
Chromosome-Level Genome Assembly and Population Genomic Analyses Reveal Geographic Variation and Population Genetic Structure of Prunus tenella



Abstract
1. Introduction
2. Results
2.1. De Novo Assembly of the P. tenella Genome
2.2. Functional Annonations, Gene Prediction, and Repetitive Sequences
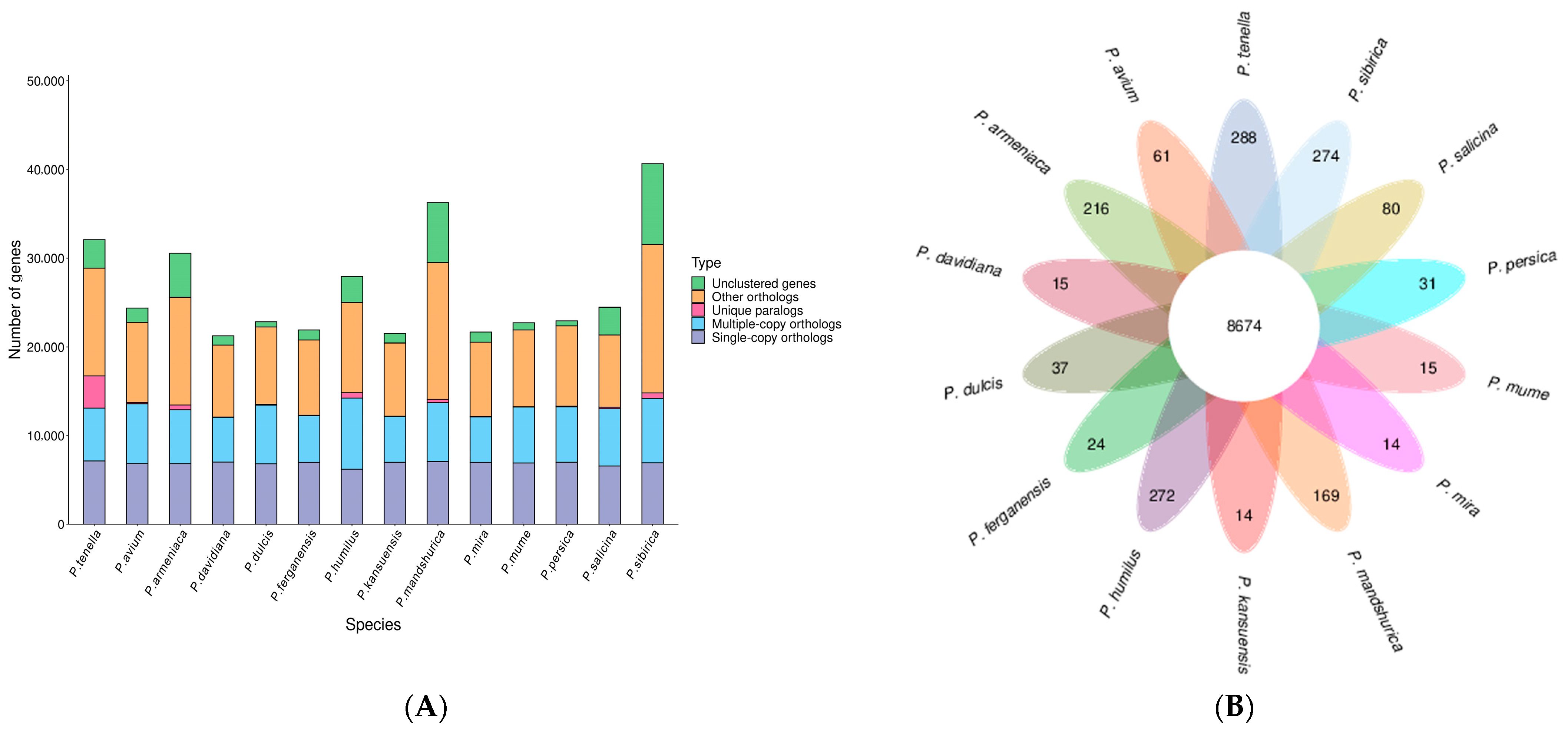
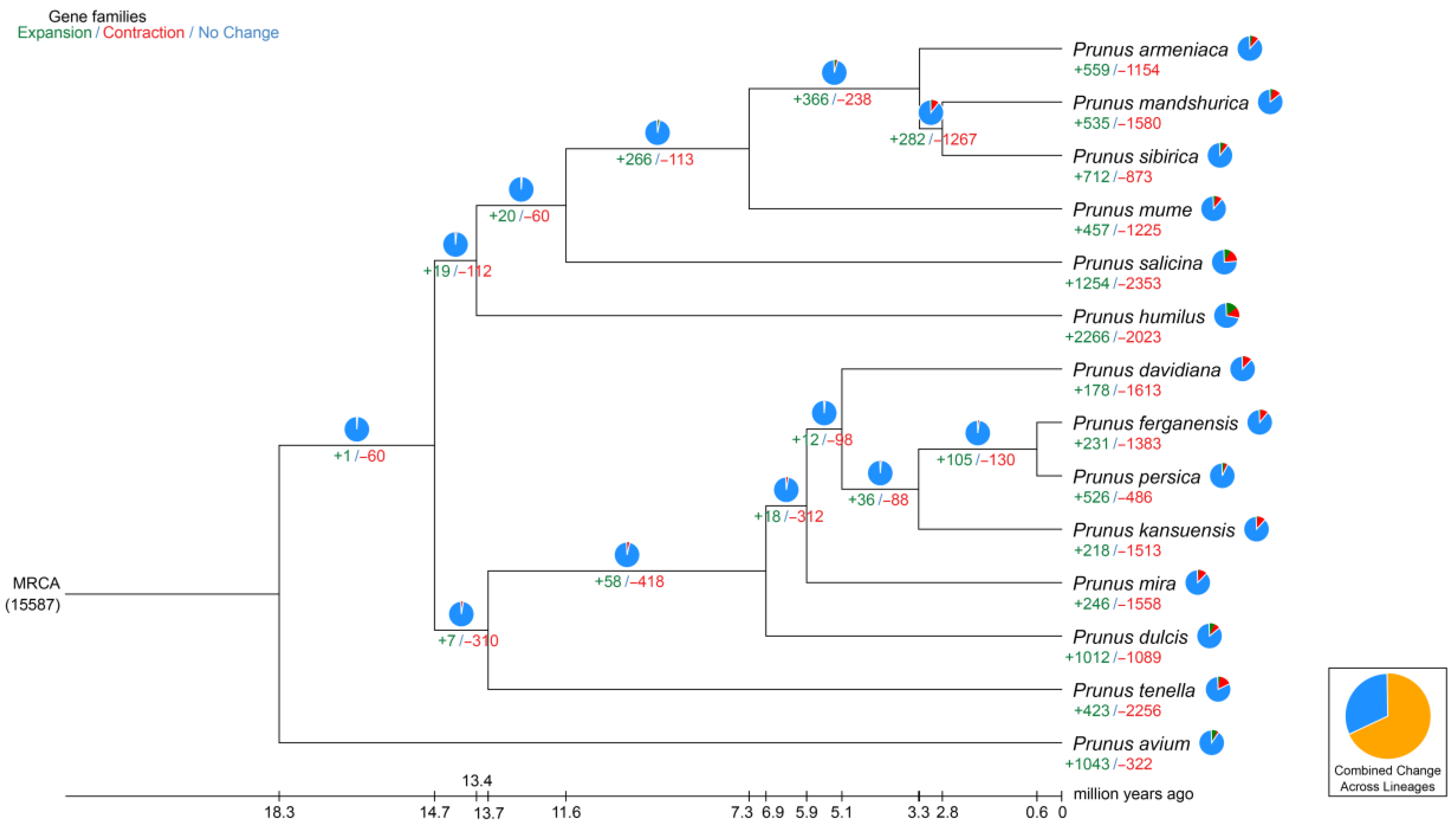
2.3. Synteny Analysis, and Genome Evolution and Phylogeny
2.4. Population Genetic Structure and Genetic Diversity Analysis
2.5. Genome-Wide Selection Signatures’ Analysis of Differentiation
3. Discussion
4. Materials and Methods
4.1. Utilized Materials
4.2. Genome Sequencing and Transcriptome Sequencing
4.3. Assurance of Sequencing Data Quality
4.4. Heterozygosity and Genome Size Estimation
4.5. Genome Assembly
4.6. Repetitive Element Annotations
4.7. Functional Annonations and Gene Prediction
4.8. Phylogenetic and Gene Family Analysis
4.9. Whole-Genome Synteny Analysis
4.10. Single-Nucleotide Polymorphism (SNP) Calling
4.11. Phylogenetic Analysis
4.12. Population Genetic Structure and Genetic Diversity Analysis
4.13. Selective Sweep Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Zeng, B.; Luo, S.P.; Li, H.L.; Madaniyati, W. Protection and propagation of Amygdalus ledebouriana Schleche in China. Xinjiang Agric. Sci. 2006, 43, 61–62. [Google Scholar]
- Yin, L.K.; Tan, L.X.; Wang, B. Rare Endangered Endemic Higher Plants in Xinjiang of China; Urumqi Xinjiang Science & Technology Publishing House: Urumqi, China, 2006. [Google Scholar]
- Zhong, H.X.; Lu, C.S.; Luo, S.P.; Li, J. The study of cold resistance test of dormancy branches and buds of Amygdalus ledebouriana Schleche in Xinjiang. Xinjiang Agric. Sci. 2016, 53, 120–125. [Google Scholar]
- Perazzolli, M.; Malacarne, G.; Baldo, A.; Righetti, L.; Bailey, A.; Fontana, P.; Velasco, R.; Malnoy, M. Characterization of resistance gene analogues (RGAs) in apple (Malus × domestica Borkh.) and their evolutionary history of the Rosaceae family. PLoS ONE 2014, 9, e83844. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, B.; Elias, M.; Azimov, R.; Turdieva, M.; Aaliev, S.; Bobokalonov, F.; Butkov, E.; Kaparova, E.; Mukhsimov, N.; Shamuradova, S.; et al. Home gardens of Central Asia: Reservoirs of diversity of fruit and nut tree species. PLoS ONE 2022, 17, e0271398. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Singh, C.; Ambika; Chandana, B.S.; Mahto, R.K.; Patial, R.; Gupta, A.; Gahlaut, V.; Gayacharan; Hamwieh, A.; et al. Exploring Chickpea Germplasm Diversity for Broadening the Genetic Base Utilizing Genomic Resourses. Front. Genet. 2022, 13, 905771. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jacob, S.R.; Mir, R.R.; Vikas, V.K.; Kulwal, P.; Chandra, T.; Kaur, S.; Kumar, U.; Kumar, S.; Sharma, S.; et al. Indian Wheat Genomics Initiative for Harnessing the Potential of Wheat Germplasm Resources for Breeding Disease-Resistant, Nutrient-Dense, and Climate-Resilient Cultivars. Front. Genet. 2022, 13, 834366. [Google Scholar] [CrossRef]
- Kefale, H.; Wang, L. Discovering favorable genes, QTLs, and genotypes as a genetic resource for sesame (Sesamum indicum L.) improvement. Front. Genet. 2022, 13, 1002182. [Google Scholar] [CrossRef]
- García-Gómez, B.E.; Salazar, J.A.; Nicolás-Almansa, M.; Razi, M.; Rubio, M.; Ruiz, D.; Martínez-Gómez, P. Molecular Bases of Fruit Quality in Prunus Species: An Integrated Genomic, Transcriptomic, and Metabolic Review with a Breeding Perspective. Int. J. Mol. Sci. 2020, 22, 333. [Google Scholar] [CrossRef]
- Filip, E.; Woronko, K.; Stępień, E.; Czarniecka, N. An Overview of Factors Affecting the Functional Quality of Common Wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2023, 24, 7524. [Google Scholar] [CrossRef]
- Li, Z.; Xue, Y.; Zhou, H.; Li, Y.; Usman, B.; Jiao, X.; Wang, X.; Liu, F.; Qin, B.; Li, R.; et al. High-resolution mapping and breeding application of a novel brown planthopper resistance gene derived from wild rice (Oryza. rufipogon Griff). Rice 2019, 12, 41. [Google Scholar] [CrossRef]
- Mamidi, S.; Healey, A.; Huang, P.; Grimwood, J.; Jenkins, J.; Barry, K.; Sreedasyam, A.; Lovell, J.T.; Feldman, M.; Wu, J.; et al. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 2020, 38, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Prahalada, G.D.; Shivakumar, N.; Lohithaswa, H.C.; Sidde Gowda, D.K.; Ramkumar, G.; Kim, S.R.; Ramachandra, C.; Hittalmani, S.; Mohapatra, T.; Jena, K.K. Identification and fine mapping of a new gene, BPH31 conferring resistance to brown planthopper biotype 4 of India to improve rice, Oryza sativa L. Rice 2017, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Laugerotte, J.; Baumann, U.; Sourdille, P. Genetic control of compatibility in crosses between wheat and its wild or cultivated relatives. Plant Biotechnol. J. 2022, 20, 812–832. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Schulthess, A.W.; Bassi, F.M.; Badaeva, E.D.; Neumann, K.; Graner, A.; Özkan, H.; Werner, P.; Knüpffer, H.; Kilian, B. Introducing Beneficial Alleles from Plant Genetic Resources into the Wheat Germplasm. Biology 2021, 10, 982. [Google Scholar] [CrossRef]
- Aleem, M.; Aleem, S.; Sharif, I.; Aleem, M.; Shahzad, R.; Khan, M.I.; Batool, A.; Sarwar, G.; Farooq, J.; Iqbal, A.; et al. Whole-Genome Identification of APX and CAT Gene Families in Cultivated and Wild Soybeans and Their Regulatory Function in Plant Development and Stress Response. Antioxidants 2022, 11, 1626. [Google Scholar] [CrossRef]
- Mk, A.; Shw, B.; Ss, A. Wheat wild germplasm: A hidden treasure. Wild Germplasm Genet. Improv. Crop Plants 2011, 2021, 55–63. [Google Scholar]
- Yumurtaci, A. Utilization of wild relatives of wheat, barley, maize and oat in developing abiotic and biotic stress tolerant new varieties. Emir. J. Food Agric. 2015, 27, 1. [Google Scholar] [CrossRef]
- Haus, M.J.; Pierz, L.D.; Jacobs, J.L.; Wiersma, A.T.; Cichy, K.E. Preliminary evaluation of wild bean (Phaseolus spp.) germplasm for resistance to Fusarium cuneirostrum and Fusarium oxysporum. Crop Sci. 2021, 61, 3264–3274. [Google Scholar] [CrossRef]
- Rostad, H.E.; Reen, R.A.; Mumford, M.H.; Zwart, R.; Thompson, J.P. Resistance to root-lesion nematode Pratylenchus neglectus identified in a new collection of two wild chickpea species (Cicer reticulatum and C. echinospermum) from turkey. Plant Pathol. 2022, 5, 71. [Google Scholar] [CrossRef]
- Jeff, E.; Olumide, S.T.; Bruce, D.; Andre, H.; Julianne, A.; Olufemi, A. Resistance in wild macadamia germplasm to Phytophthora cinnamomi and Phytophthora multivora. Ann. Appl. Biol. 2021, 178, 519–526. [Google Scholar] [CrossRef]
- Wang, B.; Yu, Z.F.; Zeng, B.; Xia, J.H.; Ma, X.X. Self-incompatibility Gene Cullin1 Cloning and Bioinformatics Analysis of Wild Almond in Xinjiang. Chin. Agric. Sci. Bull. 2017, 33, 63–68. [Google Scholar]
- Zeng, B.; Liu, N.N.; Xia, J.H.; Liu, M.W.; Wang, J.Y.; Wang, B. Molecular Cloning and Bioinformatics Analysis of SFB Genes Controlling Self-incompatibility in Xinjiang Wild Almond (Prunus tenella Batsch.). Chin. Agric. Sci. Bull. 2017, 33, 22–30. [Google Scholar]
- Yu, Z.F.; Wang, B.; Zeng, B.; Wang, J.Y. Cloning and sequence analysis of self—Incompatibility gene SBPI of wild almond in Xinjiang. Mol. Plant Breed. 2018, 16, 6955–6960. [Google Scholar]
- Zeng, B.; Li, J.; Luo, S.P.; Cheng, Y.J. Identification of Genetic Relationship of Amygdalus Plants by SSR. Xinjiang Agric. Sci. 2009, 46, 18–22. [Google Scholar]
- Lu, Z.J.; Li, J.; Omir, S.T.; Zeng, B.; Luo, S.P. ISSR analysis for genetic diversity of Amygdalus ledebouriana germplasm from Xinjiang, China. J. Fruit Sci. 2010, 27, 918–923. [Google Scholar]
- Chen, D.X.; Pan, Y.; Wang, Y.; Cui, Y.Z.; Li, L.Y. The chromosome-level reference genome of coptis chinensis provides insights into genomic evolution and berberine biosynthesis. Hortic. Res. 2021, 8, 11. [Google Scholar] [CrossRef]
- Rush, D.W.; Epstein, E. Breeding and selection for salt tolerance by the incorporation of wild germplasm into a domestic tomato. J. Am. Soc. Hortic. Sci. 1981, 106, 699–704. [Google Scholar] [CrossRef]
- D’Amico-Willman, K.M.; Ouma, W.Z.; Meulia, T.; Sideli, G.M.; Gradziel, T.M.; Fresnedo-Ramírez, J. Whole-genome sequence and methylome profiling of the almond (Prunus dulcis [mill.] d.a.webb) cultivar ‘nonpareil’. G3 Genes Genomes Genet. 2022, 12, jkac065. [Google Scholar] [CrossRef]
- Liu, J.F.; Wei, H.; Zhang, X.; Wang, D. Chromosome-level genome assembly and hazelomics database construction provides insights into unsaturated fatty acid synthesis and cold resistance in hazelnut (Corylus heterophylla). Front. Plant Sci. 2021, 12, 766548. [Google Scholar] [CrossRef]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Rokhsar, D.S. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution-Supplementary Information. Nat. Genet. 2014, 45, 486–495. [Google Scholar]
- Suguiyama, V.F.; Vasconcelos, L.A.B.; Rossi, M.M.; Biondo, C.; de Setta, N. The population genetic structure approach adds new insights into the evolution of plant LTR retrotransposon lineages. PLoS ONE 2019, 14, e0214542. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H. Decades-long phylogeographic issues: Complex historical processes and ecological factors on genetic structure of alpine plants in the Japanese Archipelago. J. Plant Res. 2022, 135, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Mori, G.M.; Zucchi, M.I.; Souza, A.P. Multiple-geographic-scale genetic structure of two mangrove tree species: The roles of mating system, hybridization, limited dispersal and extrinsic factors. PLoS ONE 2015, 10, e0118710. [Google Scholar] [CrossRef]
- Nishio, S.; Takada, N.; Terakami, S.; Takeuchi, Y.; Kimura, M.K.; Isoda, K.; Saito, T.; Iketani, H. Genetic structure analysis of cultivated and wild chestnut populations reveals gene flow from cultivars to natural stands. Sci. Rep. 2021, 11, 240. [Google Scholar] [CrossRef]
- Deb, S.; Della Lucia, M.C.; Ravi, S.; Bertoldo, G.; Stevanato, P. Transcriptome-Assisted SNP Marker Discovery for Phytophthora infestans Resistance in Solanum lycopersicum L. Int. J. Mol. Sci. 2023, 24, 6798. [Google Scholar] [CrossRef]
- Bali, S.; Robinson, B.R.; Sathuvalli, V.; Bamberg, J.; Goyer, A. Single Nucleotide Polymorphism (SNP) markers associated with high folate content in wild potato species. PLoS ONE 2018, 13, e0193415. [Google Scholar] [CrossRef]
- Roncallo, P.F.; Beaufort, V.; Larsen, A.O.; Dreisigacker, S.; Echenique, V. Genetic diversity and linkage disequilibrium using SNP (KASP) and AFLP markers in a worldwide durum wheat (Triticum turgidum L. var durum) collection. PLoS ONE 2019, 14, e0218562. [Google Scholar] [CrossRef]
- Castilla, A.R.; Méndez-Vigo, B.; Marcer, A.; Martínez-Minaya, J.; Conesa, D.; Picó, F.X.; Alonso-Blanco, C. Ecological, genetic and evolutionary drivers of regional genetic differentiation in Arabidopsis thaliana. BMC Evol. Biol. 2020, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Oh, A.; Oh, B.U. Genetic differentiation that is exceptionally high and unexpectedly sensitive to geographic distance in the absence of gene flow: Insights from the genus Eranthis in East Asian regions. Ecol. Evol. 2022, 12, e9007. [Google Scholar] [CrossRef]
- Santangelo, J.S.; Johnson, M.T.J.; Ness, R.W. Modern spandrels: The roles of genetic drift, gene flow and natural selection in the evolution of parallel clines. Proceedings. Biol. Sci. 2018, 285, 20180230. [Google Scholar] [CrossRef]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Robinson, J.; Shamim, S.; Aiden, E.L. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Loannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Doreen, W.; Thomas, P.; Ning, J.; Candice, N.H.; Hufford, M.B. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Cold Spring Harb. Lab. 2019, 1, 275–281. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Tempel, S. Using and Understanding Repeat Masker. Methods Mol. Biol. 2012, 859, 29–51. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, J.; Hannick, L.I.; Rama, M.; Ronning, C.M.; Rusch, D.B.; Town, C.D. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.; Pertea, M.; Salzberg, S. TigrScan and Glimmer HMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.D.; Handsaker, R.E.; Pulit, S.L.; Nizzari, M.M.; O’donnell, C.J.; De Bakker, P.I. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 2008, 24, 2938–2939. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using evidence modeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. Ortho Finder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Bio. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Han, M.V.; Thomas, G.W.C.; Lugo-Martinez, J.; Hahn, M.W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0:a toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Marais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLOS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, K.; Glen, S.; Michael, L.; Christina, K.; Koichiro, T. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Vilella, A.J.; Blanco-Garcia, A.; Hutter, S.; Rozas, J. VariScan: Analysis of evolutionary patterns from large-scale DNA sequence polymorphism data. Bioinformatics 2005, 21, 2791–2793. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Contig Number | Contig Size (bp) | Scaffold Number | Scaffold Size (bp) |
|---|---|---|---|---|
| N90 | 5 | 270,515 | 9 | 1,233,122 |
| N80 | 12 | 8,938,573 | 7 | 19,226,261 |
| N70 | 11 | 10,302,541 | 6 | 21,538,263 |
| N60 | 15 | 14,010,243 | 5 | 23,141,640 |
| N50 | 1 | 18,100,976 | 4 | 25,637,364 |
| Max length (bp) | 35,886,393 | 44,825,466 | ||
| Total size (bp) | 231,191,648 | 231,208,648 | ||
| Total number | 513 | 479 | ||
| Average length | 450,665.98 | 482,690.29 | ||
| Number ≥ 10 kb | 513 | 479 |
| Chr ID | Length (bp) |
|---|---|
| Chr1 | 44,825,466 |
| Chr2 | 29,024,987 |
| Chr3 | 26,387,986 |
| Chr4 | 25,637,364 |
| Chr5 | 23,141,640 |
| Chr6 | 21,538,263 |
| Chr7 | 19,226,261 |
| Chr8 | 17,664,456 |
| Total chromosome level scaffold length | 207,446,423 |
| Total scaffold length | 231,208,648 |
| Chromosome length/total length | 89.7% |
| Library | Eudicotyledons_odb10 |
|---|---|
| Fragmented BUSCOs (F) | 48 |
| Missing BUSCOs (M) | 64 |
| Complete and duplicated BUSCOs (D) | 42 |
| Complete and single-copy BUSCOs (S) | 1967 |
| Complete BUSCOs (C) | 2009 |
| Total BUSCO groups searched | 2121 |
| Summary | 94.7% |
| Class | Length (bp) | Type | Sub-Class | (%) |
|---|---|---|---|---|
| Retrotransposons | 8,454,829 | Ty1/Copia | LTR | 3.66% |
| 18,247,107 | Ty3/Gypsy | 7.89% | ||
| 15,756,928 | unknown | 6.82% | ||
| - | LINE | Non-LTR | - | |
| - | Unknown | - | ||
| DNA transposons | 1,845,596 | CACTA | TIR | 0.80% |
| 7,818,414 | Mutator | 3.38% | ||
| 4,181,524 | PIF/Harbinger | 1.81% | ||
| 278,071 | Tc1/Mariner | 0.12% | ||
| 3,345,229 | hAT | 1.45% | ||
| 7,038,992 | Helitron | Non-TIR | 3.04% | |
| Total | 66,966,690 | 28.97% |
| Database | Gene Numbers | (%) |
|---|---|---|
| GO | 10,761 | 33.54 |
| KEGG | 11,435 | 35.64 |
| KOG | 19,251 | 59.99 |
| Swissprot | 19,449 | 60.61 |
| Pfam annotation | 22,815 | 71.1 |
| Nr annotation | 31,202 | 97.24 |
| No. | Population | Location | Latitude | Longitude |
|---|---|---|---|---|
| 1 | Yumin 1 | Yumin county | 45.89251967° E | 82.52098163° N |
| 2 | Yumin 2 | Yumin county | 45.90577291° E | 82.51545937° N |
| 3 | Yumin 3 | Yumin county | 45.91605329° E | 82.50676816° N |
| 4 | Tuoli 1 | Tuoli county | 46.15794124° E | 83.56189391° N |
| 5 | Tuoli 2 | Tuoli county | 46.15213502° E | 83.53771499° N |
| 6 | Tuoli 3 | Tuoli county | 46.13930633° E | 83.55344239° N |
| 7 | Tuoli 4 | Tuoli county | 46.14667251° E | 83.57180015° N |
| 8 | Tacheng 1 | Tahcheng City | 46.95030549° E | 83.20683721° N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, Y.; Zhao, H.; Han, H.; Zhu, G.; Wang, Z.; Li, F. Chromosome-Level Genome Assembly and Population Genomic Analyses Reveal Geographic Variation and Population Genetic Structure of Prunus tenella. Int. J. Mol. Sci. 2023, 24, 11735. https://doi.org/10.3390/ijms241411735
Qin Y, Zhao H, Han H, Zhu G, Wang Z, Li F. Chromosome-Level Genome Assembly and Population Genomic Analyses Reveal Geographic Variation and Population Genetic Structure of Prunus tenella. International Journal of Molecular Sciences. 2023; 24(14):11735. https://doi.org/10.3390/ijms241411735
Chicago/Turabian StyleQin, Yue, Han Zhao, Hongwei Han, Gaopu Zhu, Zhaoshan Wang, and Fangdong Li. 2023. "Chromosome-Level Genome Assembly and Population Genomic Analyses Reveal Geographic Variation and Population Genetic Structure of Prunus tenella" International Journal of Molecular Sciences 24, no. 14: 11735. https://doi.org/10.3390/ijms241411735
APA StyleQin, Y., Zhao, H., Han, H., Zhu, G., Wang, Z., & Li, F. (2023). Chromosome-Level Genome Assembly and Population Genomic Analyses Reveal Geographic Variation and Population Genetic Structure of Prunus tenella. International Journal of Molecular Sciences, 24(14), 11735. https://doi.org/10.3390/ijms241411735