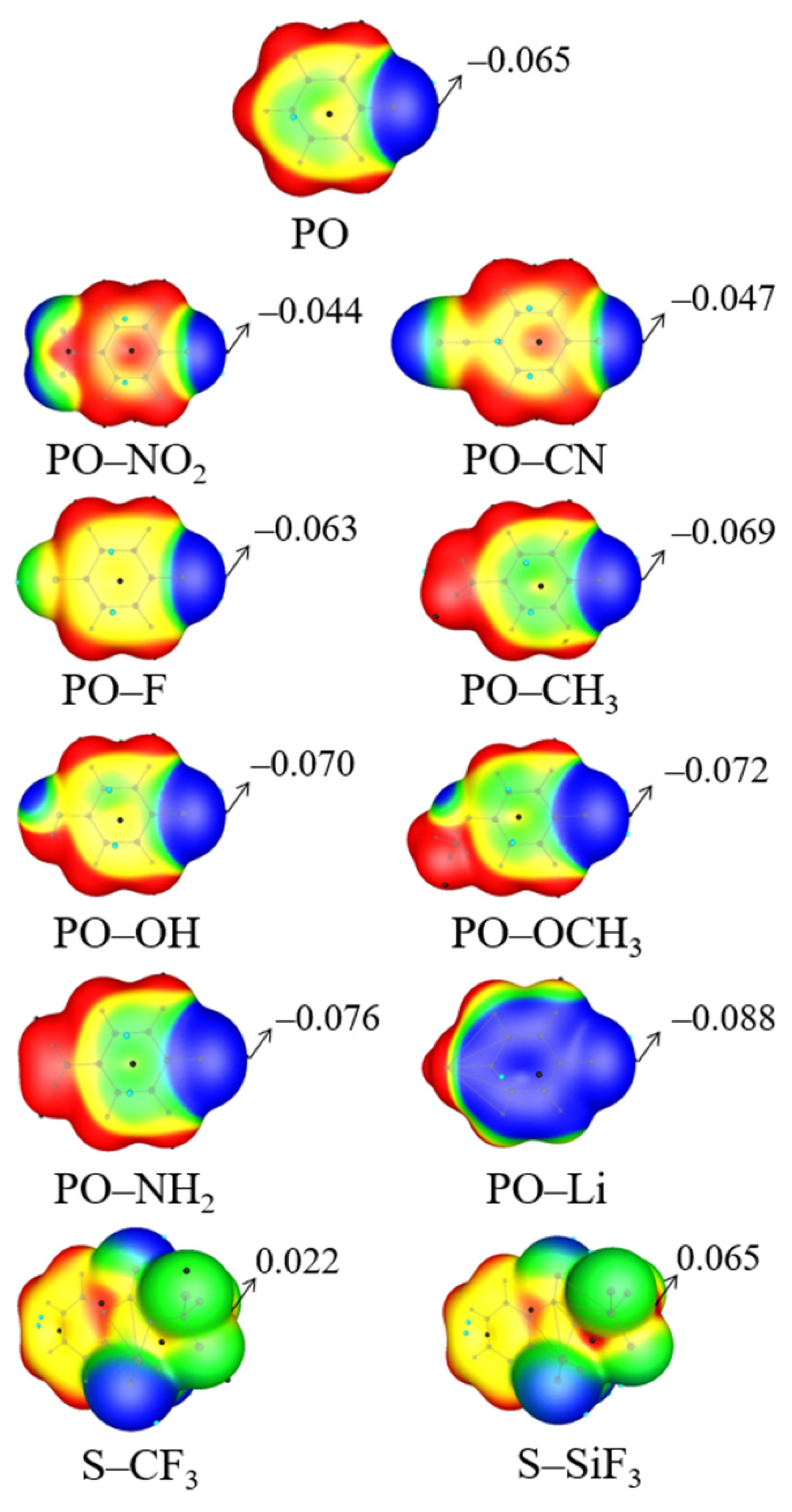{kind=link}
{kind=link}
{kind=link}
| Vmin | RO | RN | ΔRN | RO/RN | γO a | γN a | α | |
|---|---|---|---|---|---|---|---|---|
| S-CF3∙∙∙PO-NO2 | −27.6 | 3.123 | 1.431 | 0.005 | 2.183 | 0.970 | 0.441 | 110.2 |
| S-CF3∙∙∙PO | −40.8 | 3.151 | 1.436 | 0.010 | 2.193 | 0.979 | 0.442 | 109.9 |
| S-CF3∙∙∙PO-CH3 | −43.3 | 3.119 | 1.437 | 0.011 | 2.169 | 0.969 | 0.442 | 109.8 |
| S-CF3∙∙∙PO-OH | −43.9 | 3.115 | 1.437 | 0.011 | 2.169 | 0.967 | 0.442 | 109.8 |
| S-CF3∙∙∙PO-OCH3 | −45.2 | 3.144 | 1.437 | 0.011 | 2.188 | 0.976 | 0.442 | 109.8 |
| S-CF3∙∙∙PO-NH2 | −47.7 | 3.085 | 1.438 | 0.012 | 2.146 | 0.958 | 0.442 | 109.7 |
| S-CF3∙∙∙PO-Li | −55.2 | 3.097 | 1.440 | 0.014 | 2.151 | 0.962 | 0.443 | 109.6 |
| S-SiF3∙∙∙PO-NO2 | −27.6 | 1.902 | 1.819 | 0.080 | 1.045 | 0.526 | 0.499 | 94.3 |
| S-SiF3∙∙∙PO-CN | −29.5 | 1.896 | 1.821 | 0.082 | 1.042 | 0.524 | 0.499 | 94.2 |
| S-SiF3∙∙∙PO-F | −39.5 | 1.874 | 1.829 | 0.090 | 1.025 | 0.518 | 0.501 | 93.5 |
| S-SiF3∙∙∙PO | −40.8 | 1.874 | 1.831 | 0.092 | 1.024 | 0.518 | 0.502 | 93.3 |
| S-SiF3∙∙∙PO-CH3 | −43.3 | 1.866 | 1.834 | 0.095 | 1.017 | 0.515 | 0.502 | 93.1 |
| S-SiF3∙∙∙PO-OH | −43.9 | 1.860 | 1.834 | 0.095 | 1.014 | 0.514 | 0.502 | 93.0 |
| S-SiF3∙∙∙PO-OCH3 | −45.2 | 1.856 | 1.836 | 0.097 | 1.011 | 0.513 | 0.503 | 92.9 |
| S-SiF3∙∙∙PO-NH2 | −47.7 | 1.848 | 1.841 | 0.102 | 1.004 | 0.510 | 0.504 | 91.2 |
| S-SiF3∙∙∙PO-Li | −55.2 | 1.842 | 1.849 | 0.110 | 0.996 | 0.509 | 0.507 | 92.2 |
| Eint | Eb | DE | |
|---|---|---|---|
| S-CF3∙∙∙PO-NO2 | −2.69 | −2.55 | 0.14 |
| S-CF3∙∙∙PO | −2.33 | −2.12 | 0.21 |
| S-CF3∙∙∙PO-CH3 | −2.36 | −2.13 | 0.23 |
| S-CF3∙∙∙PO-OH | −2.63 | −2.35 | 0.28 |
| S-CF3∙∙∙PO-OCH3 | −2.62 | −2.36 | 0.26 |
| S-CF3∙∙∙PO-NH2 | −2.37 | −2.11 | 0.26 |
| S-CF3∙∙∙PO-Li | −2.01 | −1.62 | 0.39 |
| S-SiF3∙∙∙PO-NO2 | −46.59 | −15.12 | 31.47 |
| S-SiF3∙∙∙PO-CN | −48.12 | −16.29 | 31.83 |
| S-SiF3∙∙∙PO-F | −56.59 | −22.39 | 34.20 |
| S-SiF3∙∙∙PO | −56.45 | −21.82 | 34.63 |
| S-SiF3∙∙∙PO-CH3 | −59.29 | −23.78 | 35.51 |
| S-SiF3∙∙∙PO-OH | −61.52 | −25.64 | 35.88 |
| S-SiF3∙∙∙PO-OCH3 | −62.65 | −26.29 | 36.36 |
| S-SiF3∙∙∙PO-NH2 | −66.58 | −28.50 | 38.08 |
| S-SiF3∙∙∙PO-Li | −69.15 | −29.76 | 39.39 |
| T∙∙∙O a | N-T | |||||
|---|---|---|---|---|---|---|
| ρ | ∇2ρ | H | ρ | ∇2ρ | H | |
| S-CF3∙∙∙PO-NO2 | 0.0067 | 0.0372 | 0.0018 | 0.2926 | −0.8536 | −0.3242 |
| S-CF3∙∙∙PO | 0.0072 | 0.0390 | 0.0016 | 0.2894 | −0.8362 | −0.3191 |
| S-CF3∙∙∙PO-CH3 | 0.0074 | 0.0403 | 0.0016 | 0.2885 | −0.8316 | −0.3177 |
| S-CF3∙∙∙PO-OH | 0.0066 | 0.0361 | 0.0018 | 0.2885 | −0.8312 | −0.3176 |
| S-CF3∙∙∙PO-OCH3 | 0.0066 | 0.0357 | 0.0017 | 0.2882 | −0.8294 | −0.3169 |
| S-CF3∙∙∙PO-NH2 | 0.0078 | 0.0423 | 0.0017 | 0.2876 | −0.8271 | −0.3166 |
| S-CF3∙∙∙PO-Li | 0.0071 | 0.0388 | 0.0018 | 0.2853 | −0.8170 | −0.3142 |
| S-SiF3∙∙∙PO-NO2 | 0.0707 | 0.3233 | −0.0162 | 0.1004 | 0.4831 | −0.0279 |
| S-SiF3∙∙∙PO-CN | 0.0716 | 0.3317 | −0.0162 | 0.1000 | 0.4796 | −0.0278 |
| S-SiF3∙∙∙PO-F | 0.0758 | 0.3686 | −0.0163 | 0.0982 | 0.4656 | −0.0272 |
| S-SiF3∙∙∙PO | 0.0756 | 0.3680 | −0.0161 | 0.0976 | 0.4612 | −0.0269 |
| S-SiF3∙∙∙PO-OH | 0.0783 | 0.3917 | −0.0163 | 0.0969 | 0.4551 | −0.0267 |
| S-SiF3∙∙∙PO-NH2 | 0.0808 | 0.4139 | −0.0164 | 0.0955 | 0.4444 | −0.0263 |
| S-SiF3∙∙∙PO-OCH3 | 0.0790 | 0.3990 | −0.0162 | 0.0965 | 0.4524 | −0.0266 |
| S-SiF3∙∙∙PO-NH2 | 0.0808 | 0.4139 | −0.0164 | 0.0955 | 0.4444 | −0.0263 |
| S-SiF3∙∙∙PO-Li | 0.0815 | 0.4247 | −0.0161 | 0.0937 | 0.4308 | −0.0257 |
| CT a | E | |
|---|---|---|
| S-CF3∙∙∙PO-NO2 | 0.0013 | −0.31 |
| S-CF3∙∙∙PO | 0.0009 | −0.36 |
| S-CF3∙∙∙PO-CH3 | 0.0004 | −0.39 |
| S-CF3∙∙∙PO-OH | 0.0001 | −0.39 |
| S-CF3∙∙∙PO-OCH3 | 0.0006 | −0.39 |
| S-CF3∙∙∙PO-NH2 | 0.0005 | −0.44 |
| S-CF3∙∙∙PO-Li | 0.0002 | −0.43 |
| S-SiF3∙∙∙PO-NO2 | 0.1755 | −54.50 |
| S-SiF3∙∙∙PO-CN | 0.1780 | −55.26 |
| S-SiF3∙∙∙PO-F | 0.1900 | −58.50 |
| S-SiF3∙∙∙PO | 0.1907 | −58.67 |
| S-SiF3∙∙∙PO-CH3 | 0.1942 | −60.00 |
| S-SiF3∙∙∙PO-OH | 0.1961 | −60.58 |
| S-SiF3∙∙∙PO-OCH3 | 0.1977 | −61.20 |
| S-SiF3∙∙∙PO-NH2 | 0.2024 | −62.59 |
| S-SiF3∙∙∙PO-Li | 0.2059 | −64.08 |
| Ees | Eex | Erep | Epol | Edisp | ΔEtotal | |
|---|---|---|---|---|---|---|
| S-CF3∙∙∙PO-NO2 | −3.61 | −2.77 | 12.79 | −0.74 | −8.45 | −2.78 |
| S-CF3∙∙∙PO | −3.37 | −3.12 | 13.68 | −0.88 | −8.69 | −2.38 |
| S-CF3∙∙∙PO-CH3 | −3.50 | −3.30 | 14.17 | −1.03 | −8.76 | −2.42 |
| S-CF3∙∙∙PO-OH | −3.70 | −3.23 | 13.92 | −1.04 | −8.68 | −2.73 |
| S-CF3∙∙∙PO-OCH3 | −3.64 | −3.26 | 14.11 | −1.05 | −8.86 | −2.70 |
| S-CF3∙∙∙PO-NH2 | −3.80 | −3.69 | 15.17 | −1.14 | −8.95 | −2.42 |
| S-CF3∙∙∙PO-Li | −2.97 | −3.28 | 13.73 | −1.24 | −8.31 | −2.06 |
| S-SiF3∙∙∙PO-NO2 | −74.86 | −54.07 | 169.62 | −63.03 | −26.97 | −49.31 |
| S-SiF3∙∙∙PO-CN | −76.91 | −54.94 | 172.17 | −64.04 | −27.17 | −50.89 |
| S-SiF3∙∙∙PO-F | −88.66 | −59.35 | 185.02 | −68.96 | −27.67 | −59.62 |
| S-SiF3∙∙∙PO | −87.17 | −58.59 | 182.73 | −68.78 | −27.68 | −59.51 |
| S-SiF3∙∙∙PO-CH3 | −90.66 | −59.97 | 186.83 | −70.93 | −27.75 | −62.48 |
| S-SiF3∙∙∙PO-OH | −94.57 | −61.55 | 191.51 | −72.18 | −27.92 | −64.72 |
| S-SiF3∙∙∙PO-OCH3 | −95.80 | −62.06 | 193.06 | −73.16 | −27.90 | −65.85 |
| S-SiF3∙∙∙PO-NH2 | −100.42 | −63.69 | 197.82 | −75.37 | −28.25 | −69.91 |
| S-SiF3∙∙∙PO-Li | −100.95 | −63.45 | 196.96 | −77.16 | −28.09 | −72.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, Z.; Wu, Q.; Li, Q.; Scheiner, S. C∙∙∙O and Si∙∙∙O Tetrel Bonds: Substituent Effects and Transfer of the SiF3 Group. Int. J. Mol. Sci. 2023, 24, 11884. https://doi.org/10.3390/ijms241511884
Niu Z, Wu Q, Li Q, Scheiner S. C∙∙∙O and Si∙∙∙O Tetrel Bonds: Substituent Effects and Transfer of the SiF3 Group. International Journal of Molecular Sciences. 2023; 24(15):11884. https://doi.org/10.3390/ijms241511884
Chicago/Turabian StyleNiu, Zhihao, Qiaozhuo Wu, Qingzhong Li, and Steve Scheiner. 2023. "C∙∙∙O and Si∙∙∙O Tetrel Bonds: Substituent Effects and Transfer of the SiF3 Group" International Journal of Molecular Sciences 24, no. 15: 11884. https://doi.org/10.3390/ijms241511884
APA StyleNiu, Z., Wu, Q., Li, Q., & Scheiner, S. (2023). C∙∙∙O and Si∙∙∙O Tetrel Bonds: Substituent Effects and Transfer of the SiF3 Group. International Journal of Molecular Sciences, 24(15), 11884. https://doi.org/10.3390/ijms241511884