
Exploring the Potential of a Genome-Reduced Escherichia coli Strain for Plasmid DNA Production



Abstract
1. Introduction
2. Results and Discussion
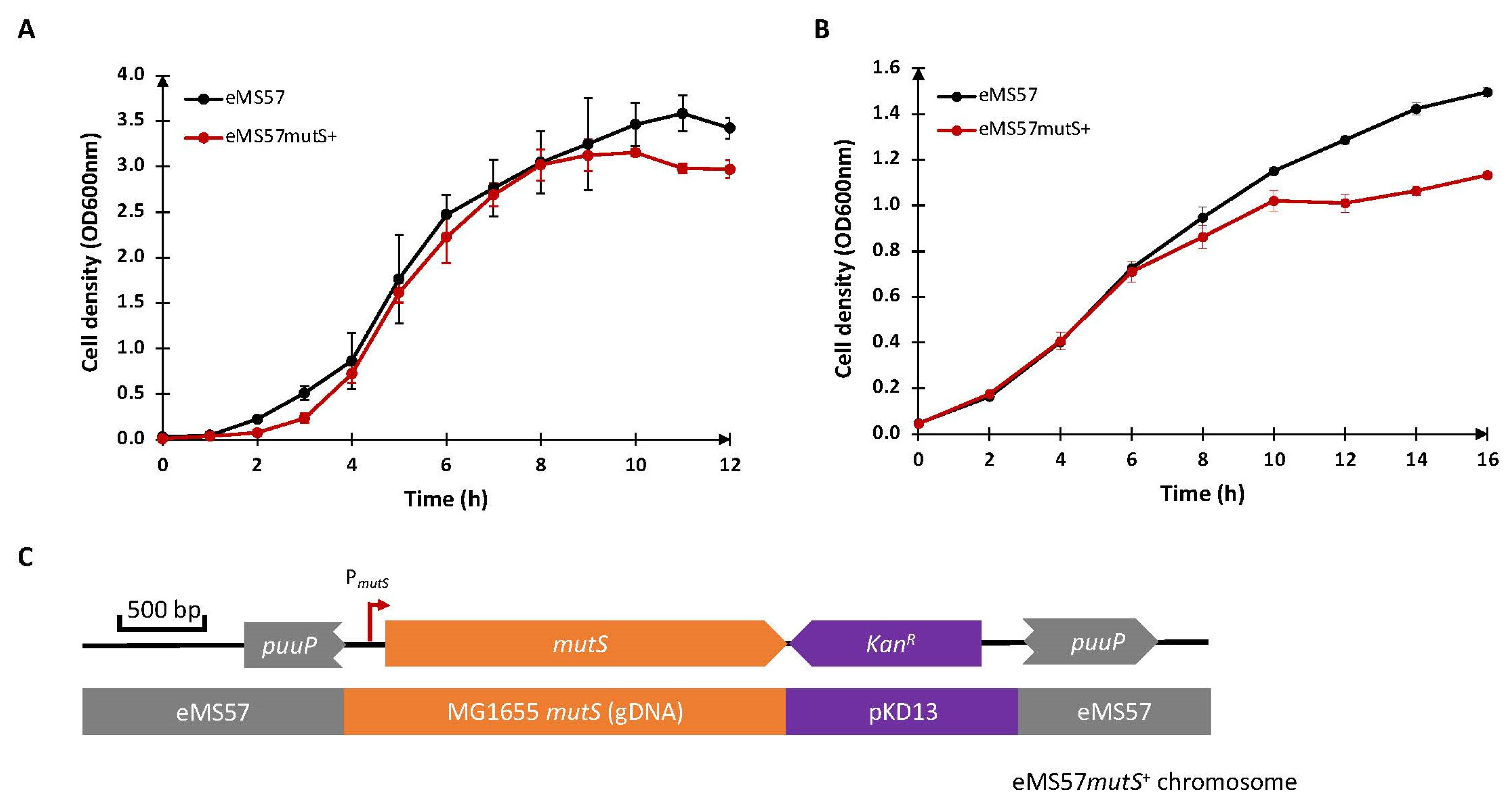
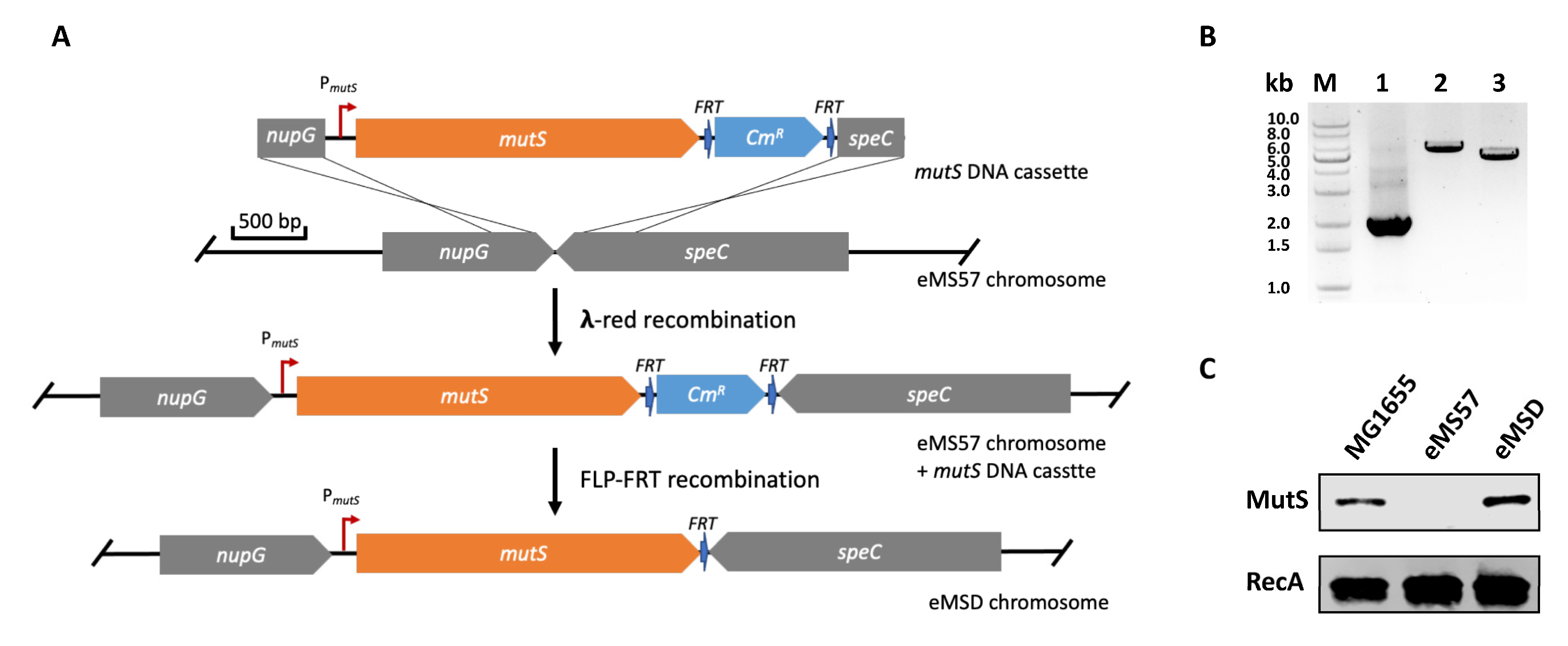
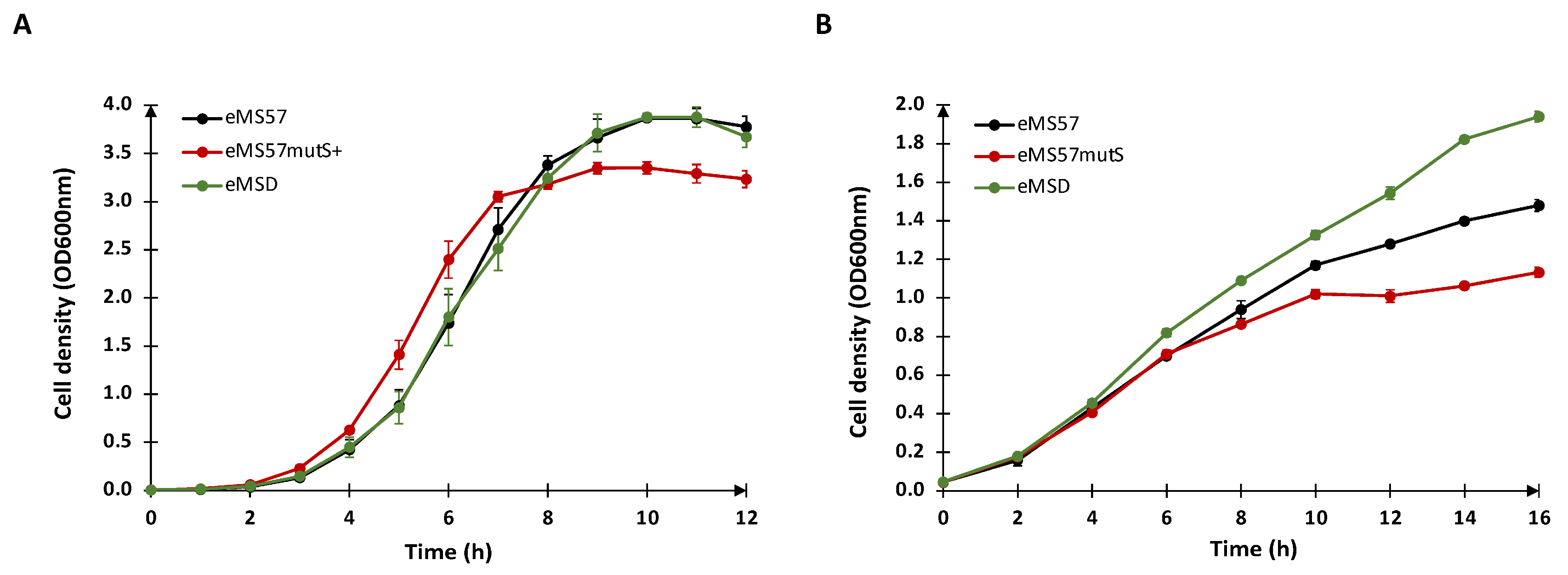
2.1. Re-Introduction of mutS Gene into eMS57 Strain
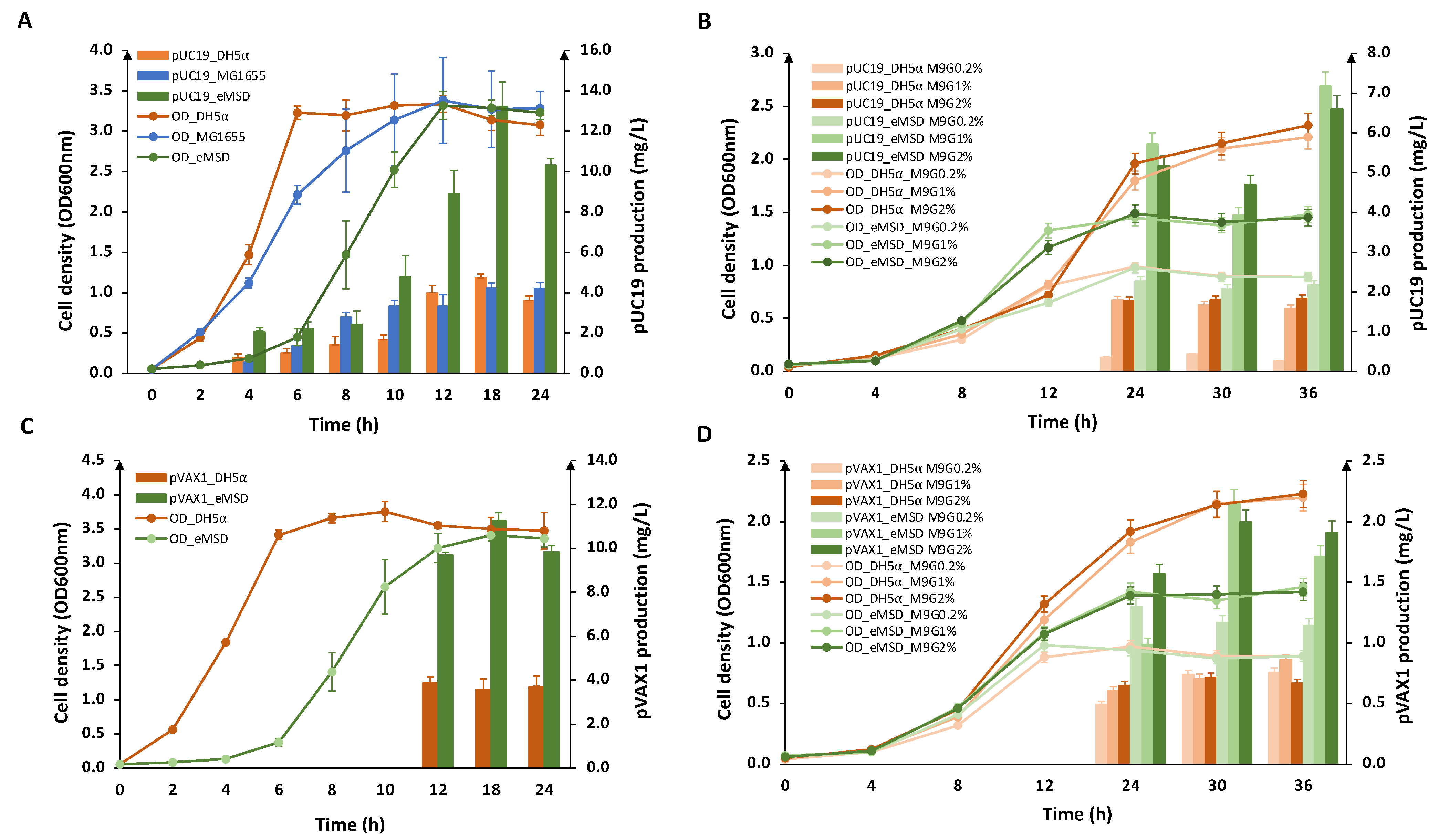
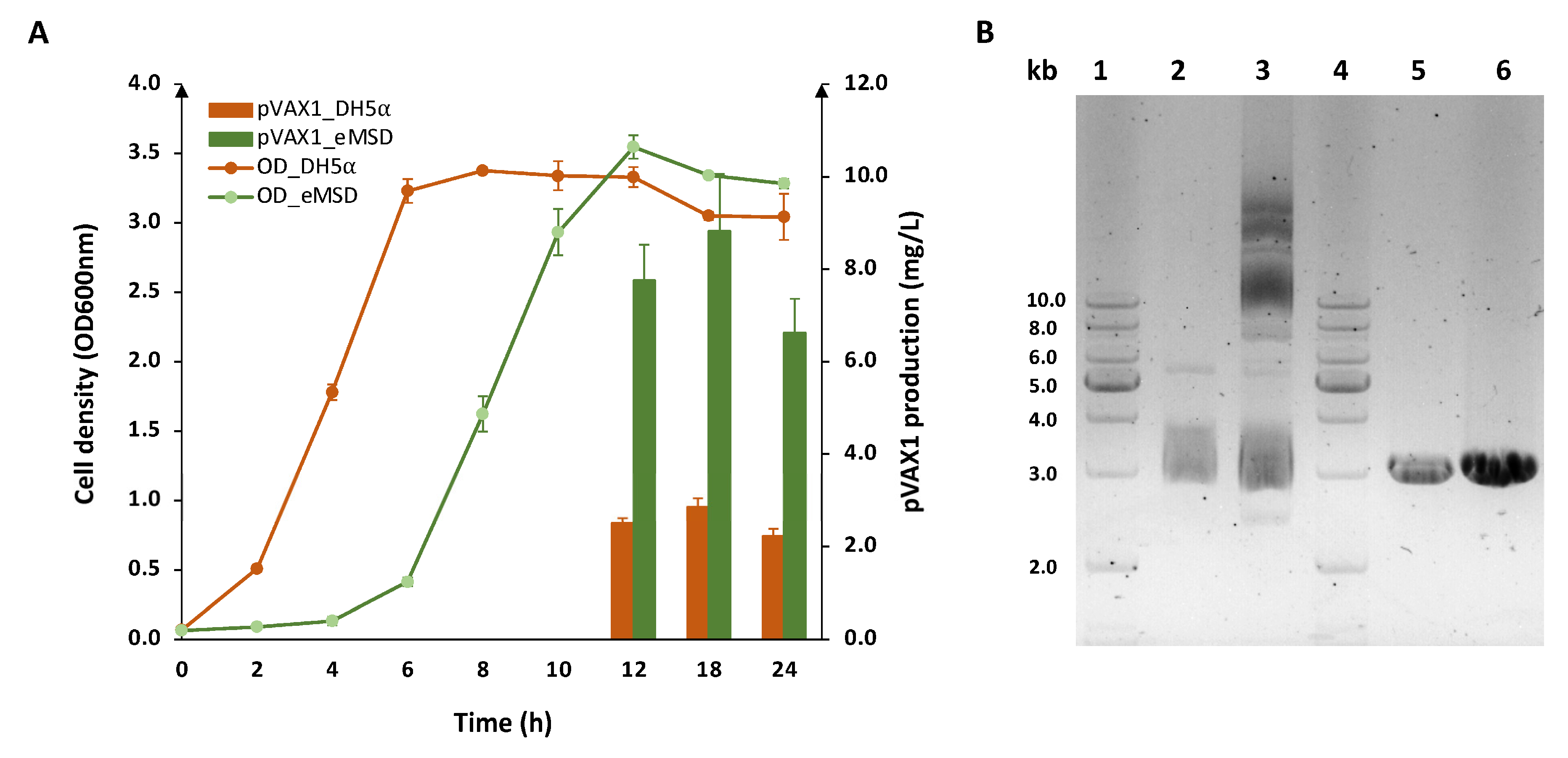
2.2. Evaluation of Plasmid DNA Production in eMSD Strain
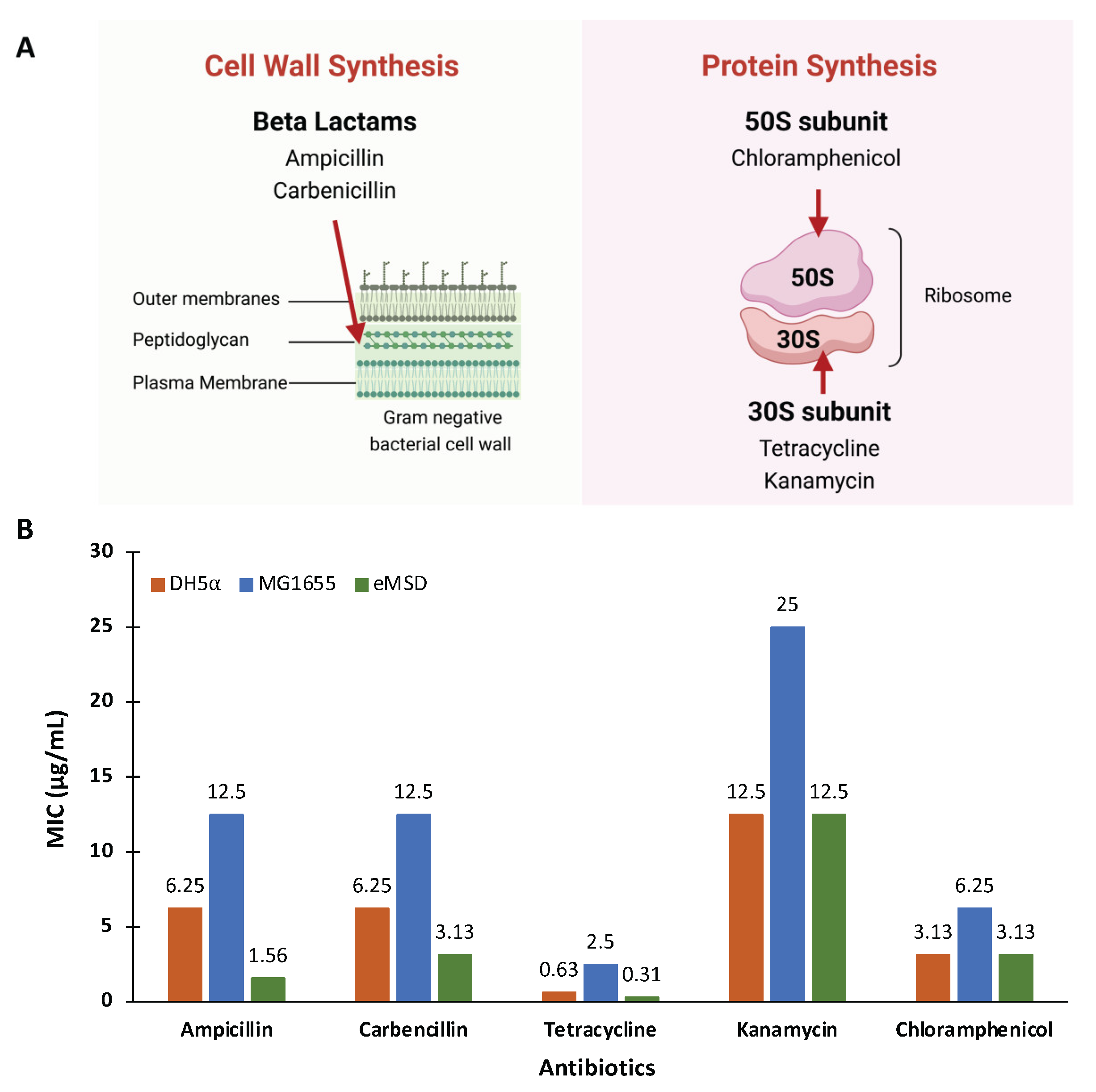
2.3. The Antibiotic Sensitivity of the eMSD Strain
3. Materials and Methods
3.1. Strains, Media, Primers, Enzymes, Chemicals, and Instruments
3.2. Construction of mutS DNA Cassette
3.3. Genome Integration of mutS Gene
3.4. Genome and Plasmid Purification
3.5. Western Blot Analysis
3.6. Cell Cultivations
3.7. Evaluation of Antibiotic Susceptibility by the Minimum Inhibitory Concentration Assay
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. Mrna Vaccines—A New Era in Vaccinology. Nat. Rev. Drug. Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Shafaati, M.; Saidijam, M.; Soleimani, M.; Hazrati, F.; Mirzaei, R.; Amirheidari, B.; Tanzadehpanah, H.; Karampoor, S.; Kazemi, S.; Yavari, B.; et al. A Brief Review on DNA Vaccines in the Era of COVID-19. Future Virol. 2022, 17, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Tavel, J.A.; Martin, J.E.; Kelly, G.G.; Enama, M.E.; Shen, J.M.; Gomez, P.L.; Andrews, C.A.; Koup, R.A.; Bailer, R.T.; Stein, J.A.; et al. Safety and Immunogenicity of a Gag-Pol Candidate Hiv-1 DNA Vaccine Administered by a Needle-Free Device in Hiv-1-Seronegative Subjects. J. Acquir. Immune Defic. Syndr. 2007, 44, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Khobragade, A.; Bhate, S.; Ramaiah, V.; Deshpande, S.; Giri, K.; Phophle, H.; Supe, P.; Godara, I.; Revanna, R.; Nagarkar, R.; et al. Efficacy, Safety, and Immunogenicity of the DNA SARS-CoV-2 Vaccine (Zycov-D): The Interim Efficacy Results of a Phase 3, Randomised, Double-Blind, Placebo-Controlled Study in India. Lancet 2022, 399, 1313–1321. [Google Scholar] [CrossRef]
- Jones, K.P.; Sagar, S.; Murphy, J.; Chartrain, M. Industrial Scale Production of Plasmid DNA for Vaccine and Gene Therapy: Plasmid Design, Production, and Purification. Enzym. Microb. Technol. 2003, 33, 865–883. [Google Scholar]
- Xenopoulos, A.; Pattnai, P. Production and Purification of Plasmid DNA Vaccines: Is There Scope for Further Innovation? Expert. Rev. Vaccines 2014, 13, 1537–1551. [Google Scholar] [CrossRef]
- Ohlson, J. Plasmid Manufacture Is the Bottleneck of the Genetic Medicine Revolution. Drug. Discov. Today 2020, 25, 1891–1893. [Google Scholar] [CrossRef] [PubMed]
- Prather, K.L.; Edmonds, M.C.; Herod, J.W. Identification and Characterization of Is1 Transposition in Plasmid Amplification Mutants of E. coli Clones Producing DNA Vaccines. Appl. Microbiol. Biotechnol. 2006, 73, 815–826. [Google Scholar] [CrossRef]
- Gonçalves, G.A.L.; Bower, D.M.; Prazeres, D.M.F.; Monteiro, G.A.; Prather, K.L.J. Rational Engineering of Escherichia coli Strains for Plasmid Biopharmaceutical Manufacturing. Biotechnol. J. 2011, 7, 251–261. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Prather, K.J.; Prazeres, D.M.; Monteiro, G.A. Structural Instability of Plasmid Biopharmaceuticals: Challenges and Implications. Trends Biotechnol. 2009, 27, 503–511. [Google Scholar] [CrossRef]
- Williams, J.A.; Carnes, A.E.; Hodgson, C.P. Plasmid DNA Vaccine Vector Design: Impact on Efficacy, Safety and Upstream Production. Biotechnol. Adv. 2009, 27, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Borja, G.M.; Mora, E.M.; Barrón, B.; Gosset, G.; Ramírez, O.T.; Lara, A.R. Engineering Escherichia coli to Increase Plasmid DNA Production in High Cell-Density Cultivations in Batch Mode. Microb. Cell. Factories 2012, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Carnes, A.E. Fermentation Design for the Manufacture of Therapeutic Plasmid DNA. BioProcess Int. 2005, 3, 36–44. [Google Scholar]
- Yau, S.Y.; Keshavarz-Moore, E.; Ward, J. Host Strain Influences on Supercoiled Plasmid DNA Production in Escherichia Coli: Implications for Efficient Design of Large-Scale Processes. Biotechnol. Bioeng. 2008, 101, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.; Mendez, J.; Storts, D.R.; Portman, E.; Patterson, B.; Frederiksen, J.; Smith, C. Effects of Bacterial Strains Carrying the Enda1 Genotype on DNA Quality Isolated with Wizard (Tm) Plasmid Purification Systems. Promega Notes 1995, 53, 12–20. [Google Scholar]
- Durfee, T.; Nelson, R.; Baldwin, S.; Plunkett, G., 3rd; Burland, V.; Mau, B.; Petrosino, J.F.; Qin, X.; Muzny, D.M.; Ayele, M.; et al. The Complete Genome Sequence of Escherichia coli Dh10b: Insights into the Biology of a Laboratory Workhorse. J. Bacteriol. 2008, 190, 2597–2606. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Inouye, M. Inactivation of the Serratia marcescens Gene for the Lipoprotein in Escherichia Coli by Insertion Sequences, Is1 and Is5; Sequence Analysis of Junction Points. Mol. Gen. Genet. MGG 1981, 183, 107–114. [Google Scholar] [CrossRef]
- Rawat, P.; Kumar, S.; Pental, D.; Burma, P.K. Inactivation of a Transgene Due to Transposition of Insertion Sequence (Is136) of Agrobacterium tumefaciens. J. Biosci. 2009, 34, 199–202. [Google Scholar] [CrossRef]
- Posfai, G.; Plunkett, G., 3rd; Feher, T.; Frisch, D.; Keil, G.M.; Umenhoffer, K.; Kolisnychenko, V.; Stahl, B.; Sharma, S.S.; de Arruda, M.; et al. Emergent Properties of Reduced-Genome Escherichia coli. Science 2006, 312, 1044–1046. [Google Scholar] [CrossRef]
- Park, M.K.; Lee, S.H.; Yang, K.S.; Jung, S.-C.; Lee, J.H.; Kim, S.C. Enhancing Recombinant Protein Production with an Escherichia coli Host Strain Lacking Insertion Sequences. Appl. Microbiol. Biotechnol. 2014, 98, 6701–6713. [Google Scholar] [CrossRef]
- Chakiath, C.S.; Esposito, D. Improved Recombinational Stability of Lentiviral Expression Vectors Using Reduced-Genome Escherichia coli. Biotechniques 2007, 43, 466–470. [Google Scholar] [CrossRef]
- Choe, D.; Lee, J.H.; Yoo, M.; Hwang, S.; Sung, B.H.; Cho, S.; Palsson, B.; Kim, S.C.; Cho, B.-K. Adaptive Laboratory Evolution of a Genome-Reduced Escherichia coli. Nat. Commun. 2019, 10, 935. [Google Scholar] [CrossRef]
- Cunningham, D.S.; Koepsel, R.R.; Ataai, M.M.; Domach, M.M. Factors Affecting Plasmid Production in Escherichia coli from a Resource Allocation Standpoint. Microb. Cell Factories 2009, 8, 27. [Google Scholar]
- Kurihara, S.; Tsuboi, Y.; Oda, S.; Kim, H.G.; Kumagai, H.; Suzuki, H. The Putrescine Importer Puup of Escherichia coli K-12. J. Bacteriol. 2009, 191, 2776–2782. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-Step Inactivation of Chromosomal Genes in Escherichia coli K-12 Using Pcr Products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Yu, B.J.; Kang, K.H.; Lee, J.H.; Sung, B.H.; Kim, M.S.; Kim, S.C. Rapid and Efficient Construction of Markerless Deletions in the Escherichia coli Genome. Nucleic Acids Res. 2008, 36, e84. [Google Scholar] [CrossRef]
- Bryant, J.A.; Sellars, L.E.; Busby, S.J.W.; Lee, D.J. Chromosome Position Effects on Gene Expression in Escherichia coli K-12. Nucleic Acids Res. 2014, 42, 11383–11392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ding, Q.; Jiang, G.-Z.; Liu, Z.-N.; Wang, H.-Y.; Zhao, G.-R. Chromosome Engineering of Escherichia coli for Constitutive Production of Salvianic Acid A. Microb. Cell Factories 2017, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Phue, J.N.; Lee, S.J.; Trinh, L.; Shiloach, J. Modified Escherichia coli B (Bl21), a Superior Producer of Plasmid DNA Compared with Escherichia coli K (Dh5alpha). Biotechnol. Bioeng. 2008, 101, 831–836. [Google Scholar] [CrossRef]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and Resistance Mechanisms of Antibiotics: A Guide for Clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300–305. [Google Scholar] [CrossRef]
- Seregina, T.A.; Petrushanko, I.Y.; Shakulov, R.S.; Zaripov, P.I.; Makarov, A.A.; Mitkevich, V.A.; Mironov, A.S. The Inactivation of Lps Biosynthesis Genes in E. coli Cells Leads to Oxidative Stress. Cells 2022, 11, 2667. [Google Scholar] [CrossRef] [PubMed]
- Pichon, C.; du Merle, L.; Lequeutre, I.; Le Bouguenec, C. The Afar Small Rna Controls Expression of the Afad-Viii Invasin in Pathogenic Escherichia coli Strains. Nucleic Acids Res. 2013, 41, 5469–5482. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [PubMed]
- Hwang, Y.E.; Im, S.; Kim, H.; Sohn, J.-H.; Cho, B.-K.; Cho, J.H.; Sung, B.H.; Kim, S.C. Adhesive Antimicrobial Peptides Containing 3,4-Dihydroxy-L-Phenylalanine Residues for Direct One-Step Surface Coating. Int. J. Mol. Sci. 2021, 22, 11915. [Google Scholar] [CrossRef]
- Hubbard, A.T.M.; Jafari, N.V.; Feasey, N.; Rohn, J.L.; Roberts, A.P. Effect of Environment on the Evolutionary Trajectories and Growth Characteristics of Antibiotic-Resistant Escherichia coli Mutants. Front. Microbiol. 2019, 10, 2001. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmids | Descriptions | Reference |
| pUC19 | pUC ori; AmpR | Enzynomics |
| pVAX1 | pUC ori; KmR | Invitrogen |
| pACYCDuet-1 | p15A ori; CmR | Novagen |
| pCP22 | FLP expression vector; AmpR | [32] |
| pREDI | Vector expresses λ-Red protein and I-SceI endonuclease; AmpR | [26] |
| pUC19-nupG | For construction of mutS DNA cassette; AmpR | This study |
| pUC19-nupG-FRT-CmR1 | For construction of mutS DNA cassette; AmpR | This study |
| pUC19-nupG-FRT-CmR-FRT-speC | For construction of mutS DNA cassette; AmpR; CmR | This study |
| pUC19-nupG-mutS-FRT-CmR-FRT-speC | Vector contains mutS DNA cassette; AmpR; CmR | This study |
| Strains | Descriptions | Reference |
| XL1-Blue | Stratagene | |
| DH5α | Enzynomics | |
| MG1655 | E. coli K-12 strain | [33] |
| eMS57 | Adaptive evolution strain in M9 glucose medium | [22] |
| eMS57mutS+ | eMS57, puuP::mutS-kanR | [22] |
| eMSD | Reintroduction of mutS at the locus between nupG and speC in the eMS57 genome | This study |
| Name | Sequence (5′-3′) |
| XhoI_mutS_F | CGCTCGAGCCAACCGATACAATTTTGCGT |
| mutS_EcoRI_R | GCGAATTCGTTATTACACCAGGCTCTTCAAGC |
| nupG_out_F | GCCAACGCTTGGGTTAATCAACAC |
| speC_out_R | CCGCTGTTTGCTGCACTGGATG |
| XbanupGF | GACTCTAGAATCTGACCATCCCGTTCTTCTTAAG |
| nupGXENNR | GACCATATGCCATGGGAATTCCTCGAGCGGTAAAAAAAACGGGTCACCTTCTGGC |
| EcoRIFRTCmR1F | GTAACGAATTCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCCTTTTGGCGAAAATGAGACGTTGATCGGC |
| CmR1NcoR | TGCCCATGGTGAAAACGGGGGCG |
| CmR2NcoF | GTTTTCACCATGGGCAAATATTATACGC |
| CmR2FRTR | GCGAGCCAACGGCGGCCCGCAAAAAAGAAGTTCCTATACTTTCTAGAGAATAGGAACTTCGGAATAGGAACTTCTTACGCCCCGCCCTGCCACTCATCG |
| oopspeCF | TTTTTTGCGGGCCGCCGTTGGCTCGCTTCTTACTTCAACACATAACCGTACAACC |
| SpeCNdeR | CACCATATGGTGTTGCCGAGCGTTTATAACAAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.; Bui, L.M.; Byun, J.-Y.; Cho, B.-K.; Kim, S.C. Exploring the Potential of a Genome-Reduced Escherichia coli Strain for Plasmid DNA Production. Int. J. Mol. Sci. 2023, 24, 11749. https://doi.org/10.3390/ijms241411749
Nguyen TT, Bui LM, Byun J-Y, Cho B-K, Kim SC. Exploring the Potential of a Genome-Reduced Escherichia coli Strain for Plasmid DNA Production. International Journal of Molecular Sciences. 2023; 24(14):11749. https://doi.org/10.3390/ijms241411749
Chicago/Turabian StyleNguyen, Thi Thuy, Le Minh Bui, Ji-Young Byun, Byung-Kwan Cho, and Sun Chang Kim. 2023. "Exploring the Potential of a Genome-Reduced Escherichia coli Strain for Plasmid DNA Production" International Journal of Molecular Sciences 24, no. 14: 11749. https://doi.org/10.3390/ijms241411749
APA StyleNguyen, T. T., Bui, L. M., Byun, J.-Y., Cho, B.-K., & Kim, S. C. (2023). Exploring the Potential of a Genome-Reduced Escherichia coli Strain for Plasmid DNA Production. International Journal of Molecular Sciences, 24(14), 11749. https://doi.org/10.3390/ijms241411749